

Abstract
In previous study, we synthesized a novel combi-molecule, JDF-12, with superior cytotoxicity against prostate cancer cells, but it has a poor stability in liquid after preparation with traditional method and is susceptible to hydrolysis and binding to organs highly expressing epidermal growth factor receptor (EGFR), resulting in side effects. In this study, the nanotechnology was employed to prepare JDF-12 aiming to increase its anti-tumor effect and reduce its systemic side effects. The JDF-12 loaded nanoparticles were formulated with biocompatible and biodegradable poly (D,L-lactic-co-glycolic acid)-block-poly(ethyleneglycol) (PLGA-b-PEG) copolymer and surface functionalized with a single-chain antibody that recognizes the extracellular domain of prostate stem cell antigen (PSCA), enabling a controlled release, “stealth” property, and cell-specific targeting. The targeted nanoparticles exhibited a sustained drug release in vitro and were specifically endocytosed by prostate cancer cells though the receptor-mediated endocytosis resulting in enhanced cellular toxicity in vitro. Moreover, a better outcome with reduced drug toxicity was observed in a PC3M xenograft animal model after treatment with these nanoparticles. Our results demonstrate the feasibility of nanoparticle-based technology in the development of pharmaceutically suboptimal chemotherapeutics.
Keywords: Prostate cancer, epidermal growth factor receptor, nanoparticle
Introduction
Prostate cancer (PCa) is the most common malignancy in men, and has been the second cause of cancer related death [1]. Although the early-stage prostate cancer may be successfully treated by surgery and/or radiotherapy, the therapeutic options for hormone refractory prostate cancer (HRPC) are very limited [2]. Drug resistance and target deficiency are the challenges in the treatment of HRPC. Our previous study reported that combi-molecule, JDF-12, could specifically bind to the epidermal growth factor receptor (EGFR) which is highly expressed in PCa, block the EGFR-mediated signaling pathway, and further decompose into another EGFR tyrosine kinase (TK) inhibitor and a DNA alkylating moiety (mitozolomide, MTZ); the later two exerted synergistic anti-tumor effect on HRPC [3]. JDF12 has a more potent anti-tumor effect on the PCa as compared to currently used drugs, but its clinical application is compromised by its hydrophobic nature, lack of target selectivity and severe side effects [3].
Nanotechnology has been gained increasing attention in the cancer chemotherapies because it is able to improve the solubility and pharmacokinetics of hydrophobic drugs, increase the therapeutic efficacy, lower the drug toxicity, overcome the multi-drug resistance, and maintain a relatively high drug concentration in the tumor [4-7]. The nanotechnology is expected to revisit pharmaceutically suboptimal but biologically active new molecular entities, which were previously considered undevelopable by conventional approaches. After systematic administration, the drug-loaded nanoparticles (NPs) circulating in the vascular system extravasate out of the vasculature and accumulate in the tumor tissues due to its passive targeting, which takes advantage of the so-called enhanced permeability and retention (EPR) effect [8]. Their retention and uptake by cancer cells are further facilitated by its active targeting, which refers to the active moieties (for example, monoclonal antibodies, aptamer, ligand, etc) on the surface of nanoparticles directing against the tumor-related molecules and triggering the receptor-mediated endocytosis [9]. By passive and active targeting, the drug reaching the tumor significantly increases, and systemic toxicity reduces [10].
In our previous study, our results demonstrated that PC3M cells expressed prostate stem cell antigen (PSCA) on the cell membrane and could internalize the single-chain PSCA conjugated nanoparticles through the scAbPSCA-mediated endocytosis mechanism [11]. In this study, core-shell delivery system was prepared by integrating three distinct functional components, a core constituted by poly(D,L-lactic-co-glycolic acid) (PLGA) and JDF-12, a “stealth” shell formed by poly(ethylene glycol) (PEG) molecules and a single-chain antibody conjugated to the nanoparticle surface for targeted delivery. We hypothesized that the targeted nanoparticles could be specifically endocytosed by PCa cells though the receptor-mediated endocytosis, resulting in a increased cellular cytotoxicity.
Materials and methods
Synthesis of PLGA-PEG-NH2 block copolymer
A carboxylic acid terminal group of PLGA was activated by using (N,N’-dicyclohexylcarbodiimide) DCC and (N-hydroxysuccinimide)NHS, and reacted with PEG-bis-amine to prepare the PLGA-PEG-NH2 as previously described [10].
Development of PEG-PLGA/JDF-12 NPs
JDF-12 was synthesized according to the method previously described in our study [3]. In cell experiment, JDF-12 was dissolved in dimethyl sulfoxide (DMSO) and diluted with sterile RPMI-1640 containing 10% fetal bovine serum (FBS). For NP preparation, the drug was dissolved in DMSO, giving a final concentration of 5 mg/ml.
JDF12-encapsulated NPs were prepared by using a nanoprecipitation method. In brief, 100 mg of PLGA-PEG-NH2 was dissolved in 5 ml of (tetrahydrofuran) THF, and JDF-12 in DMSO was added. The mixture was homogenized and dropped into water containing 0.2% (polyvinyl alcohol) PVA under mechanical stirring. The NPs were stirred overnight at room temperature to allow the organic solvent to evaporate. The suspension was centrifuged at 1200 rpm to remove the agglomerates. The NPs were harvested by centrifugal ultrafiltration with a molecular weight cut-off of 50 kDa and then re-suspended for use.
Synthesis of scAb-PEG3400-COOH
Single chain antibody (scAb) bearing free sulphydryl groups were prepared according to previously reported [11]. Then, scAb-PEG3400-COOH was synthesized by conjugating scAb with free sulphydryl groups to mal-PEG-COOH [11]. The scAb-functionalized PEG (scAb-PEG-COOH) was purified through a Sepharose CL-4B column (GE Healthcare UK Limited, Buckinghamshire, UK).
Preparation of scAb-PEG-PLGA/JDF-12 NPs
scAb-PEG-COOH aqueous solution was mixed with 50 mg/ml EDC and 50 mg/ml NHS. After incubation for 20 min at room temperature, 0.5 ml of PEG-PLGA/JDF-12 NPs (3.2 mg/ml) was added and incubated overnight at 4°C after homogenization. The scAb-PEG-PLGA/JDF-12 NPs were purified through a filter with a 100 kDa molecular weight cutoff (Millipore, MA). scAb-PEG-PLGA and PEG-PLGA/JDF-12 NPs were prepared in a similar manner and used as a control. scAb-PEG-PLGA/coumarin, PEG-PLGA/coumarin, PEG-PLGA/rodamine, scAb-PEG-PLGA/rodamine NPs were synthesized by using coumarin or rodamine instead of JDF-12. The scAb on the NP surface was confirmed by flow cytometry, confocal laser scanning microscopy (CLSM) and protein assay as described previously [11].
NP characterization
The particle size and surface charge of NPs were evaluated by Quasi-elastic laser light scattering (QELS) with a ZetaPALS dynamic light-scattering detector (laser at 15 mW and incident beam at 676 nm; Brookhaven Instruments, Holtsville, NY).
The morphological examination of NPs was performed by transmission electron microscope (TEM) (JEOL JEM-200CX) at an acceleration voltage of 200 kV. A dispersion of NPs was dropped onto a carbon-coated copper grid, dried in air at room temperature, and imaged within 24 h.
To determine the JDF-12 content, lyophilized NPs were dissolved in acetonitrile and underwent a constant shaking at room temperature before the addition of methanol. After centrifugation, the JDF-12 content in the supernatant was measured by high-pressure liquid chromatography (HPLC; Agilent 1100, Palo Alto, CA) with a pentafluorophenyl column (Curosil-PFP, 250×4.6 mm, 5 μm, Phenomenex, Torrance, CA) with a non-gradient mobile phase of water and acetonitrile (v/v 50/50) at a constant flow rate of 1 mL/min. Absorbance was measured at 315 nm to determine the JDF12 content using a previously established calibration.
Characterization of release kinetics
JDF-12 loaded NPs were aliquoted and then added into a series of semipermeable dialysis, minidialysis tubes (100 μl in volume, molecular weight cut-off of 10 kD, Pierce) which were then sealed in 40 L of PBS at 37°C with gentle shaking to mimic the infinite sink condition. At a predetermined time point, a fraction of NPs samples was collected from the dialysis tubes. The JDF-12 remaining in the NPs at each time point was extracted using acetonitrile and quantified by reverse-phase HPLC with a C18 column, using a linear gradient of acetonitrile and water eluents.
Growth inhibition assay
Growth inhibitory activities were evaluated by the sulforhodamine B (SRB) assay as previously described [3]. PC3M cells were seeded in 96-well plates (2.5×103 cells/well) and incubated for 24 h prior to drug addition. The cells were then treated with free JDF-12, PEG-PLGA/JDF-12 and scAb-PEG-PLGA/JDF-12 at different concentrations (0.75-200 μM) for 2 h. Then, these cells were washed with PBS thrice and incubated for another 5 days. The culture medium without addition of above drugs served as a control. The concentration of a drug at which the growth of 50% (IC50) of cells was inhibited was determined by nonlinear regression analysis (sigmoidal dose-response curve).
NP endocytosis in vitro
For the detection of NPs uptake, PC3M cells were seeded into 6-well plates (5×104 cells/well) followed by incubation for 24 h. The cells were rinsed twice with PBS (pH 7.2) and pre-incubated at 37°C for 1 h in serum-free medium. The medium was then refreshed with 2 ml of medium containing void-NPs (scAb-PEG-PLGA), PEG-PLGA/rodamine or scAb-PEG-PLGA/rodamine at 5 μg/mL. After incubation for 2 h, cells were washed with PBS five times, fixed in 4% paraformaldehyde for 20 min. The nuclei were stained with DAPI, and cells were imaged under a confocal microscope (LSM 510 Meta, Carl Zeiss, Gottingen, Germany). Fluorescence intensity was analyzed by using Image J 1.43.
Intracellular uptake of NPs
Detection of intracellular uptake of NPs was conducted as previously reported [12]. In brief, PC3M cells were seeded into 6-well plates (2×105 cells/well). Then, cells were washed with pre-warmed PBS before the addition of PEG-PLGA/JDF-12 or scAb-PEG-PLGA/JDF-12 (equivalent to 20 μM JDF-12). To examine the specificity of scAb-mediated uptake, PC3M cells were incubated with excess free PSCA monoclonal antibodies before incubation with scAb-PEG-PLGA/JDF-12. After incubation for 2 h, cells were washed six times with PBS, collected, and lysed in 100 μL of cell lysis buffer for 10 min. An aliquot was used for the determination of total protein (bicinchoninic acid protein assay kit, Pierce, Rockford, IL). JDF-12 in the remaining cell suspension was extracted by acetonitrile and then quantified by HPLC after centrifugation at 14,000 rpm for 10 min using a previously established calibration. The data were normalized to per milligram cell protein.
In vivo antitumor study
The detection of in vivo antitumor activity was conducted as previously reported [3]. In brief, dose finding was done with 5 mice per group, and the maximum tolerated dose (MTD) was defined as the dose at which the drug failed to induce >15% weight loss in at least 14 days. For the establishment of xenograft animal model, PC3M cells (3×105) were suspended in media and matrigel at 1:1 and then inoculated into the flank of male 8-week old BALB/c nude mice. Treatment was initiated when the tumors reached 50 mm3 in volume. Each formulation was prepared, quantified and diluted so that 100 μL of drug solution was equivalent to 100 mg/kg JDF-12. Tumor-bearing mice were treated by tail vein injection of PBS, scAb-PEG-PLGA, JDF-12, PEG-PLGA/JDF-12 or scAb-PEG-PLGA/JDF-12 six times every five days (a total of 600 mg JDF-12/kg body weight) (n=10 per group). Animals were killed at predesigned time points, and tumors were collected. Simultaneously, 1 ml of blood was collected from the orbital sinus and analyzed for a toxicity profile of the treatment regimens.
Statistical analysis
One-way ANOVA with Fisher’s LSD post hoc comparisons at 95% confidence interval (CI) was used for statistical comparisons.
Results
Preparation of scAb-PEG-PLGA/JDF-12 NPs
A schematic diagram of the scAb-PEG-PLGA/JDF-12 preparation is shown in Figure 1. To construct the biomaterials that can self-assemble into NPs, a diblock copolymer PLGA-PEG-NH2 consisting of PLGA-COOH and PEG-bis-amine was synthesized. Then, nanoprecipitation method was employed to encapsulate the hydrophobic JDF-12. In the aqueous solution, the hydrophobic PLGA provided a biodegradable matrix for the encapsulation of JDF-12 while an amine-terminated hydrophilic PEG of the diblock copolymer was oriented toward the aqueous medium to form the antibiofouling coat of NPs. For the conjugation of targeting moiety to the surface of PEG-PLGA/JDF-12 NPs, the antibody was first pretreated with 2-mercaptoethylamine to yield scAb-bearing free sulfhydryl groups, and then scAb was conjugated with mal-PEG3400-COOH in aqueous solution. The resulting scAb-PEG3400-COOH was linked to the amine terminal on the surface of PEG-PLGA/JDF-12 NPs in aqueous medium which endows NPs the targeting capability.
Figure 1.
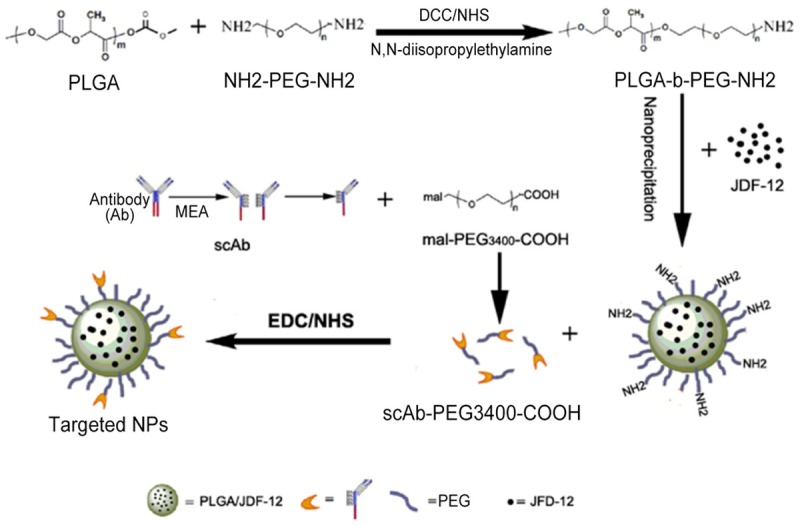
Schematic diagram of the preparation of targeted nanoparticles.
Determination of scAb on the scAb-PEG-PLGA/JDF-12NPs
scAb on the NP surface was evaluated by FCM, CLSM and protein assay. Compared to the PEG-PLGA/JDF-12 NPs, a substantial shift of PE fluorescence was demonstrated in the scAb-PEG-PLGA/JDF-12, indicating that NPs were scAb-coated (Figure 2A). Moreover, the binding of scAb to the NPs surface was also confirmed by CLSM. As shown in Figure 2B and 2C, scAb-PEG-PLGA/coumarin showed merged red/green fluorescence while PEG-PLGA/coumarin only showed green fluorescence, indicating the presence of scAb on the NPs surface. The protein assay was used to quantify the amount of scAb binding to the NPs surface. According to the protein assay, the amount of scAb conjugated to the NPs surface was approximately 22.6±4.7 μg scAb/mg NPs.
Figure 2.
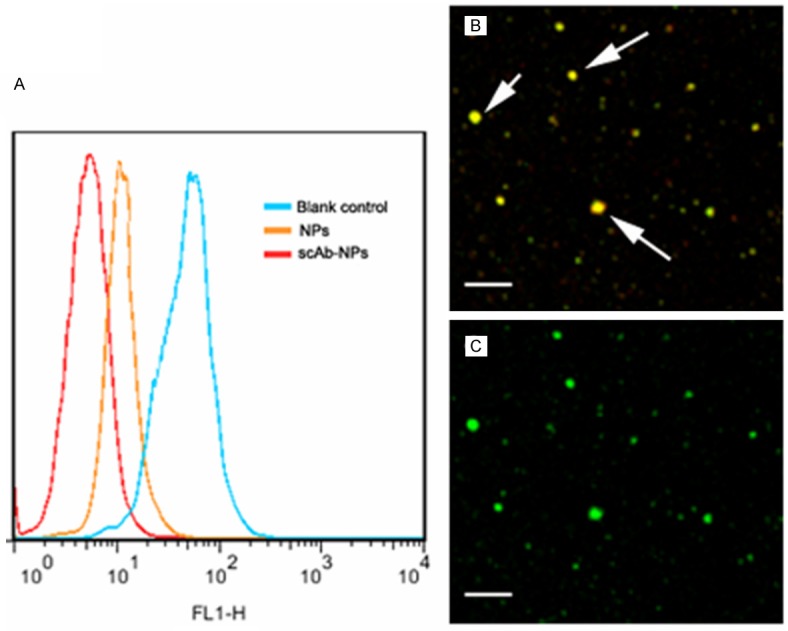
Determination of scAb on the nanoparticle surface. (A) Significant shift of PE fluorescence intensity was observed for scAb-PEG-PLGA/JDF-12 (scAb-NPs) as compared to blank control and PEG-PLGA/JDF-12 (NPs), indicating the presence of scAb on the nanoparticle surface. Confocal microscopic images of scAb-PEG-PLGA/coumarin showed merged red/green fluorescence (B); However, PEG-PLGA/coumarin showed only green fluorescence (C).
Biophysicochemical characteristics
As shown in Figure 3, the hydrodynamic particle size of scAb-PEG-PLGA/JDF-12 was 152.2± 38.4 nm in PBS when measured with the dynamic laser light scattering technique. The scAb-PEG-PLGA/JDF-12 exhibited a negative zeta potential of -16.8±2.7 mV, which contributed to the dispersion. SEM and TEM were used to examine the morphology of scAb-PEG-PLGA/JDF-12 NPs. As shown in Figure 3B and 3C, the ultrastructure was similar to a biological cell (a nuclear core was surrounded by a hydrophilic shell). Drug loading efficacy plays an important role in the drug delivery system and directly affects the therapeutic effects of the system. The JDF-12 loading of scAb-PEG-PLGA/JDF-12 NPs was 5.16±1.03% w/w.
Figure 3.
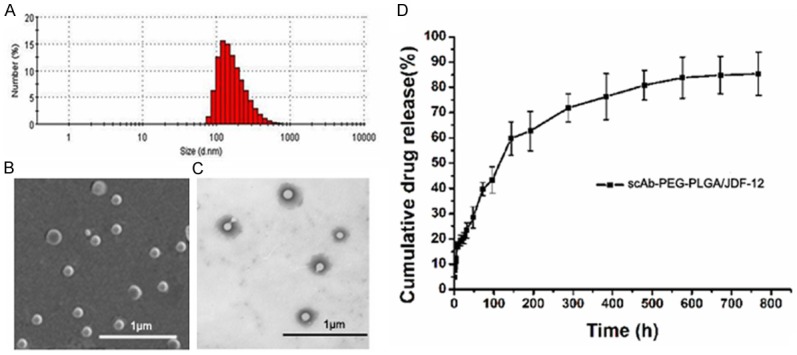
A. Hydrodynamic particle size of scAb-PEG-PLGA/JDF-12 NPs measured using dynamic laser light scattering. B. Representative scanning electron microscopic image of scAb-PEG-PLGA/JDF-12 NPs; C. Representative transmission electron microscopic image of scAb-PEG-PLGA/JDF-12 NPs; D. Kinetics of physicochemical release showed the controlled release of JDF-12.
The in vitro release of JDF-12 was determined as the percentage of JDF-12 released to the total JDF-12 loaded in NPs. A rapid release of JDF-12 was observed with significant release (17.5%) within 8 h. Subsequently, JDF-12 release became relatively rapid (62.8% cumulative release of JDF-12 within 8 days) followed by a slow and continuous release (85.4% cumulative release of JDF-12 within 32 days).
Growth inhibitory activity
The in vitro cytotoxic activity of scAb-PEG-PLGA/JDF-12, PEG-PLGA/JDF-12, JDF-12 and Void-NPs (scAb-PLGA-PEG) was evaluated by SRB assay in PC3M cells. PC3M cells were incubated with scAb-PEG-PLGA/JDF-12, PEG-PLGA/JDF-12, JDF-12 and Void-NPs for 2 h for the specific particle uptake, followed by further incubation in medium for a total of 5 days before the measurement of cell viability by SRB assay. To eliminate to possibility that the NPs were cytotoxic, the scAb-PLGA-PEG at equivalent dose served as a control. The range of JDF-12 concentration was designed according to previously reported [3].
As shown in Figure 4B, the inhibitory potency of scAb-PEG-PLGA/JDF-12 was 2.43-fold greater than that of PEG-PLGA/JDF-12 in PC3M cells. Moreover, scAb-PEG-PLGA/JDF-12 showed a greater inhibitory capability than free JDF-12.
Figure 4.
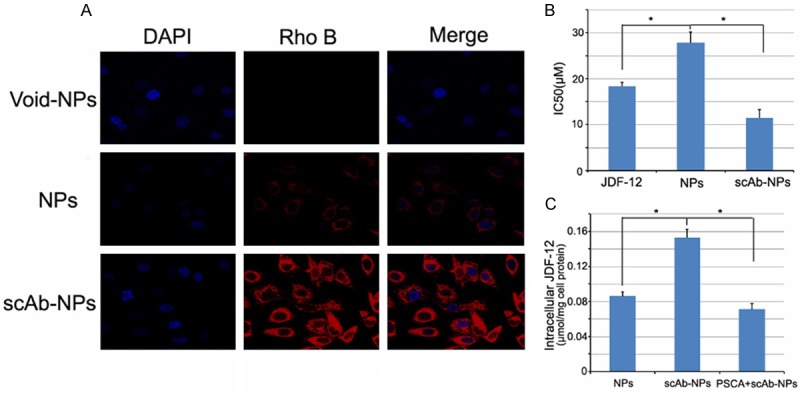
A. Confocal laser scanning microscopy of PC3M cells after 2-h incubation with scAb-PEG-PLGA/rodamine NPs, PEG-PLGA/rodamine and void NPs. The nucleus was stained with DAPI. B. IC50 of JDF-12, PEG-PLGA/JDF-12, and scAb-PEG-PLGA/JDF-12 in PC3M cells. Data are expressed as means ± standard error (n = 3). C. Intracellular drug concentration of PC3M cells treated with PEG-PLGA/JDF-12, scAb-PEG-PLGA/JDF-12 and scAb-PEG-PLGA/JDF-12 plus saturated free antibodies. Data are expressed as mean ± standard error (n = 4). *P<0.05.
Since JDF-12 was a hydrophobic and membrane-permeable drug, free JDF-12 could rapidly penetrate the cell membrane to exert effects. However, the scAb-PEG-PLGA/JDF-12 NPs were inactive and had to be endocytosized by cells in which free drug was released to inhibit the cell proliferation. Our results showed scAb-PLGA-PEG had no significant cytotoxic effect on PC3M cells.
Cell-specific endocytosis and intracellular JDF-12 concentration
CLSM was used to evaluate the targeting effect of scAb-PEG-PLGA/JDF-12 on the cellular uptake and to examine differential endocytosis by PC3M cells. The NPs were visualized by the encapsulating red fluorescent dye, rodamine, instead of JDF-12. As shown in Figure 4A, the red fluorescence was in the cytosol and surrounded the nuclei. The fluorescence intensity of scAb-PEG-PLGA/rodamine was significantly higher than that of PEG-PLGA rodamine in PC3M cells after 2-h incubation, confirming a relatively rapid endocytosis of scAb-PEG-PLGA/rodamine by PC3M cells.
To verify the enhanced uptake of scAb-PEG-PLGA/JDF-12 in PC3M cells, the intracellular JDF-12 concentration was detected after 2-h incubation. As shown in Figure 4C, the intracellular JDF-12 concentration after scAb-PEG-PLGA/JDF-12 treatment was 1.78-fold higher than that after PEG-PLGA/JDF-12 treatment. Moreover, the intracellular JDF-12 concentration significantly reduced in presence of saturated free antibodies in the medium, indicating that the JDF-12 uptake was blocked by free PSCA antibody. Intracellular JDF-12 were ascribed to the cellular uptake of NPs and/or the release of JDF-12 into the medium and subsequent cellular uptake during incubation. Because the amount of JDF-12 released from NPs during incubation was almost identical between scAb-PEG-PLGA/JDF-12 and PEG-PLGA/JDF-12 according to the in vitro release profile (data not shown), the higher intracellular JDF-12 concentration was more likely to be attributable to the enhanced endocytosis of scAb-PEG-PLGA/JDF-12 by PC3M cells.
The results suggest that the scAb-PEG-PLGA/JDF-12 NPs are internalized by PC3M cells though the scAbPSCA-mediated endocytosis.
In vivo efficacy
To validate the anti-tumor effects of scAb-PEG-PLGA/JDF-12, tumor bearing mice after inoculation of PC3M cells were divided into five groups and animals were treated independently with PBS, scAb-PEG-PLGA, JDF-12, PEG-PLGA/JDF-12 or scAb-PEG-PLGA/JDF-12. There were no marked differences in the weight and tumor size among groups. As shown in Figure 5A, mice in PBS and scAb-PEG-PLGA group showed tumor enlargement. The tumor size in JDF-12, PEG-PLGA/JDF-12 and scAb-PEG-PLGA/JDF-12 groups reduced significantly as compared to PBS-treated group. The tumor size remained unchanged in PEG-PLGA/JDF-12 group and JDF-12 group, and no marked difference was observed in the tumor size between them. In contrast, scAb-PEG-PLGA/JDF-12 induced a greater inhibition on the tumor growth than PEG-PLGA/JDF-12 and JDF-12, indicating a superior outcome. One possible explanation for the enhanced antitumor effects in scAb-PEG-PLGA/JDF-12 group was that scAb-PEG-PLGA/JDF-12 accumulated in the tumor via “passive targeting” activity and further facilitated the cellular uptake via “active targeting” activity.
Figure 5.
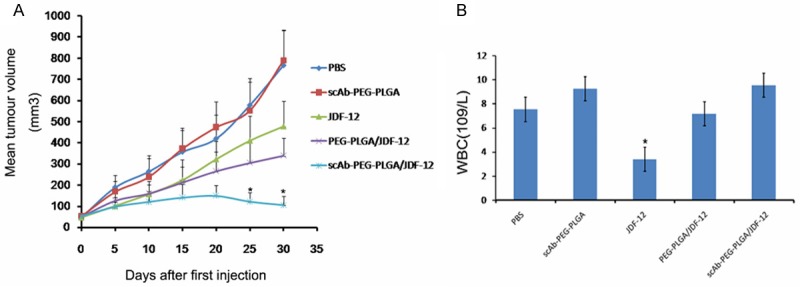
Tumor size and drug toxicity in PC3M xenograft mose model. Mice treated with scAb-PEG-PLGA/JDF-12 showed decreased tumor volume and reduced drug toxicity. A. Tumor volume in different groups. B. White blood cell (WBC) count in different groups. Data are expressed as mean ± standard error. *P<0.05.
In vivo toxicity was also assessed by the detection of white blood cells, an indicator sensitive to the cytotoxicity of cytotoxic agents. As shown in Figure 5B, white blood cell count in JDF-12 group was significantly lower than that in other groups, suggesting that JDF-12 induced leukopenia. However, no difference was observed in the white blood cell count among PBS, PEG-PLGA/JDF-12 and scAb-PEG-PLGA/JDF-12 groups, indicating JDF-12 encapsulated in the NPs reduced the JDF-12 associated cytotoxicity in vivo.
Discussion
Our previous study demonstrated that JDF-12 engineered to possess properties associated with those of MTZ and an EGFR inhibitor represented a new chemotherapeutic, and showed better anti-tumor effects against EGFR over-expressing PCa cells [3]. However, it has a poor solubility and a lack of targeted property, resulting in systemic toxicity.
Targeted drug delivery system is helpful to solve these problems. In this study, NPs were developed using the materials approved by the Food and Drug Administration (FDA) for clinical use. The targeted NPs were synthesized with serial chemical procedures. The drug-loaded PEG-PLGA NPs were first prepared using the nanoprecipitation method to render NPs “stealth” capacity by decreasing their non-specific clearance and biofouling with plasma proteins, followed by the conjugation of target moiety to the NPs surface. In addition, the negative zeta potential of NPs improved their stability owing to the charge repulsion.
It is well known that the particle size plays an important role in the EPR effect, which results from the hypervascular permeability and impaired lymphatic drainage in the tumor [8,12]. However, the optimal particle size is heterogeneous in a solid tumor because the vascular structure of tumor differs among tumors [13]. Some studies have suggested 100-200 nm of particle size as the suitable range for EPR effect, but that was determined by the animal model [14]. In our study, the particle size was 152.2±38.4 nm, which was within this range.
The antiproliferative evaluation showed that scAb-PEG-PLGA/JDF-12 was more cytotoxic than PEG-PLGA/JDF-12 in PC3M cells. The greater inhibitory effect was partly attributed to a higher intracellular drug level, which was confirmed by the measurement of intracellular JDF-12 concentration. The higher intracellular drug level was related to the scAbPSCA-mediated endocytosis, which was also known as the active targeting confirmed by CLSM. Moreover, scAb-PEG-PLGA/JDF-12 also demonstrated a greater inhibitory capability than JDF-12. The possible explanation is that, though JDF-12 as a membrane-permeable drug can penetrate the cell membrane rapidly, JDF-12 is degraded gradually after washing while scAb-PEG-PLGA/JDF-12 endocytosized by tumor cell may continuously release JDF-12 to exert a sustained inhibitory effect. Furthermore, some studies demonstrate that the drug-loaded polymeric NPs may avoid the multi-drug resistance effect [15], which may be also attributed to the better anti-tumor effect of scAb-PEG-PLGA/JDF-12.
Evaluation of in vivo anti-tumor effect showed that scAb-PEG-PLGA/JDF-12 was more effective to inhibit tumor growth than other drugs. The possible explanations for the enhanced anti-tumor capacity of scAb-PEG-PLGA/JDF-12 are as follows: a) After intravenous administration, the preferential accumulation of scAb-PEG-PLGA/JDF-12 in the tumor arises from the prolonged stay in the circulation as a result of pegylation-induced reduction in immunogenicity [16]. b) Because the tumor has hypervascular permeability and impaired lymphatic drainage, scAb-PEG-PLGA/JDF-12 may extravasate out of the tumor vasculature, which is unlike the physiological vasculature with the pore sizes of 400-600 nm [17]. Thus, the dug-loaded NPs accumulate in the tumor through EPR effect. c) The targeted NPs are designed to bind to PSCA protein on the surface of PCa cells and can be specifically internalized into tumor cells via the receptor-mediated endocytosis. d) scAb-PEG-PLGA/JDF-12 has a sustained release profile and may exert a prolonged anti-tumor effect. Thus, the anti-tumor effect of scAb-PEG-PLGA/JDF-12 is significantly enhanced, and systemic drug toxicity is reduced.
Conclusion
In summary, a targeted NP is successfully developed by overcoming the obstacles in development of JDF-12 as a novel chemotherapeutic. The preliminary findings demonstrate the better anti-tumor effect and the reduced systemic toxicity of this targeted delivery system against HRPC in vitro and in vivo. We postulate that a similar approach may be used to develop other drugs if there are difficulties in their development with conventional approaches.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81202012 to You-Qiang Fang, 81201694 to Yun Luo), Specialized Research Fund for the Doctoral Program of Higher Education of China (20110171120088 to You-Qiang Fang, 20120171120059 to Yun Luo), Guangdong Science and Technology Project (2013B021800084 to You-Qiang Fang), The Fundamental Research Funds for the Central Universities (14YKPY25 to You-Qiang Fang), Medical Scientific Research Foundation of Guangdong Province, China (B2011097 to You-Qiang Fang), Research Program of the 3rd Affiliated Hospital of Sun Yat-sen University (Yun Luo).
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Heidegger I, Massoner P, Eder IE, Pircher A, Pichler R, Aigner F, Bektic J, Horninger W, Klocker H. Novel therapeutic approaches for the treatment of castration-resistant prostate cancer. J Steroid Biochem Mol Biol. 2013;138:248–256. doi: 10.1016/j.jsbmb.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fang Y, Qiu Q, Domarkas J, Larroque-Lombard AL, Rao S, Rachid Z, Gibbs BF, Gao X, Jean-Claude BJ. “Combi-targeting” mitozolomide: conferring novel signaling inhibitory properties to an abandoned DNA alkylating agent in the treatment of advanced prostate cancer. Prostate. 2012;72:1273–1285. doi: 10.1002/pros.22475. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Wu W, Jiang X. Nanoscaled boron-containing delivery systems and therapeutic agents for cancer treatment. Nanomedicine (Lond) 2015;10:1149–1163. doi: 10.2217/nnm.14.213. [DOI] [PubMed] [Google Scholar]
- 5.Patel NR, Pattni BS, Abouzeid AH, Torchilin VP. Nanopreparations to overcome multidrug resistance in cancer. Adv Drug Deliv Rev. 2013;65:1748–1762. doi: 10.1016/j.addr.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koo H, Min KH, Lee SC, Park JH, Park K, Jeong SY, Choi K, Kwon IC, Kim K. Enhanced drug-loading and therapeutic efficacy of hydrotropic oligomer-conjugated glycol chitosan nanoparticles for tumor-targeted paclitaxel delivery. J Control Release. 2013;172:823–831. doi: 10.1016/j.jconrel.2013.08.297. [DOI] [PubMed] [Google Scholar]
- 7.Hoang B, Ernsting MJ, Murakami M, Undzys E, Li SD. Docetaxel-carboxymethylcellulose nanoparticles display enhanced anti-tumor activity in murine models of castration-resistant prostate cancer. Int J Pharm. 2014;471:224–233. doi: 10.1016/j.ijpharm.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65:71–79. doi: 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66:6732–6740. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- 10.Yoon JJ, Chung HJ, Lee HJ, Park TG. Heparin-immobilized biodegradable scaffolds for local and sustained release of angiogenic growth factor. J Biomed Mater Res A. 2006;79:934–942. doi: 10.1002/jbm.a.30843. [DOI] [PubMed] [Google Scholar]
- 11.Gao X, Luo Y, Wang Y, Pang J, Liao C, Lu H, Fang Y. Prostate stem cell antigen-targeted nanoparticles with dual functional properties: in vivo imaging and cancer chemotherapy. Int J Nanomedicine. 2012;7:4037–4051. doi: 10.2147/IJN.S32804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 13.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95:4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charrois GJ, Allen TM. Rate of biodistribution of STEALTH liposomes to tumor and skin: influence of liposome diameter and implications for toxicity and therapeutic activity. Biochim Biophys Acta. 2003;1609:102–108. doi: 10.1016/s0005-2736(02)00661-2. [DOI] [PubMed] [Google Scholar]
- 15.Fabbri F, Brigliadori G, Carloni S, Ulivi P, Tesei A, Silvestrini R, Amadori D, Zoli W. Docetaxel-ST1481 sequence exerts a potent cytotoxic activity on hormone-resistant prostate cancer cells by reducing drug resistance-related gene expression. Prostate. 2010;70:219–227. doi: 10.1002/pros.21055. [DOI] [PubMed] [Google Scholar]
- 16.Sapra P, Tyagi P, Allen TM. Ligand-targeted liposomes for cancer treatment. Curr Drug Deliv. 2005;2:369–381. doi: 10.2174/156720105774370159. [DOI] [PubMed] [Google Scholar]
- 17.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]