

Abstract
By removing modified nucleotides that block reverse transcriptase, two methods have now made tRNAs amenable to RNA-seq.
The use of high-throughput RNA sequencing (RNA-seq) has revealed an extraordinarily diverse population of functional RNAs present in cells. However, current approaches poorly detect RNAs that are highly modified and/or extensively folded, such as tRNAs. The complementary methods of Zheng et al.1 and Cozen et al.2 have now overcome many of the technical hurdles for globally sequencing tRNAs by biochemically removing select tRNA modifications before RNA-seq library preparation. These clever strategies have revealed new insights into tRNA regulation and should be useful for identifying other modified RNAs.
Although tRNAs have often been considered abundant ‘housekeeping’ RNAs, recent work has revealed that tRNAs are tightly regulated and play a role in controlling cell physiology3. Differences in tRNA expression patterns allow distinct translational programs to be expressed in proliferating versus differentiated cells4, and even modest overexpression of the initiator methionine tRNA is sufficient to induce cell proliferation5. In addition, tRNAs can be cleaved into smaller RNAs, some of which regulate cell proliferation and are more abundant than well-characterized microRNAs6. There is thus a pressing need to better understand how tRNAs are globally regulated by cellular cues.
Standard RNA-seq methods rely on ligating adaptor oligonucleotides to the ends of RNAs followed by reverse transcription using a primer complementary to the 3′ adaptor7. This works wells for most transcripts, but tRNAs are notable exceptions for two key reasons. First, mature tRNAs adopt a compact tertiary structure, which limits the efficiency of adaptor ligation and sterically interferes with cDNA synthesis. Second, an average of 8 or more nucleotides per tRNA are post-transcriptionally modified to ensure proper tRNA folding and/or translational fidelity, with the changes often occurring on the Watson-Crick face of the base8. These base alterations include so-called ‘hard-stop’ modifications—such as N1-methyladenosine (m1A) and N1-methylguanosine (m1G)—that block extension by reverse transcriptase (RT). As a result, current RNA-seq methods often yield only truncated sequences from a large subset of tRNAs (Fig. 1).
Figure 1.
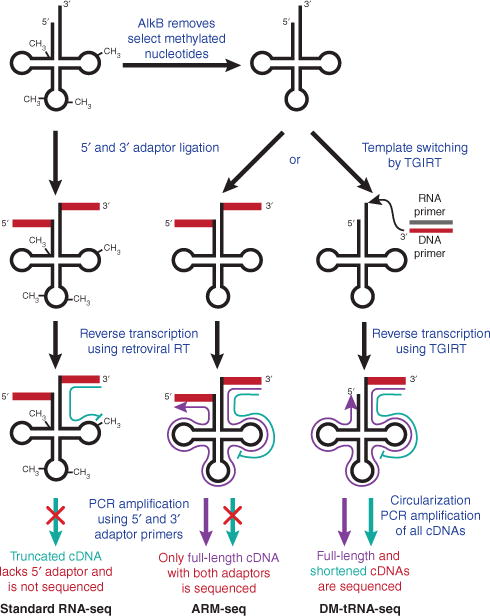
Removing methylated nucleotides to sequence tRNAs. Whereas reverse transcriptase (RT) cannot extend through a fully modified tRNA (left), treatment with AlkB removes select methylated nucleotides to allow efficient reverse transcription and deep sequencing (center and right). Full-length and truncated cDNAs are shown in purple and green, respectively.
For RNA-seq to accurately quantitate tRNAs, as well as distinguish between tRNAs that differ by a single nucleotide, hard-stop modifications must somehow be removed (or altered) prior to reverse transcription. To overcome this hurdle, both groups1,2 took advantage of the Escherichia coli dealkylating enzyme AlkB, which naturally functions in DNA and RNA repair by removing the methyl groups from m1A and m3C to revert the bases to their unmodified forms9. Zheng et al.1 further expanded the utility of AlkB by rationally designing a mutant enzyme that targets m1G. Treatment with a combination of wild-type and mutant AlkB allowed >70% of m1A, m3C and m1G to be demethylated from cellular tRNAs. Although this represents an important step forward, it should be noted that AlkB does not remove all hard stop–modified nucleotides, such as N2,N2-dimethylguanosine, which is present in ~20% of tRNAs.
After demethylation, the DM-tRNA-seq method of Zheng et al.1 takes advantage of a thermostable group II intron reverse transcriptase (TGIRT) rather than the usual retroviral RT. This thermostable enzyme is highly processive and synthesizes cDNA via a template-switching mechanism, allowing the adaptor ligation step to be eliminated from the sequencing protocol10. Consistent with previous results, untreated (fully modified) tRNAs resulted in sequencing reads that were predominantly truncated owing to RT stops. In contrast, AlkB treatment significantly increased the amount of longer and full-length cDNAs obtained, allowing the authors to quantitate closely related tRNAs and distinguish between them1.
The modification status of tRNAs can further be obtained by ARM-seq (AlkB-facilitated RNA methylation sequencing), which compares sequencing read counts between untreated and AlkB-treated samples2. Unlike DM-tRNA-seq, the method of Cozen et al.2 requires the ligation of adaptors to both ends of the mature RNAs. This is not as efficient as template switching, but it ensures that only full-length transcripts (mature tRNAs, tRNA precursors and tRNA-derived small RNAs) are PCR amplified and sequenced. In contrast, DM-tRNA-seq cannot distinguish an RT stop from a bona fide small RNA.
ARM-seq was 94% accurate at capturing known modification sites and identified many new sites in tRNAs that were then validated by primer extension assays. Interestingly, tRNA precursors have been thought to largely lack modified nucleotides, but the data from Cozen et al.2 suggest that many m1A modifications may be added before the pre-tRNA 5′ leader and 3′ trailer sequences have been removed. In addition, it is now clear that most tRNA-derived small RNAs are modified and that their levels have likely been underestimated in prior RNA-seq experiments by approximately threefold.
Both sequencing methods enhance our ability to globally monitor tRNA levels and to capture the dynamics of nucleotide modifications. They should provide new insights into how tRNA processing and stability are regulated, as well as reveal how tRNA misregulation affects diseases such as cancer. Broadly speaking, these methods can also be applied to look for previously undetected hard-stop modifications present in other transcripts, including mRNAs and noncoding RNAs. Considering that there are more than 100 different modified nucleotides known in nature, these AlkB-based sequencing methods serve as promising examples for how to identify and sequence RNAs containing other important base modifications.
Acknowledgments
J.E.W. is supported by US National Institutes of Health grant R00-GM104166 and the Rita Allen Foundation.
Footnotes
COMPETING FINANCIAL INTERESTS
The author declares no competing financial interests.
References
- 1.Zheng G, et al. Nat Methods. 2015;12:835–837. doi: 10.1038/nmeth.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cozen AE, et al. Nat Methods. 2015;12:879–884. doi: 10.1038/nmeth.3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilusz JE. Wiley Interdiscip Rev RNA. 2015;6:453–470. doi: 10.1002/wrna.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gingold H, et al. Cell. 2014;158:1281–1292. doi: 10.1016/j.cell.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Pavon-Eternod M, Gomes S, Rosner MR, Pan T. RNA. 2013;19:461–466. doi: 10.1261/rna.037507.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson P, Ivanov P. FEBS Lett. 2014;588:4297–4304. doi: 10.1016/j.febslet.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z, Gerstein M, Snyder M. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Machnicka MA, et al. Nucleic Acids Res. 2013;41:D262–D267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 10.Mohr S, et al. RNA. 2013;19:958–970. doi: 10.1261/rna.039743.113. [DOI] [PMC free article] [PubMed] [Google Scholar]