

Supplemental Digital Content is available in the text.
Keywords: heart failure, inflammation, macrophages, monocytes, myocardial infarction
Background—
Healing after myocardial infarction (MI) involves the biphasic accumulation of inflammatory Ly-6Chigh and reparative Ly-6Clow monocytes/macrophages. Excessive inflammation disrupts the balance between the 2 phases, impairs infarct healing, and contributes to left ventricle remodeling and heart failure. Lipoprotein-associated phospholipase A2 (Lp-PLA2), a member of the phospholipase A2 family of enzymes, produced predominantly by leukocytes, participates in host defenses and disease. Elevated Lp-PLA2 levels associate with increased risk of cardiovascular events across diverse patient populations, but the mechanisms by which the enzyme elicits its effects remain unclear. This study tested the role of Lp-PLA2 in healing after MI.
Methods and Results—
In response to MI, Lp-PLA2 levels markedly increased in the circulation. To test the functional importance of Lp-PLA2, we generated chimeric mice whose bone marrow–derived leukocytes were Lp-PLA2–deficient (bmLp-PLA2−/−). Compared with wild-type controls, bmLp-PLA2−/− mice subjected to MI had lower serum levels of inflammatory cytokines tumor necrosis factor-α, interleukin (IL)-1β, and IL-6, and decreased number of circulating inflammatory myeloid cells. Accordingly, bmLp-PLA2−/− mice developed smaller and less inflamed infarcts with reduced numbers of infiltrating neutrophils and inflammatory Ly-6Chigh monocytes. During the later, reparative phase, infarcts of bmLp-PLA2−/− mice contained Ly-6Clow macrophages with a skewed M2-prone gene expression signature, increased collagen deposition, fewer inflammatory cells, and improved indices of angiogenesis. Consequently, the hearts of bmLp-PLA2−/− mice healed more efficiently, as determined by improved left ventricle remodeling and ejection fraction.
Conclusions—
Lp-PLA2 augments the inflammatory response after MI and antagonizes healing by disrupting the balance between inflammation and repair, providing a rationale for focused study of ventricular function and heart failure after targeting this enzyme acutely in MI.
Myocardial infarction (MI) is a leading cause of death worldwide.1 Although the case fatality rate of MI has declined, survival with development of long-term left ventricular (LV) dysfunction because of cumulative ischemic myocardial damage has added to the growing epidemic burden of chronic heart failure. The human, social, and economic consequences of chronic ischemic cardiomyopathy present a major challenge and unmet medical need. Patients who initially survive MI must overcome a major obstacle: ischemia damages the heart, and effective cardiac repair likely requires a precise balance between removal of debris and formation of a scar that is compatible with heart function. MI survivors frequently develop heart failure; although many therapeutics in current use have proven beneficial, the high residual morbidity and mortality presents an urgent problem that requires a better understanding of the disease’s pathophysiology.
Clinical Perspective on p 987
During the past several years, neutrophils, monocytes, and macrophages have emerged as consequential to the inflammatory and healing process that occurs after MI.2 We now understand that ischemic injury triggers the accumulation of these myeloid cells in the infarcted myocardium.3,4 Shortly after onset of ischemia, large numbers of neutrophils and inflammatory Ly-6Chigh monocytes infiltrate the infarcted myocardium and produce interleukin (IL)-1β, IL-6, and tumor necrosis factor-α. Within 4 to 5 days, Ly-6Chigh monocytes give rise to Ly-6Clow reparative macrophages,3 which potentiate healing via vascular endothelial growth factor, transforming growth factor-β, and IL-10. The 2 phases comprising inflammatory Ly-6Chigh monocyte recruitment and reparative Ly-6Clow macrophage differentiation are essential to post-MI recovery; their perturbation (ie, in the context of comorbidities) leads to impaired heart function and heart failure.3,4
Lipoprotein-associated phospholipase A2 (Lp-PLA2), a member of the phospholipase A2 family of enzymes, hydrolyses glycerophospholipids. The ensuing enzymatic reactions frequently generate metabolic signaling molecules with a multitude of biological actions. For example, by hydrolyzing phosphotidylcholine at C2 of the glycerol backbone, Lp-PLA2 produces lysophosphatidylcholine,5 which fosters oxidative stress, affects vascular smooth muscle cell proliferation, and increases tissue accumulation of macrophages.6 Several observational studies showed that Lp-PLA2 levels correspond with future cardiovascular events, such as acute MI and sudden cardiac death.7–9 Although preliminary studies reported reduced development of advanced coronary atherosclerosis10 or stabilization of the necrotic core size11 with selective inhibition of Lp-PLA2 by the inhibitor darapladib, 2 recently completed phase III trials did not provide evidence in favor of inhibiting Lp-PLA2 in cardiovascular disease.12 Neither study focused on LV function nor chronic heart failure end points. Moreover, it remains unclear whether Lp-PLA2 participates in the inflammatory and reparative phases that characterize the innate immune response shortly after MI; these pathways likely influence LV remodeling and the development of chronic ischemic cardiomyopathy. This study sought to evaluate whether Lp-PLA2 participates in infarct healing and HF after MI.
Methods
Further details are available in the Data Supplement.
Animals and Animal Experiments
Eight- to ten-week-old female C57BL6/J (wild-type [WT]) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Lp-PLA2–deficient mice (Lp-PLA2−/−) were kindly provided by Glaxosmithkline Pharmaceuticals Ltd (King of Prussia, PA). All protocols were approved by the Animal Review Committee at Massachusetts General Hospital. C57BL6/J mice were lethally irradiated and reconstituted with WT and Lp-PLA2−/− bone marrow to generate respective chimeric mice. The chimeras had normal leukocyte counts and exhibited no obvious abnormalities, consistent with the Lp-PLA2–deficient mice.13 MI was induced by permanent ligation of the left anterior descending artery. We observed no differences in mortality between the groups.
Lp-PLA2/PAF Acetyl-Hydrolase Activity Assay
The PAF hydrolase activity assay was performed as previously described14 with modifications using [3H]PAF (Platelet Activating Factor, 1-O-Hexadecyl-[Acetyl-3H(N)]-, Hexadecyl PAF) as a substrate. Unlabeled PAF (1-O-Hexadecyl-2-O-acetyl-sn-glycero-3-phophorylcholine) was purchased from Enzo Life Sciences, 1-O-Hexadecyl-2-O-[Acetyl-3H(N)]-, Hexadecyl PAF, [acetyl-3H]-, (250μCi (9.25MBq) was purchased from Perkin Elmer, and Bio-Safe II was purchased from Research Products International Corp, Mount Prospect, IL.
Histology
Murine hearts were embedded in Tissue-Tek O.C.T compound (Sakura Finetek) and prepared for sectioning and staining.
Flow Cytometry and Flow-Assisted Cell Sorting
Antibodies used for flow cytometry are listed in the Data Supplement. Data were acquired on a BD LSRII and analyzed with FlowJo. Cells were sorted with BD AriaII.
Reverse Transcription Polymerase Chain Reaction
RNA was isolated from sorted cells with the RNeasy Micro Kit (Qiagen). Quantitative real-time TaqMan polymerase chain reaction was run on a 7500 PCR thermal cycler (Applied Biosystems).
Magnetic Resonance Imaging
Magnetic resonance imaging was performed on days 1 and 21 after permanent coronary ligation as described previously.3 We obtained cine images of the LV short axis by using a 7 Tesla horizontal bore Pharmascan (Bruker) and a custom-built mouse cardiac coil (Rapid Biomedical). Late gadolinium enhancement was performed on day 1 to determine infarct size. Acquisition was done as described previously.15 Images were analyzed using the software Segment (http://segment.heiberg.se). The end-diastolic volume, end-systolic volume, ejection fraction, LV volume, heart rate, and cardiac output were measured.
Statistics
Results are shown as mean±SEM. Unpaired Student t test was applied to evaluate differences between 2 groups. One-way ANOVA with post hoc Tukey multiple comparisons test was performed when comparing >2 groups between days, because different mice were euthanized on each time points for organ harvest. P values ≤0.05 denote significant changes.
Results
Expression of Lp-PLA2 After MI and Its Role on Healing
To elucidate whether Lp-PLA2 participates in healing after MI, we first measured Lp-PLA2 expression and serum activity in steady state and after MI. Lp-PLA2 mRNA expression by real-time polymerase chain reaction increased in the infarcts of WT mice as early as 1 day after MI (Figure 1A), suggesting Lp-PLA2 may participate in myocardial ischemic injury. Concordantly, Lp-PLA2 activity was also increased shortly after MI (Figure 1B). To define the role of Lp-PLA2 in hematopoietic cells in the pathophysiology of acute MI, we generated chimeric mice by irradiating and reconstituting WT mice with bone marrow either from WT or from Lp-PLA2−/− (bmLp-PLA2−/−) mice. In comparison with WT mice, bmLp-PLA2−/− mice had lower Lp-PLA2 activity at steady state, and this activity did not change after MI (Figure 1B). These findings establish leukocytes as major sources of Lp-PLA2 in response to MI. We then evaluated infarct healing and demonstrated a significant decrease in infarct size in bmLp-PLA2−/− mice 7 days after MI compared with WT (Figure 1C and 1D).
Figure 1.
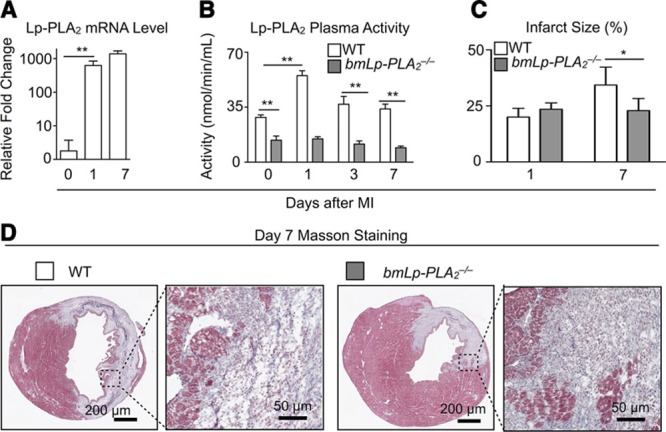
Lipoprotein-associated phospholipase A2 (Lp-PLA2) activity and mRNA expression during myocardial infarction. A, mRNA levels quantified by real-time polymerase chain reaction in wild-type (WT) mice at the indicated time points after myocardial infarction (MI). Day 0 represents the steady-state mice. Results are presented as mean±SD, **P≤0.01, n=5 to 10 per group. B, Plasma Lp-PLA2 activity in chimera WT and bmLp-PLA2−/− mice at steady state and in bmLp-PLA2−/− mice at indicated time points after MI. **P≤0.01, n=5 to 10 per group. C, Quantification of infarct size on days 1 and 7 after MI in WT and bmLp-PLA2−/− mice (left). *P≤0.05, n=4 to 6 per group. D, Representative images of infarct size on day 7 after MI were shown in both WT and bmLp-PLA2−/− mice.
Lp-PLA2 Influences Systemic Inflammation
Coronary occlusion stimulates an inflammatory response characterized by cytokine and chemokine production, and leukocyte recruitment to the heart. Because Lp-PLA2 participates in inflammation, we assessed the effect of Lp-PLA2 deficiency after MI. Serum concentrations of inflammatory cytokines tumor necrosis factor-α, IL-1β, and IL-6 increased dramatically in WT mice on day 1 after MI, and eventually declined to undetectable amounts on day 7. In contrast, bmLp-PLA2−/− mice showed only moderately elevated tumor necrosis factor-α and IL-6, and negligibly increased IL-1β, demonstrating a diminished inflammatory response in the absence of Lp-PLA2 (Figure 2A). Time-course profiling of circulating leukocytes after MI revealed that both WT and bmLp-PLA2−/− mice augmented inflammatory myeloid cells (neutrophils and Ly-6Chigh monocytes), but compared with WT mice, bmLp-PLA2−/− mice had fewer neutrophils and Ly-6Chigh cells in blood at days 3 and 7, indicating Lp-PLA2 contributes to the systemic inflammation after MI (Figure 2B and 2C).
Figure 2.
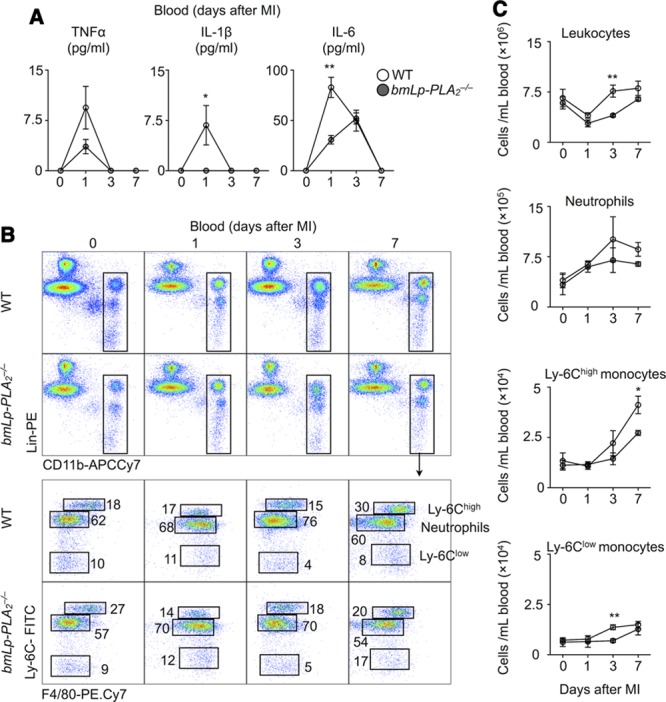
Lipoprotein-associated phospholipase A2 (LP-PLA2) moderately affects systemic inflammation. A, ELISA analysis of plasma levels of tumor necrosis factor-α (TNF-α), interleukin (IL)-1β and IL-6 in wild-type (WT) and bmLp-PLA2−/− mice at the indicated time points after myocardial infarction (MI). B, Representative flow cytometric images of leukocyte profiling of blood in WT and bmLp-PLA2−/− mice, C, Quantification of total leukocytes, neutrophils, Ly-6Chigh monocytes, Ly-6Clow monocytes in the blood. Results in 1 of 3 experiments with similar patterns are presented as mean±SEM, *P≤0.05, **P≤0.01 (WT vs bmLp-PLA2−/−) vs n=5 per group.
Lp-PLA2 Impairs the Appearance of Reparative Macrophages
The myocardium displays a biphasic monocyte and macrophage response during MI.3,4 In the first phase, inflammatory Ly-6Chigh monocytes infiltrate the ischemic myocardium from the blood and participate in inflammation. In the second phase, reparative Ly-6Clow macrophages contribute to collagen deposition and scar formation. We profiled leukocytes in the myocardium in the steady state and 1, 3, and 7 days after MI in both WT and bmLp-PLA2−/− mice. The infarcts of both strains accumulated neutrophils, which peaked on day 1, Ly-6Chigh monocytes, which peaked on day 3, and Ly-6Clow macrophages, which peaked on day 7 (Figure 3A and 3B). This finding agrees with our previous observations.3,4 Yet, for nearly every peak, infarcts of bmLp-PLA2−/− mice accumulated only half the number of cells (neutrophils on day 1 and Ly-6Chigh monocytes on day 3) compared with WT controls, consistent with our observations in the blood (Figure 2), and affirming that Lp-PLA2 aggravates inflammation.
Figure 3.
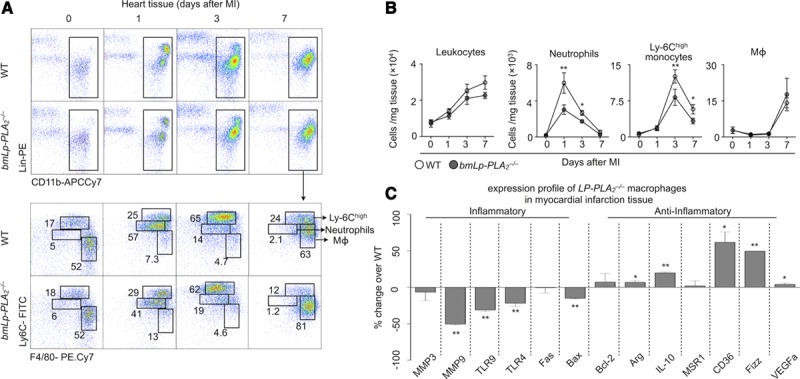
Attenuated inflammatory response in bmLp-PLA2−/− myocardial infarct tissue. A, Representative images for flow cytometric analysis of myocardial infarction (MI) tissue cell suspensions at the indicated time points after MI in wild-type (WT) and bmLp-PLA2−/− mice. B, Flow cytometry based quantification of neutrophil, monocyte and MΦ (macrophage) numbers in MI tissue of WT versus bmLp-PLA2−/− mice before and 1, 3, and 7 days post MI. Results in one of three experiments with similar patterns are presented as mean±SEM, * P≤0.05, n = 4 per group. C, Gene expression profiling of WT and bmLp-PLA2−/− macrophages sorted from MI tissue 7 days after permanent left anterior descending artery ligation. Results are presented as mean±SEM percent change of marker expression in Lp-PLA2−/− compared with WT control mice, *P≤0.05, **P≤0.01, n=5 per group. IL indicates interleukin; and VEGF, vascular endothelial growth factor.
Aside from determining the number of cells that accumulate (quantity), macrophage activity (quality) is an essential measure of the cells’ impact on inflammation and repair. We asked whether Lp-PLA2 shapes macrophage function by measuring expression of signature M1/M2 genes in sorted cardiac macrophages. In comparison with WT macrophages, Lp-PLA2−/− macrophages exhibited higher expression of mRNAs that encode M2-associated genes (Arg, IL-10, CD36, and Fizz) and lower levels of those corresponding to M1-associated genes (MMP-3, MMP-9, TLR9, and TLR4; Figure 3C). These data are consistent with the idea that lysophosphotidylcholine, a product of Lp-PLA2, potentiates an M1-like macrophage phenotype.16 Together, these data show that Lp-PLA2 promotes recruitment of inflammatory myeloid cells and delays the appearance of reparative macrophages in the ischemic myocardium.
Lp-PLA2 Retards Healing After MI
The differences in leukocyte recruitment between WT and bmLp-PLA2−/− mice prompted us to determine whether the absence of Lp-PLA2 affects the repair of the ischemic myocardium. To this end, we profiled myeloid cell infiltration, extracellular matrix deposition, neovascularization, and smooth muscle cell accumulation by immunohistochemistry (Figure 4). Compared with WT controls, the myocardium of bmLp-PLA2−/− mice accumulated fewer myeloid CD11b+ cells, indicating less severe inflammation. Infarcts of bmLp-PLA2−/− mice also had larger regions of extracellular matrix deposition, as evidenced by higher percentage of collagen I+ areas (24% versus 32%), larger CD31+ areas (5% versus 11%), suggesting improved neovascularization of the heart, but no changes in the number of smooth muscle actin+ myofibroblasts (α- smooth muscle actin+area). Collectively, the results of histological examination demonstrate more effective healing in the absence of Lp-PLA2. These results demonstrate that Lp-PLA2 inhibits the resolution of inflammation after MI.
Figure 4.
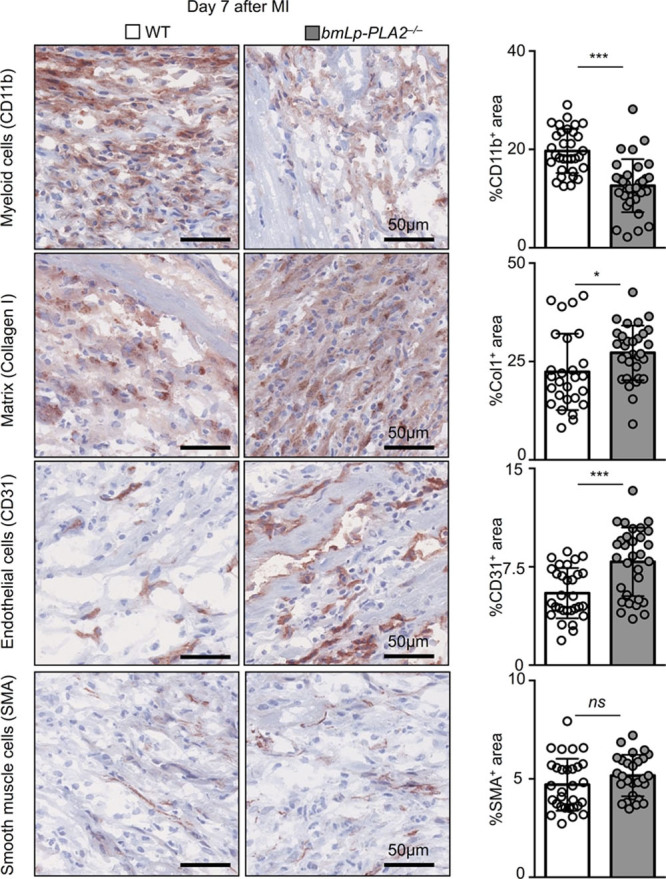
Improved cardiac remodeling in bmLp-PLA2−/− mice 7 days after myocardial infarction. Immunohistochemical staining of myocardial infarction (MI) tissue for CD11b, Collagen 1 (Col1), CD31, and nonvascular smooth muscle α actin (SMA) in wild-type (WT) and bmLp-PLA2−/− chimeric mice 7 days post MI. Quantification of 10 randomly selected fields of view per mouse (5 mice). Results are presented as mean±SEM, *P≤0.05, ***P≤0.001, n=50 fields of view per group.
Improved Heart Function in the Absence of Lp-PLA2
To test whether inflammation mediated by Lp-PLA2 after MI translated to impaired heart function, we performed magnetic resonance imaging in vivo in WT and bmLp-PLA2−/− mice. In the steady state, we detected no differences in cardiac function between WT and bmLp-PLA2−/− mice. After permanent coronary artery ligation, the end-diastolic volume and end-systolic volume, ejection fraction, LV volume, heart rate, and cardiac output were measured in individual mice on days 1 and 21 after MI. Late gadolinium enhancement was performed on day 1 to determine infarct size (Figure 5A and 5B; Table I in the Data Supplement). The infarct sizes were similar on day 1 in both groups, excluding a potential surgical bias (Figure 5B). Although LV volume increased similarly between the 2 groups on day 21, increased end-diastolic volume was only observed in WT mice, indicating more favorable remodeling in mice lacking Lp-PLA2. Moreover, compared with day 1, ejection fraction at day 21 diminished in WT mice but increased modestly in bmLp-PLA2−/− mice, suggesting improved recovery of heart function in the absence of Lp-PLA2. Overall, the data show that Lp-PLA2 aggravates LV remodeling and impairs LV function after MI.
Figure 5.
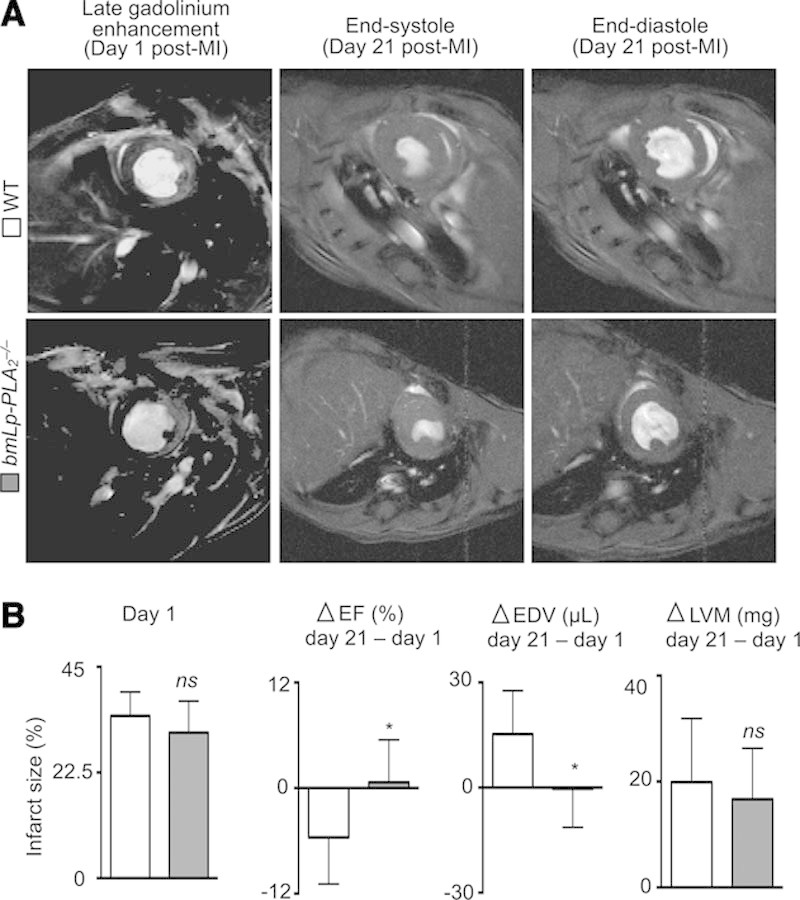
Lp-PLA2 aggravates LV dysfunction after myocardial infarction. A, Representative MRI with late gadolinium enhancement (left panels) on day 1 after permanent left anterior descending artery ligation, and end-systole (middle panels) and end-diastole (right panels) on day 21 of infarcted hearts from wild-type (WT) and bmLp-PLA2−/− mice, respectively. B, Quantification of individual changes (Δ) in heart parameters from day 1 to day 21 post myocardial infarction (MI; day 21−day 1). Results are presented as mean±SD percent change of marker expression in WT and bmLp-PLA2−/− mice, respectively. * P≤0.05, n ≥7 per group. EDV indicates end-diastolic volume; EF, ejection fraction; and LVM, left ventricle volume.
Discussion
Recruitment of neutrophils and Ly-6Chigh monocytes into the infarcted myocardium and the subsequent generation of reparative macrophages from Ly-6Chigh monocytes contribute to necrotic debris clearance, matrix deposition, granulation tissue formation, and angiogenesis. Perturbations in the inflammatory response impair infarct healing and promote heart failure.17–19 This study shows that deficiency of Lp-PLA2 on hematopoietic cells attenuates systemic inflammation after MI, impairs leukocyte infiltration into infarcts, and enhances generation of reparative Ly-6Clow macrophages, leading to less adverse LV remodeling and improved recovery of LV function. Together, the data show that Lp-PLA2 modulates inflammation after MI and suggest that targeting of Lp-PLA2 might lessen LV dysfunction and the development of chronic heart failure after MI.
Monocyte-derived macrophages can produce substantial Lp-PLA2,20 and in the mouse, Lp-PLA2 is expressed almost exclusively by myeloid cells (www.immgen.org). Previous studies have identified macrophage Lp-PLA2 expression at both the mRNA and the protein levels in human and rabbit aortic lesions.21 Plaques with characteristics of vulnerable and ruptured atheromata, but not early lesions, contain abundant Lp-PLA2.22 Therefore, extensive efforts have been taken to prevent atherosclerosis-related coronary heart disease by inhibiting Lp-PLA2 and thus promoting plaque stability.12 But Lp-PLA2 may participate in coronary heart disease by modulating inflammation independent of effects on the plaque itself. For example, our data showed that Lp-PLA2 rose dramatically during MI. As a phospholipase, increased Lp-PLA2 hydrolyzes phospholipids of oxidatively damaged cells or lipoproteins. Lp-PLA2 hydrolyzes oxidized phosphatidylcholine, generating lysophosphatidylcholine and oxidized fatty acids that are capable of acting as monocyte chemoattractants as well as maintaining macrophages in a proinflammatory phenotype.6,16,23 Lp-PLA2 can also contribute to inflammation by interfering with phagocytic clearance of apoptotic cells, for example, neutrophils, via cleavage and removal of oxidized phosphatidylserine, a known eat-me signal.24 Consistent with this hypothesis, we found that bmLp-PLA2−/− mice had milder inflammatory responses as well as fewer neutrophils and Ly-6Chigh monocytes accumulating in the myocardium. This reduced number of inflammatory leukocytes ameliorated adverse LV remodeling and improved heart function recovery. Therefore, our data strongly support a role for Lp-PLA2 in increased risk of heart failure by directly promoting systemic and local myocardial inflammatory responses after MI, functions distinct from effects on the atherosclerotic plaques. Our experiments, performed with bone marrow chimeras, demonstrate an effect on MI healing with a ≈80% knockdown of Lp-PLA2, indicating this is a sufficient reduction to elicit positive effects. Future studies will need to elucidate in more detail the full scope of Lp-PLA2 function not only on leukocyte behavior but also on effects attributed to other cells in the heart, such as fibroblasts and endothelial cells.
Recently, 2 large multicenter phase III trials completed evaluation of the Lp-PLA2 inhibitor darapladib on the reduction of cardiovascular adverse events in >28 000 patients with documented coronary heart disease. These 2 complementary trials covered both chronic and acute coronary heart disease and evaluated traditional coronary heart disease end points. Thus, these studies did not formally focus on LV function or long-term development of heart failure.25,26 The Stabilization of pLaques usIng Darapladib-Thrombolysis in Myocardial Infarction (SOLID-TIMI 52) trial enrolled patients within 30 days of acute coronary syndrome and the STabilization of Atherosclerotic plaque By Initiation of darapLadIb TherapY (STABILITY) trial enrolled patients with stable chronic coronary heart disease. Despite promising preclinical results,10 both clinical trials concluded that direct inhibition of Lp-PLA2 with darapladib failed to reduce major adverse cardiovascular events.12,27 Crucially, the trials did not test whether blockade of Lp-PLA2 per se was beneficial: patients receiving darapladib or placebo were already receiving ≤4 different therapeutics against heart disease, including statins and ACE inhibitors. The trials, therefore, showed that under the specific parameters of the study, darapladib did not provide benefit above that afforded by current treatment regimens.
Our results provide an alternative explanation as to why the clinical trials might not have improved the tested end points. Our data show that, as early as day 1 after MI, expression of Lp-PLA2 was increased. In the absence of Lp-PLA2, inflammatory leukocyte recruitment was blunted, yielding a smaller inflammatory response that correlated with improved healing and heart function 7 and 21 days later, respectively. Lp-PLA2 might elicit its most detrimental effects in the acute phase after MI. If so, blocking the enzyme earlier than the SOLID or STABILITY trials might benefit end points related to LV function. Several human studies have shown data in support of the idea that healing of the infarcted myocardium involves the biphasic accumulation of monocytes and macrophages. In 1 study, a cohort of 36 patients monitored >2 weeks after MI demonstrated a peak of circulating inflammatory CD16− monocytes on day 2.6 after MI, followed by another peak of CD16+ monocytes on day 4.8.19 Because CD16– monocytes resemble inflammatory Ly-6Chigh monocytes, whereas CD16+ monocytes (and in particular CD16+ CD14dim) resemble Ly-6Clow monocytes, these findings suggest that acute inflammation may likewise peak in humans within 1 week after MI. Immediate inhibition of Lp-PLA2 might be the optimal time window for improving outcomes related to LV function. The SOLID trial, which enrolled patients within 30 days, did not test whether patients receiving darapladib within the first 3 days post MI benefited from treatment. Hence, the clinical trial findings do not argue against the role of Lp-PLA2 in inflammation after MI.
In summary, the data demonstrate that Lp-PLA2 regulates the host response after MI through modulation of inflammation. By enhancing inflammation and impairing repair, the induced Lp-PLA2 negatively regulates recovery of LV function. In addition to stimulating atherosclerotic plaque vulnerability, the effect of Lp-PLA2 after MI reveals a novel role of this enzyme in modulating the myocardial response to ischemic injury. These mechanistic insights have implications for developing effective therapeutics against ischemic cardiomyopathy and chronic heart failure post MI.
Acknowledgments
We thank Michael Waring and Nathalie Bonheur for sorting cells. We also thank Dr Partha Dutta for assistance with mice irradiation.
Sources of Funding
This work was supported, in part, by GSK funding (GlaxoSmithKline: Targeting Lp-PLA2 in Inflammatory Cardiovascular Disease) and by National Institutes of Health grants 1R01HL095612 and R56AI104695 (to Dr Swirski).
Disclosures
None.
Supplementary Material
Footnotes
Drs He and Chousterman contributed equally to this work.
The Data Supplement is available at http://circheartfailure.ahajournals.org/lookup/suppl/doi:10.1161/CIRCHEARTFAILURE.115.002334/-/DC1.
CLINICAL PERSPECTIVE
Myocardial infarction involves balanced accumulation of monocytes and macrophages in tissue. This study shows that lipoprotein-associated phospholipase A2 (Lp-PLA2) is involved in the regulation of host response after myocardial infarction through modulating inflammatory responses. Elevated Lp-PLA2 after myocardial infarction associated with heightened tumor necrosis factor-α, interleukin (IL)-1β and IL-6 levels in serum. Accordingly, increased numbers of neutrophils and inflammatory Ly-6Chigh monocytes accumulated into the infarcted myocardium. Depletion of Lp-PLA2 gave rise to Ly-6Clow macrophages with a skewed M2-prone gene expression signature in the reparative phase, leading to increased collagen deposition, fewer inflammatory cells, and improved indices of angiogenesis. Consequently, the hearts of bmLp-PLA2−/− mice healed more efficiently, as observed by improved left ventricle remodeling and ejection fraction. Lp-PLA2 thus may serve as a therapeutic target for heart failure after myocardial infarction.
References
- 1.Bloom DE, Cafiero ET, Jane-Llopis E, Abrahams-Gessel S, Bloom LR, Fathima S, Feigl AB, Gaziano T, Mowafi M, Pandya A, Prettner K, Rosenberg L, Seligman B, Stein AZ, Weinstein C. The Global Economic Burden of Noncommunicable Diseases. Cologny, Geneva: World Economic Forum; 2011. [Google Scholar]
- 2.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–166. doi: 10.1126/science.1230719. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114:1611–1622. doi: 10.1161/CIRCRESAHA.114.303204. doi: 10.1161/CIRCRESAHA.114.303204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacPhee CH, Moores KE, Boyd HF, Dhanak D, Ife RJ, Leach CA, Leake DS, Milliner KJ, Patterson RA, Suckling KE, Tew DG, Hickey DM. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase, generates two bioactive products during the oxidation of low-density lipoprotein: use of a novel inhibitor. Biochem J. 1999;338(pt 2):479–487. [PMC free article] [PubMed] [Google Scholar]
- 6.Shi Y, Zhang P, Zhang L, Osman H, Mohler ER, III, Macphee C, Zalewski A, Postle A, Wilensky RL. Role of lipoprotein-associated phospholipase A2 in leukocyte activation and inflammatory responses. Atherosclerosis. 2007;191:54–62. doi: 10.1016/j.atherosclerosis.2006.05.001. doi: 10.1016/j.atherosclerosis.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Gerber Y, McConnell JP, Jaffe AS, Weston SA, Killian JM, Roger VL. Lipoprotein-associated phospholipase A2 and prognosis after myocardial infarction in the community. Arterioscler Thromb Vasc Biol. 2006;26:2517–2522. doi: 10.1161/01.ATV.0000240406.89440.0c. doi: 10.1161/01.ATV.0000240406.89440.0c. [DOI] [PubMed] [Google Scholar]
- 8.Herrmann J, Mannheim D, Wohlert C, Versari D, Meyer FB, McConnell JP, Gössl M, Lerman LO, Lerman A. Expression of lipoprotein-associated phospholipase A(2) in carotid artery plaques predicts long-term cardiac outcome. Eur Heart J. 2009;30:2930–2938. doi: 10.1093/eurheartj/ehp309. doi: 10.1093/eurheartj/ehp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oei HH, van der Meer IM, Hofman A, Koudstaal PJ, Stijnen T, Breteler MM, Witteman JC. Lipoprotein-associated phospholipase A2 activity is associated with risk of coronary heart disease and ischemic stroke: the Rotterdam Study. Circulation. 2005;111:570–575. doi: 10.1161/01.CIR.0000154553.12214.CD. doi: 10.1161/01.CIR.0000154553.12214.CD. [DOI] [PubMed] [Google Scholar]
- 10.Wilensky RL, Shi Y, Mohler ER, III, Hamamdzic D, Burgert ME, Li J, Postle A, Fenning RS, Bollinger JG, Hoffman BE, Pelchovitz DJ, Yang J, Mirabile RC, Webb CL, Zhang L, Zhang P, Gelb MH, Walker MC, Zalewski A, Macphee CH. Inhibition of lipoprotein-associated phospholipase A2 reduces complex coronary atherosclerotic plaque development. Nat Med. 2008;14:1059–1066. doi: 10.1038/nm.1870. doi: 10.1038/nm.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serruys PW, García-García HM, Buszman P, Erne P, Verheye S, Aschermann M, Duckers H, Bleie O, Dudek D, Bøtker HE, von Birgelen C, D’Amico D, Hutchinson T, Zambanini A, Mastik F, van Es GA, van der Steen AF, Vince DG, Ganz P, Hamm CW, Wijns W, Zalewski A Integrated Biomarker and Imaging Study-2 Investigators. Effects of the direct lipoprotein-associated phospholipase A(2) inhibitor darapladib on human coronary atherosclerotic plaque. Circulation. 2008;118:1172–1182. doi: 10.1161/CIRCULATIONAHA.108.771899. doi: 10.1161/CIRCULATIONAHA.108.771899. [DOI] [PubMed] [Google Scholar]
- 12.White HD, Held C, Stewart R, Tarka E, Brown R, Davies RY, Budaj A, Harrington RA, Steg PG, Ardissino D, Armstrong PW, Avezum A, Aylward PE, Bryce A, Chen H, Chen MF, Corbalan R, Dalby AJ, Danchin N, De Winter RJ, Denchev S, Diaz R, Elisaf M, Flather MD, Goudev AR, Granger CB, Grinfeld L, Hochman JS, Husted S, Kim HS, Koenig W, Linhart A, Lonn E, Lopez-Sendon J, Manolis AJ, Mohler ER, Nicolau JC, Pais P, Parkhomenko A, Pedersen TR, Pella D, Ramos-Corrales MA, Ruda M, Sereg M, Siddique S, Sinnaeve P, Smith P, Sritara P, Swart HP, Sy RG, Teramoto T, Tse HF, Watson D, Weaver WD, Weiss R, Viigimaa M, Vinereanu D, Zhu J, Cannon CP, Wallentin L. Darapladib for preventing ischemic events in stable coronary heart disease. N Engl J Med. 2014;370:1702–1711. doi: 10.1056/NEJMoa1315878. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Z, Fehrenbach ML, Ravaioli G, Kokalari B, Redai IG, Sheardown SA, Wilson S, Macphee C, Haczku A. The effect of lipoprotein-associated phospholipase A2 deficiency on pulmonary allergic responses in Aspergillus fumigatus sensitized mice. Respir Res. 2012;13:100. doi: 10.1186/1465-9921-13-100. doi: 10.1186/1465-9921-13-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tew DG, Southan C, Rice SQ, Lawrence MP, Li H, Boyd HF, Moores K, Gloger IS, Macphee CH. Purification, properties, sequencing, and cloning of a lipoprotein-associated, serine-dependent phospholipase involved in the oxidative modification of low-density lipoproteins. Arterioscler Thromb Vasc Biol. 1996;16:591–599. doi: 10.1161/01.atv.16.4.591. [DOI] [PubMed] [Google Scholar]
- 15.Courties G, Heidt T, Sebas M, Iwamoto Y, Jeon D, Truelove J, Tricot B, Wojtkiewicz G, Dutta P, Sager HB, Borodovsky A, Novobrantseva T, Klebanov B, Fitzgerald K, Anderson DG, Libby P, Swirski FK, Weissleder R, Nahrendorf M. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol. 2014;63:1556–1566. doi: 10.1016/j.jacc.2013.11.023. doi: 10.1016/j.jacc.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qin X, Qiu C, Zhao L. Lysophosphatidylcholine perpetuates macrophage polarization toward classically activated phenotype in inflammation. Cell Immunol. 2014;289:185–190. doi: 10.1016/j.cellimm.2014.04.010. doi: 10.1016/j.cellimm.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 17.Maekawa Y, Anzai T, Yoshikawa T, Asakura Y, Takahashi T, Ishikawa S, Mitamura H, Ogawa S. Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction:a possible role for left ventricular remodeling. J Am Coll Cardiol. 2002;39:241–246. doi: 10.1016/s0735-1097(01)01721-1. [DOI] [PubMed] [Google Scholar]
- 18.Panizzi P, Swirski FK, Figueiredo JL, Waterman P, Sosnovik DE, Aikawa E, Libby P, Pittet M, Weissleder R, Nahrendorf M. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol. 2010;55:1629–1638. doi: 10.1016/j.jacc.2009.08.089. doi: 10.1016/j.jacc.2009.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T, Kitabata H, Okochi K, Arita Y, Ishibashi K, Komukai K, Kataiwa H, Nakamura N, Hirata K, Tanaka A, Akasaka T. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol. 2009;54:130–138. doi: 10.1016/j.jacc.2009.04.021. doi: 10.1016/j.jacc.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 20.Tsimikas S, Tsironis LD, Tselepis AD. New insights into the role of lipoprotein(a)-associated lipoprotein-associated phospholipase A2 in atherosclerosis and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2007;27:2094–2099. doi: 10.1161/01.ATV.0000280571.28102.d4. doi: 10.1161/01.ATV.0000280571.28102.d4. [DOI] [PubMed] [Google Scholar]
- 21.Häkkinen T, Luoma JS, Hiltunen MO, Macphee CH, Milliner KJ, Patel L, Rice SQ, Tew DG, Karkola K, Ylä-Herttuala S. Lipoprotein-associated phospholipase A(2), platelet-activating factor acetylhydrolase, is expressed by macrophages in human and rabbit atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1999;19:2909–2917. doi: 10.1161/01.atv.19.12.2909. [DOI] [PubMed] [Google Scholar]
- 22.Kolodgie FD, Burke AP, Skorija KS, Ladich E, Kutys R, Makuria AT, Virmani R. Lipoprotein-associated phospholipase A2 protein expression in the natural progression of human coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:2523–2529. doi: 10.1161/01.ATV.0000244681.72738.bc. doi: 10.1161/01.ATV.0000244681.72738.bc. [DOI] [PubMed] [Google Scholar]
- 23.Zalewski A, Macphee C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis: biology, epidemiology, and possible therapeutic target. Arterioscler Thromb Vasc Biol. 2005;25:923–931. doi: 10.1161/01.ATV.0000160551.21962.a7. doi: 10.1161/01.ATV.0000160551.21962.a7. [DOI] [PubMed] [Google Scholar]
- 24.Tyurin VA, Balasubramanian K, Winnica D, Tyurina YY, Vikulina AS, He RR, Kapralov AA, Macphee CH, Kagan VE. Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic ‘eat-me’ signals: cleavage and inhibition of phagocytosis by Lp-PLA2. Cell Death Differ. 2014;21:825–835. doi: 10.1038/cdd.2014.1. doi: 10.1038/cdd.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Donoghue ML, Braunwald E, White HD, Serruys P, Steg PG, Hochman J, Maggioni AP, Bode C, Weaver D, Johnson JL, Cicconetti G, Lukas MA, Tarka E, Cannon CP. Study design and rationale for the Stabilization of pLaques usIng Darapladib-Thrombolysis in Myocardial Infarction (SOLID-TIMI 52) trial in patients after an acute coronary syndrome. Am Heart J. 2011;162:613–619.e1. doi: 10.1016/j.ahj.2011.07.018. doi: 10.1016/j.ahj.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 26.White H, Held C, Stewart R, Watson D, Harrington R, Budaj A, Steg PG, Cannon CP, Krug-Gourley S, Wittes J, Trivedi T, Tarka E, Wallentin L. Study design and rationale for the clinical outcomes of the STABILITY Trial (STabilization of Atherosclerotic plaque By Initiation of darapLadIb TherapY) comparing darapladib versus placebo in patients with coronary heart disease. Am Heart J. 2010;160:655–661. doi: 10.1016/j.ahj.2010.07.006. doi: 10.1016/j.ahj.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 27.O’Donoghue ML, Braunwald E, White HD, Steen DP, Lukas MA, Tarka E, Steg PG, Hochman JS, Bode C, Maggioni AP, Im K, Shannon JB, Davies RY, Murphy SA, Crugnale SE, Wiviott SD, Bonaca MP, Watson DF, Weaver WD, Serruys PW, Cannon CP, Steen DL SOLID-TIMI 52 Investigators. Effect of darapladib on major coronary events after an acute coronary syndrome: the SOLID-TIMI 52 randomized clinical trial. JAMA. 2014;312:1006–1015. doi: 10.1001/jama.2014.11061. doi: 10.1001/jama.2014.11061. [DOI] [PubMed] [Google Scholar]