

Abstract
The voltage-gated Na+ channel Nav1.5 is essential for action potential (AP) formation and electrophysiological homoeostasis in the heart. The ubiquitin–proteasome system (UPS) is a major degradative system for intracellular proteins including ion channels. The ubiquitin protein ligase E3 component N-recognin (UBR) family is a part of the UPS; however, their roles in regulating cardiac Nav1.5 channels remain elusive. Here, we found that all of the UBR members were expressed in cardiomyocytes. Individual knockdown of UBR3 or UBR6, but not of other UBR members, significantly increased Nav1.5 protein levels in neonatal rat ventricular myocytes, and this effect was verified in HEK293T cells expressing Nav1.5 channels. The UBR3/6-dependent regulation of Nav1.5 channels was not transcriptionally mediated, and pharmacological inhibition of protein biosynthesis failed to counteract the increase in Nav1.5 protein caused by UBR3/6 reduction, suggesting a degradative modulation of UBR3/6 on Nav1.5. Furthermore, the effects of UBR3/6 knockdown on Nav1.5 proteins were abolished under the inhibition of proteasome activity, and UBR3/6 knockdown reduced Nav1.5 ubiquitylation. The double UBR3–UBR6 knockdown resulted in comparable increases in Nav1.5 proteins to that observed for single knockdown of either UBR3 or UBR6. Electrophysiological recordings showed that UBR3/6 reduction-mediated increase in Nav1.5 protein enhanced the opening of Nav1.5 channels and thereby the amplitude of the AP. Thus, our findings indicate that UBR3/6 regulate cardiomyocyte Nav1.5 channel protein levels via the ubiquitin–proteasome pathway. It is likely that UBR3/6 have the potential to be a therapeutic target for cardiac arrhythmias.
Keywords: Nav1.5 channel, UBR, cardiomyocyte, degradation, ubiquitin, proteasome
Introduction
The Nav1.5 channel, which is the cardiac isoform of the voltage-gated Na+ channel, is critical for the generation of the cardiac action potential (AP) and the conduction of electrical impulses 1. As a membrane protein, the function of Nav1.5 in cell electrical excitability not only depends on its own activation but also on the number of Na+ channels in the cardiomyocyte plasma membrane 2,3. Although multiple lines of evidence have documented the molecular mechanisms underlying the Nav1.5 channel activation, there is a substantial lack of understanding concerning its degradative regulation.
The rapid internalization and degradation of Nav1.5 channels have been reported to be associated with the ubiquitin–proteasome system (UPS) 4,5. The UPS, which consists of ubiquitin (Ub), Ub-activating enzyme (E1), Ub-conjugating enzyme (E2), Ub-protein ligase (E3) and the proteasome, is the major degradation pathway of intracellular proteins including voltage-gated channels 4,6. Ub can bind to Nav1.5 via the E1–E2–E3 enzyme cascade and post-translationally regulate the expression of Nav1.5 channels. This process is also a prerequisite for ion channel protein internalization and degradation 7. Ubiquitylation of plasma membrane proteins generally induce their endocytosis, followed by lysosomal or proteasomal degradation 8,9. As a plasma membrane protein, Nav1.5 has been observed to be ubiquitylated by a ubiquitin E3 ligase Nedd4-2, which leads to Nav1.5 internalization rather than degradation 1,8,10. It remains unclear whether the UPS is involved in the regulation of Nav1.5.
In the UPS system, the E3 protein superfamily is the largest family which consists of more than 500 distinct members 11. Among these members, a subfamily termed Ub-protein ligase E3 component N-recognins (UBRs) shares a conserved zinc finger-like 70-residue domain with mammalian E3s and contains at least seven members (UBR1–UBR7). UBR1, UBR2, UBR4 and UBR5 were captured by N-terminal degradation determinants, whereas UBR3, UBR6 and UBR7 were not 12. These UBRs may be involved in Johanson–Blizzard syndrome 13, the sensory system 14 and neurogenesis 15. In addition, UBR1, UBR2, UBR4 and UBR5 in the N-end rule pathway play an important role in cardiac proliferation and hypertrophy 16, angiogenesis 17 and cardiovascular development 15. However, whether UBRs affect the electrical activity of cardiomyocytes remains unknown.
In the present study, we identified the transcript expression profiles of all of the UBR members present in rat cardiomyocytes. Gene knockdown analysis revealed the distinct regulative effects of UBR3 and UBR6 on Nav1.5 channels. Further studies showed that UBR3 and UBR6 mediated Nav1.5 degradation through the UPS. The UBR3/6-mediated regulation of Nav1.5 could change the opening of Nav1.5 channels and thereby the amplitude of the APs.
Materials and methods
RNA interference
Rat UBR1–UBR7 and human UBR3 and UBR6 were knocked down by specific small interference RNAs (siRNAs) (Jima, Shanghai, China). The scramble control RNA (Shanghai GenePharma Co., Ltd, Shanghai, China) was set up using the 21-nucleotide RNA oligonucleotide that corresponded to the coding sequence of luciferase. All of the siRNA sequences are listed in Table1. Lipofectamine RNAiMAX Reagent (Invitrogen, Carlsbad, CA, USA) was used to transfect the siRNAs into rat cardiomyocytes or HEK293T cells according to the manufacturer’s protocols. The suppression efficiency of UBR1–UBR7 was determined by immunoblotting.
Table 1.
The siRNA sequences in this study
| Species | Target gene symbol | Sequence (5′–3′) |
|---|---|---|
| Rat | UBR1 | S-GGCCCGACAUCUUAUUGAATT |
| A-UCAAUAAGAUGUCGGGCCTT | ||
| UBR2 | S-GCGCCACAGAUGAAAUCAATT | |
| A-UUGAUUUCAUCUGUGGCGCTT | ||
| UBR3 | S-GCGGCACUUUAUAAAUUAUTT | |
| A-AUAAUUUAUAAAGUGCCGCTT | ||
| UBR4 | S-CUCCACCACAGAUGAAGAATT | |
| A-UUCUUCAUCUGUGGUGGAGTT | ||
| UBR5 | S-GGGCCUUAUUCCUAAGUAUTT | |
| A-AUACUUAGGAAUAAGGCCCTT | ||
| UBR6 | S-GUCCAAUCCUUGUACAUUATT | |
| A-UAAUGUACAAGGAUUGGACTT | ||
| UBR7 | S-GACUGAACUUAAGGAUUAUTT | |
| A-AUAAUCCUUAAGUUCAGUCTT | ||
| Homo | UBR3 | S-CCGUCUUUGAAAGAUUUAATT |
| A-UUAAAUCUUUCAAAGACGGTT | ||
| UBR6 | S-GCAGACUGGAGGAAUAUAUTT | |
| A-AUAUAUUCCUCCAGUCUGCTT | ||
| Negative control | S-UUCUCCGAACGUGUCACGUTT | |
| A-ACGUGACACGUUCGGAGAATT |
S: sense; A: antisense.
Isolation of primary neonatal rat ventricular myocytes
All of the animal experiments were approved by the Animal Experiment Committee of Tongji University School of Medicine and conformed to the Guide for the Care and Use of Laboratory Animals (1996) from the U.S. National Institutes of Health. Neonatal rat ventricular myocytes (NRVM) were isolated from the hearts of 1- to 2-day-old Sprague–Dawley rat pups as previously described 18. In brief, after the hearts of the neonatal rats were excised and washed, the ventricles were minced and incubated in a PBS solution containing trypsin (0.25%), collagenase (0.1%) and DNAase (1%) for 5 min. at 37°C. Next, the isolated cells were transferred into a tube containing DMEM supplemented with 10% foetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin. The same procedure was repeated five times to collect a sufficient number of cells. To wash the impurities, the cell pellet was centrifuged and re-suspended. The isolated cells were then purified by differential adhesion for 2 hrs. Finally, the purity of the cardiomyocytes was approximately 98%, as assessed by myocardial troponin immunofluorescence staining. Unsettled cells were washed after 24 hrs, and the medium was replaced daily.
Cell culture and transfection
The NRVMs and human embryonic kidney 293T (HEK293T) cells were maintained in DMEM supplemented with 10% foetal calf serum (Gibco, Waltham, MA, USA), 1% penicillin and streptomycin in a humidified incubator at 37°C with 5% CO2. All of the cells were cultured to 70–90% confluence before transfection. The NRVMs were transfected using the siRNAs. The siRNA of human UBR3/6 as well as Na+ channel plasmids (OriGene, Rockville, MD, USA) were cotransfected into HEK293T cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Exactly, 0.003% SDS and 2 μmol/l MG132 [N-[(phenylmethoxy)carbonyl]-l-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-l-leu-cinamide were employed to activate or inhibit the proteasome activity respectively.
RNA extraction, reverse transcription PCR and quantitative real-time PCR
Total RNA was extracted from neonatal rat myocardium and siRNA-treated cells at 48 hrs using TRIzol reagent (Invitrogen) as previously described 19. Reverse transcription PCR (RT-PCR) was conducted using the 1st Strand cDNA Synthesis Kit (TAKARA, Dalian, China) following procedures outlined in the manufacturer’s instructions. Real-time PCR was performed with the SYBR® Premix Ex Taq™ II kit (TAKARA) to identify gene expression. Relative expression levels of target genes were calculated by calibrating to the house-keeping gene β-actin as 2-(target gene Ct-reference gene Ct). All of the RT-PCR primer sequences are shown in Table2.
Table 2.
The RT-PCR primer sequences in this study
| Species | Target gene symbol | Sequence (5′–3′) |
|---|---|---|
| Rat | UBR1 | F-CTTAGCGTTCCCGTCCTTGT |
| R-GCCATGGTGACCAGATGGAA | ||
| UBR2 | F-TACCAACCAACCTCATCCGC | |
| R-AGTTTGTTGGCTCCTCTCGG | ||
| UBR3 | F-AGGCATGCAGAACAAGGGAA | |
| R-GGAACCTTGGTGCAGACACT | ||
| UBR4 | F-AGTGCAATGGACTCCTTCCG | |
| R-GCGCAGGAAAAGCAGTTTGA | ||
| UBR5 | F-CTGTCGGCAAGGTGTGCTTA | |
| R-GCTCTCTGGAGACCGAAGTT | ||
| UBR6 | F-CCCACAGTGGTTCGATGTGA | |
| R-ATCCATACGCCTGCGAAGTT | ||
| UBR7 | F-GCCACCTATTGGCCCTTGAA | |
| R-TGTCAGTTGCCTGGTCACTC | ||
| β-actin | F-CTGGAACGGTGAAGGTGACA | |
| R-AAGGGACTTCCTGTAACAATGCA |
F: forward; R: reverse.
Western blot analysis
The cells were lysed with radioimmunoprecipitation assay (RIPA) lysis buffer [150 mM NaCl, 50 mM Tris–HCl (pH 7.4), 1% sodium deoxycholate, 1% NP-40, 1 mM PMSF and 1 mM ethylenediaminetetraacetic acid] at 4°C for 20 min. After centrifugation at 20,000 × g and 4°C for 10 min., 4× SDS gel sample buffer (Invitrogen) was added to the cleared lysate. The proteins were fractionated based on their molecular weight by SDS-PAGE (Invitrogen), transferred onto a polyvinylidene fluoride membrane (Invitrogen) at a constant current of 350 mA for 90 min. at 4°C and immunoblotted using the corresponding antibodies (anti-UBR3 and anti-UBR6 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-Nav1.5 antibodies were purchased from Sigma-Aldrich, St. Louis, MO, USA; anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were obtained from Cell Signaling (Danvers, MA, USA); anti-Ubiquitin antibodies were obtained from Abcam (Cambridge, MA, USA); horseradish peroxidase-conjugated secondary antibodies were obtained from Santa Cruz Biotechnology).
Determination of Nav1.5 protein ubiquitination
Total proteins were extracted with RIPA buffer from HEK293 cells transfected with Nav1.5 plasmids, either alone or combined with UBR3/6 siRNA. They were incubated for 12 hrs by rotation at 4°C with either anti-Nav1.5 antibodies or the isotype control antibodies, added with protein-A/G-Sepharose beads (Beyotime, Shanghai, China), and incubated for a further 3 hrs to precipitate the protein-antibody complexes. Then, the precipitates were electrophoresed on a SDS gel, and immunoblotted with anti-Nav1.5 and anti-ubiquitin antibodies.
Cycloheximide blocking assay
Cycloheximide (CHX), a specific inhibitor of protein synthesis that works by preventing the translocation of ribosomes, can effectively inhibit de novo protein synthesis in eukaryotic cells 20,21. This process was accomplished by treating UBR3/6 knockdown cells with or without CHX to test the additional translational inhibition. More specifically, NRVMs were transfected with the siRNA of UBR3/6 for 24 hrs. Next, we added 100 μg/ml CHX (Sigma-Aldrich). Aliquots of cells were collected at the 24-hr time-point and then subsequently at 4, 8 and 12 hrs after CHX addition. Protein was then extracted, and Western blot analysis was performed to analyse the levels of Nav1.5 channel proteins.
Electrophysiological measurements
Standard voltage and current clamp techniques were used to assess the cardiac Na+ current and AP properties respectively 22,23. Whole-cell patch-clamp recordings were performed at room temperature (24°C) using an EPC-10 amplifier and pulse software (HEKA, Ludwigshafen, Germany) on the 2nd or 3rd day. Single cell cardiac electrophysiological properties were acquired from healthy NRVM. The extracellular solution (pH 7.4, titrated with NaOH) consisted of the following (in mM): NaCl 140, CsCl 10, CaCl2 2, MgCl 21, glucose 5 and 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) 10. The intracellular solution (pH 7.2, titrated with CsOH) contained the following (in mM): CsF 110, CsCl 10, NaF 10, ethylene glycol tetraacetate (EGTA) 11, CaCl2 1, MgCl 21, Na2ATP 2 and HEPES 10.
Statistical analysis
All of the data are presented as the mean ± SEM. Statistical differences among multiple groups were compared by one-way anova followed by the Fisher’s least significant difference test, using SPSS 13.0 (SPSS Inc., Chicago, IL, USA). P < 0.05 was considered statistically significant. Voltage-clamp data were compiled and analysed using Pulsefit (HEKA) and Origin 7.5 (OriginLab, Northampton, MA, USA). Single cell current amplitudes were normalized for differences in cell size by whole-cell membrane capacitance. Data were collected from at least three independent batches of experiments.
Results
Knockdown of UBR 3 and 6 elevates Nav1.5 channel protein expression
To analyse the potential roles of UBR1–7 in Nav1.5 expression, we first performed RT-PCR to test the mRNA expression of UBR1–UBR7 in rat cardiomyocytes. As shown in Figure1, all of the UBR members were expressed. UBR6 displayed the highest expression level, while UBR5 and UBR7 had the lowest expression. Next, using the gene silencing approach, we aimed to determine whether UBRs affected the protein expression of intracellular Nav1.5 channels. Considering the sequence similarity of UBR genes, we examined the specificity and efficiency of siRNA knockdown in NRVMs. After 48 hrs of transfection, the gene expression of the UBR family was tested by RT-PCR, and the results showed that UBR1–UBR7 siRNAs specifically interfered with their respective UBRs (Table3). Then, we measured the protein expression of Nav1.5 channels in UBR-deficient NRVMs. The Western blot data demonstrated that knockdown of UBR3 and UBR6 but not UBR1, UBR2, UBR4, UBR5 or UBR7 significantly increased the protein levels of intracellular Nav1.5 channels (Fig.2A and B). In another set of experiments with HEK293T cells expressing Nav1.5 channels, down-regulation of UBR3/6 showed effects on Nav1.5 that were similar to those observed in NRVMs (Fig.2C). These findings suggest that both UBR3 and UBR6 may play an important role in the regulation of intracellular Nav1.5 channel expression. Next, we examined the effects of UBR3–UBR6 double knockdown. The increase in Nav1.5 channel protein was also observed for UBR3–UBR6 double knockdown, which was comparable to that for the individual knockdown of either UBR3 or UBR6 (Fig.2D).
Figure 1.
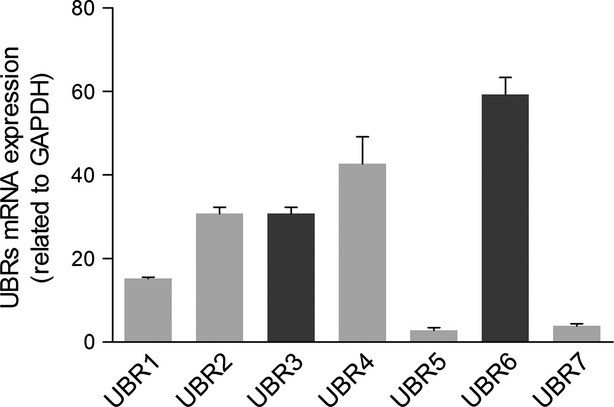
Quantitative PCR analysis of UBRs in NRVMs. The mRNA expression of every member of the UBR family could be detected in NRVMs. UBR2, UBR3, UBR4 and UBR6 had a relatively higher expression, while UBR5 and UBR7 were lower. All of the UBR1–7 primer pairs are listed in Table2. GAPDH served as a control (n = 3).
Table 3.
The specificity and the efficiency of siRNA knockdown of UBR members
| UBR1 | UBR2 | UBR3 | UBR4 | UBR5 | UBR6 | UBR7 | |
|---|---|---|---|---|---|---|---|
| SiRNA of UBR1 | ++++ | − | + | − | − | − | − |
| SiRNA of UBR2 | − | ++++ | + | + | − | − | − |
| SiRNA of UBR3 | + | + | +++ | − | + | − | − |
| SiRNA of UBR4 | − | − | − | ++ | − | − | − |
| SiRNA of UBR5 | − | − | − | − | ++++ | − | − |
| SiRNA of UBR6 | + | − | + | + | − | ++++ | + |
| SiRNA of UBR7 | + | + | + | + | + | − | ++++ |
++++ means the knockdown efficiency (decrease in protein level) >70%, +++ means 50–70%, ++ means 35–50%, + means 35–25%, − means <25%.
Figure 2.
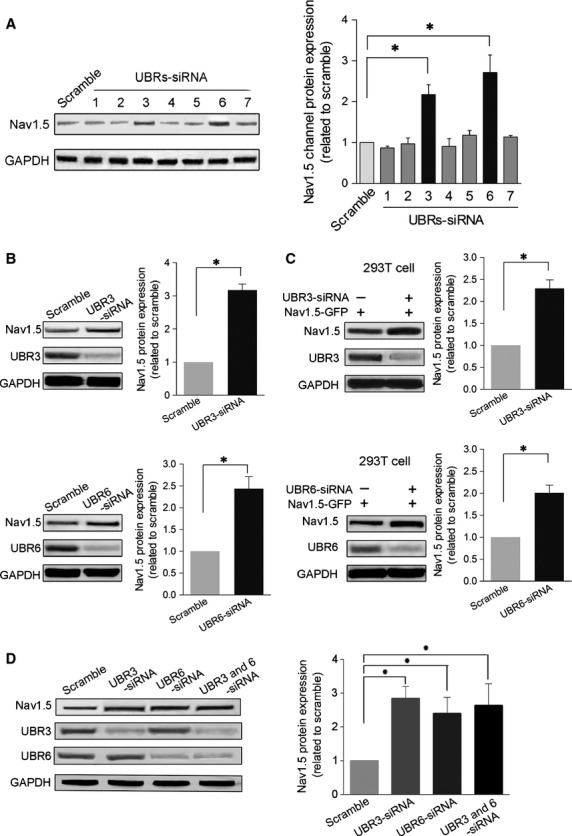
Protein expression of Nav1.5 channels in UBR knockdown cells. (A) Left. A typical example of a Western blot is shown using anti-Nav1.5 antibodies to assess the expression of Nav1.5 channel protein in NRVMs receiving different UBR siRNAs. Only UBR3/6 knockdown significantly changed Nav1.5 channel expression. GAPDH served as a loading control. Right. The pooled and quantified Western blot data. (B) Western blot analysis of Nav1.5 channels in UBR3/6 knockdown NRVMs. UBR3/6 significantly increased Nav1.5 expression in NRVMs. Upper. Western blot analysis and pooled data for Nav1.5 channels and UBR3 protein expression with or without transfection of UBR3 siRNA. GAPDH served as a loading control. Lower. Western blot analysis and pooled data using anti-Nav1.5 and anti-UBR6 antibodies to analyse Nav1.5 channel expression with or without transfection of UBR6 siRNA. GAPDH served as a loading control. (C) Western blot analysis in HNK293T cells expressing Nav1.5 channels after UBR3/6 knockdown. UBR3/6 could significantly increase Nav1.5 expression. Upper. Western blot and pooled data for Nav1.5 channels and UBR3 proteins level in HNK293T cells expressing Nav1.5 channels with or without UBR3 siRNA. GAPDH served as a loading control. Lower. Western blot analysis and pooled data using anti-Nav1.5 and anti-UBR6 antibodies to analyse Nav1.5 channel expression in HNK293T cells expressing Nav1.5 channels with or without UBR6 siRNA. GAPDH served as a loading control. (D) Western blot analysis of Nav1.5 channels in NRVMs. Cell were transfected with UBR3 and UBR 6 siRNA either alone or combined together. The scramble control RNA served as the control. Left. A typical example of a Western blot image for Nav1.5 channels and UBR3/6 protein expression. GAPDH served as a loading control. Right. Quantitative data for Western blots. All the data are from three independent experiments, *P < 0.01.
UBR3/6 regulate the protein expression of Nav1.5 channels via degradation
The orchestration of mRNA transcription, protein translation, post-translational modification and protein degradation determines protein levels in cells. To understand the potential reasons underlying the increased Nav1.5 channel expression in UBR3/6-deficient cells, we first measured the mRNA expression of Nav1.5 channels. The RT-PCR results showed that UBR3/6 knockdown did not significantly change the mRNA level of Nav1.5 (Fig.3A). Next, CHX, an effective inhibitor of protein synthesis, was used to explore whether enhanced Nav1.5 protein synthesis contributed to its elevation in response to reduced levels of UBR3/6. Western blot data revealed that CHX treatment resulted in a time-dependent decrease in endogenous Nav1.5 levels in normal cells. Conversely, the pharmacological treatment did not counteract the increase in Nav1.5 protein caused by UBR3/6 knockdown (Fig.3B and C). Then, a proteasome activator SDS (0.003%) and a proteasome inhibitor MG132 (2 μM) were used to examine whether the proteasome pathway is involved in Nav1.5 protein degradation. As shown in Figure3D, SDS reduced Nav1.5 proteins, whereas MG132 increased Nav1.5 proteins. The additional application of UBR3/6 knockdown did not modify the increase in Nav1.5 proteins caused by MG132. These results suggest that UBR3 and UBR6 may degrade Nav1.5 channels through the proteasome way. To test whether ubiquitination mediated Nav1.5 degradation through proteasomes, we examined Nav1.5 ubiquitylation by immunoprecipitating Nav1.5 proteins followed by anti-ubiquitin immunoblotting, using HEK293T cells overexpressing Nav1.5. In Western blots for ubiquitylation, both unmodified Nav1.5 and ubiquitinated Nav1.5 were detected in the samples after immunoprecipitation (IP) with anti-Nav1.5 (Fig.3E). The ubiquitinated Nav1.5 displayed a larger molecular weight compared to the unmodified Nav1.5, and long-chain ubiquitylation of Nav1.5 is indicated by the molecular weight difference. In contrast to the very diffuse ubiquitylation band of whole-cell lysate (Fig.3E, upper panel, right lane), the bands of ubiquitinated Nav1.5 are concentrated (Fig.3E, upper panel, left 3 lanes), indicating that Nav1.5 proteins carry homogenous amounts of ubiquitin moieties. The amount of ubiquitinated Nav1.5 was remarkably reduced by the knockdown of either UBR3 or UBR6. The results showed that UBR3/6 knockdown reduced Nav1.5 ubiquitylation level, suggesting the stimulative effects of UBR3/6 on Nav1.5 ubiquitylation (Fig.3E).
Figure 3.
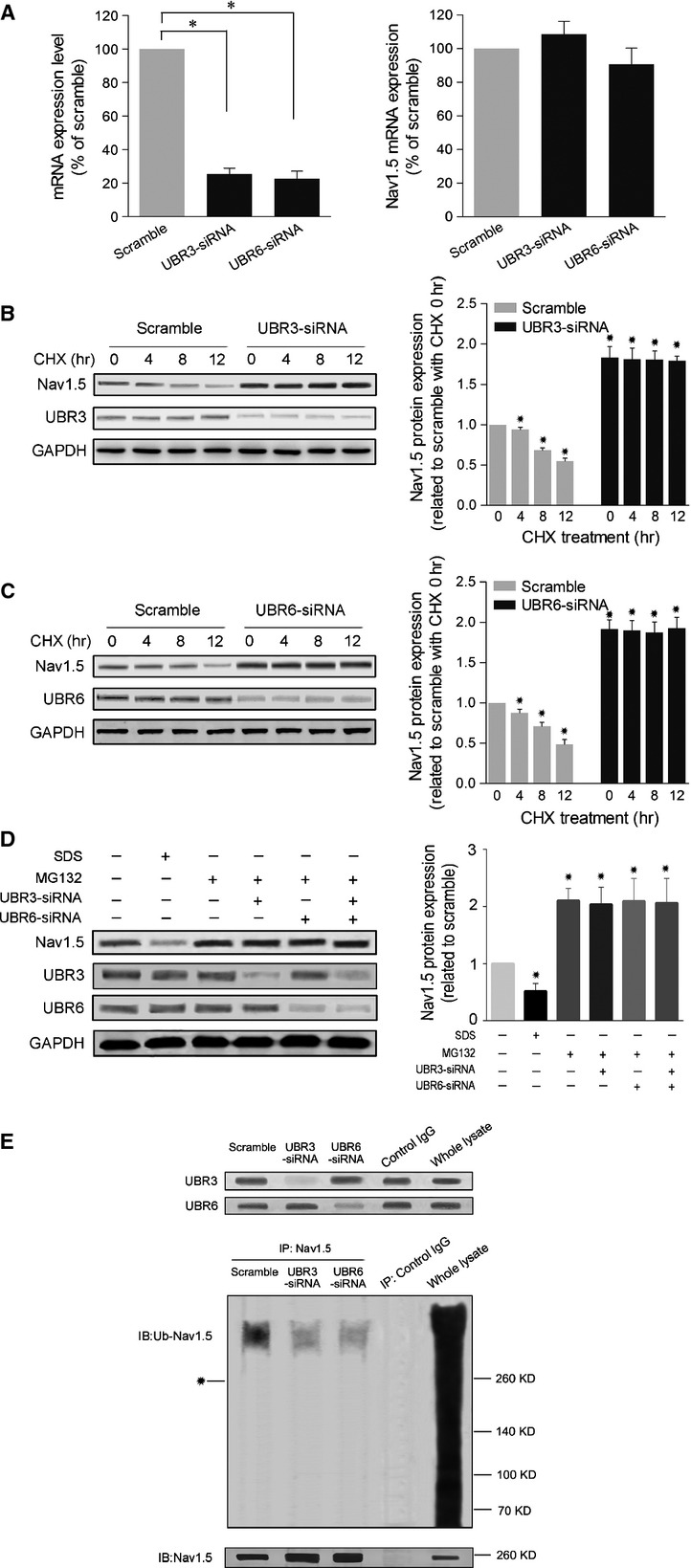
De novo synthesis inhibiting assay and proteasome inhibiting assay of UBR3/6 knockdown-induced increases in Nav1.5 channels. (A) mRNA expression of Nav1.5 channels after UBR3/6 knockdown in NRVMs. UBR3/6 did not affect the mRNA level of Nav1.5 channels. Left. The efficiency of UBR3/6-siRNA knockdown in NRVMs. Right. The mRNA expression of Nav1.5 channels in UBR3/6 knockdown NRVMs. β-actin served as a control. (B) Effect of cycloheximide, an inhibitor for protein synthesis, on Nav1.5 channel expression in UBR3 knockdown NRVMs. NRVMs were transfected with or without UBR3 siRNA for 24 hrs prior to subsequent addition with cycloheximide (CHX) (100 μg/ml). Aliquots of the cells were collected at 4, 8 and 12 hrs after CHX treatment for Western blot analysis. A typical example of a Western blot analysis (left panel) and the summarized data (right panel) are shown (n = 3, *P < 0.01). Following CHX treatment, the endogenous Nav1.5 protein level showed a time-dependent decrease in cells receiving scramble control RNA. The increase in Nav1.5 protein in UBR3 knockdown cells was not counteracted. (C) Effect of cycloheximide on Nav1.5 channel expression in UBR6 knockdown NRVMs. NRVMs were transfected with or without UBR6 siRNA for 24 hrs prior to subsequent treatment with cycloheximide (CHX) (100 μg/ml). Aliquots of the cells were collected at 4, 8 and 12 hrs after CHX treatment for Western blot analysis. A typical example of a Western blot analysis (left panel) and the summarized data (right panel) are provided (n = 3, *P < 0.01). Following CHX treatment, the endogenous Nav1.5 protein level showed a time-dependent decrease in scramble control cells. The UBR3/6 reduction-induced increase in Nav1.5 protein was not counteracted. (D) Effect of proteasome activation and inhibition on Nav1.5 protein levels in NRVMs. SDS (0.003%) and MG132 (2 μM) were used to activate and inhibit proteasomes respectively. SDS was incubated for 24 hrs before the harvest of NRVMs. Individual or combined siRNAs against UBR3 and UBR6 were transfected for 12 hrs prior to subsequent addition with MG132 for a further 24 hrs. A typical example of a Western blot analysis (left panel) and the summarized data (right panel) are provided (n = 3, *P < 0.01). (E) Examination of Nav1.5 protein ubiquitination in Nav1.5-overexpressing HEK293 cells receiving UBR3/6 knockdown. Cell lysates were immunoprecipitated with anti-Nav1.5 antibodies (IP: Nav1.5), then immunoblotted with anti-ubiquitin (IB: Ub-Nav1.5) and anti-Nav1.5 (IB: Nav1.5) antibodies. *Position of unmodified Nav1.5. The image is a representative from three independent experiments.
Reduced levels of UBR3/6 enhance the opening of Nav1.5 channel and thereby the amplitude of the action potential
Given the putative roles of Nav1.5 in the generation of an AP and the effects of UBR3/6 on Nav1.5 expression, we further measured Na+ currents and APs in NRVMs with or without UBR3/6 siRNA (Fig.4). We found that knockdown of UBR3/6 did not change the shape of the I–V curve of the Nav1.5 channel but significantly increased the peak current density (P < 0.01) (Fig.4A and B). The peak current density generated by the Nav1.5 channels in UBR3 knockdown cells increased from (−51.49 ± 7.735) pA/pF to (−75.94 ± 13.43) pA/pF (n > 10; P < 0.01). The peak current density of the Nav1.5 channels in UBR6 knockdown cells could be elevated from (−51.49 ± 7.735) pA/pF to (−87.06 ± 10.22) pA/pF (n > 10; P < 0.01). However, there was no change in the activation curve in UBR3/6-deficient and normal cells (Fig.4C). Moreover, both UBR3 and UBR6 knockdown significantly increased the amplitude of the AP (Fig.4D and E). These results suggest that UBR3/6 regulate Nav1.5 channel opening and AP in cardiomyocytes.
Figure 4.
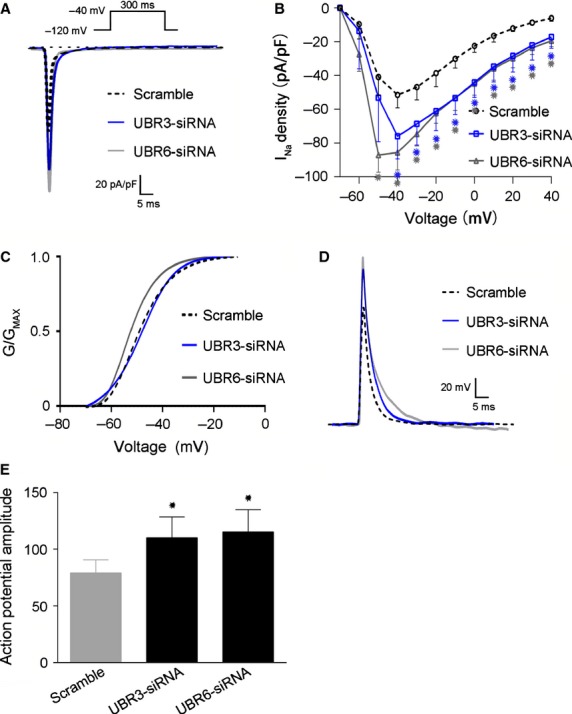
Effects of UBR3/6 knockdown on Nav1.5 channel currents and action potentials. (A) Representative tracings of Nav1.5 currents from rat cardiomyocytes. The amplitude of the UBR3/6 knockdown cells was increased. (B) Current–voltage (I–V) relationship of transient INa from rat cardiomyocytes (n > 10 per group *P < 0.01). The current traces were recorded at Vm in the range of −70 to +40 mV from a holding potential of −120 mV. The INa density of the UBR3/6 knockdown cells was elevated compared to the NC. (C) The activation curve for Nav1.5 channels from NC (negative control) and UBR3/6 knockdown rat cardiomyocytes (n > 10 per group). There were no significant differences between them. (D) Representative AP (action potential) recordings from NC (negative control) and UBR3/6 knockdown rat cardiomyocytes. (E) Statistical analysis of the amplitude of the APA (action potential amplitude) (n > 10 per group, *P < 0.01). The amplitude was increased in UBR3/6 knockdown cells.
Discussion
In the present study, we investigated the effects of UBR isoforms on the expression of cardiac Nav1.5 channels. We found that only UBR3 and UBR6 affected the protein levels of Nav1.5 channels. The UBR3/6-mediated regulation of Nav1.5 channels was not transcriptionally mediated but was associated with the ubiquitin–proteasome degradation system. Furthermore, UBR3/6 knockdown enhanced the opening of Nav1.5 channels and thereby the amplitude of APs. Overall, we identified a new regulatory function of UBR3 and UBR6 in cardiac electrophysiology by the degradation of Nav1.5 protein.
Ubiquitination is a key step in the ubiquitin–proteasome pathway, which serves as an important regulator for plasma membrane proteins in their trafficking, internalization and degradation 24. Once ubiquitylated, the internalized membrane proteins can be targeted for lysosomal or proteasomal degradation 8. In the UPS, E3 ligase is the key enzyme in recognizing the specificity degradation signals of substrate ubiquitylation 11. UBR box family is a unique class of E3 ligase that recognize N-degrons or structurally related determinants for ubiquitin-dependent proteolysis and perhaps other processes as well. The mammalian genome encodes at least seven UBR box-containing proteins, called UBR1 to UBR7 25. UBR1 is a 225-kD RING-type E3 ligase containing at least three substrate-binding sites 25. The mouse UBR1 and UBR2 are 200-kD N-recognins with 47% identity and 68% similarity to each other, and are functionally overlapped 26. UBR3 is a 213-kD RING finger E3, shares weak but detectable homology to UBR1 and UBR2 25. UBR4 is an exceptionally large protein with a MW of 570-kD, and UBR5 (300-kD) is also known as EDD/Hhyd. Phylogenetic analysis of the UBR box motif sequences from yeast to mouse classified them into the following subfamilies: UBR1/2/3 (with the RING finger), UBR4, UBR6 (with an F-box), UBR5 (with the HECT domain) and UBR7 (with the plant homeodomain) 25. In recent studies, Nav1.5 has been reported to be internalized by the ubiquitin E3 ligase Nedd4-2, and thus down-regulated from its functional expression 27,28. However, the internalization of Nav1.5 by Nedd4-2 does not seem to be linked to degradation as there was no reduction in the total level of Nav1.5 protein upon co-expression with Nedd4-2 1,8,10. In contrast, another study found that pharmacological inhibition of the proteasome was linked to an increase in Nav1.5 expression and INa in neonatal rat cardiomyocytes 5. However, it is unclear whether any ubiquitin E3 ligases target Nav1.5 for the ubiquitin–proteasome degradation pathway. The UBR family has been described to be involved in the function of olfactory and other sensory systems 14, neurogenesis and cardiovascular development 15, and genome stability 17,29. Nevertheless, the roles of UBR family members in cardiomyocytes remain unknown. In the present study, we sought to elucidate the relationships between UBRs and Nav1.5 expression. Our results showed that individual knockdown of UBR3 and UBR6, but not other UBR isoforms, increased the protein levels of Nav1.5 channels in neonatal rat cardiomyocytes. Next, we confirmed that this regulation of Nav1.5 channels by UBR3/6 was not transcriptionally mediated as the inhibition of de novo protein synthesis did not affect the increase in Nav1.5 proteins upon UBR3/6 knockdown. We further observed that the inhibition of proteasomes with MG132 abolished the effects of UBR3/6 knockdown (Fig.3D), and that UBR3/6 knockdown reduced Nav1.5 ubiquitylation (Fig.3E). These results strongly suggest that UBR3/6 promote Nav1.5 ubiquitylation and degradation through proteasomes. Interestingly, we observed complete stabilization of Nav1.5 proteins with single knockdown of either UBR3 or UBR6. Besides, double UBR3–UBR6 knockdown resulted in similar increases of Nav1.5 proteins, compared to those obtained with single knockdown (Figs2D and 3D). It is likely that UBR3 and UBR6 act in concert to promote degradation of Nav1.5. Another possibility is that UBR3 and UBR6 may take effect through similar mechanisms. The covalent binding sites on Nav1.5 for ubiquitylation are theoretically limited. UBR3 and UBR6 may induce the same ubiquitylation level at the same ubiquitylation sites. Thus, single or combined knockdown of UBR3 and UBR6 may take the same effect. Notably, unlike UBR1, UBR2 or UBR4, although UBR3 and UBR6 belong to the UBR family, they are not bound to the known N-end rule substrates 25,30. UBR3 and UBR6 may have other non-N-end rule physiological ligands. The details of UBR3 and UBR6 on Nav1.5 protein ubiquitylation remain to be elucidated. Despite that, our observations revealed the regulation of UBR3 and UBR6 on Nav1.5 protein levels through the ubiquitin–proteasome degradation pathway. To our knowledge, UBR3 and UBR6 are the first E3 members observed to be linked to the degradation of Nav1.5 through proteasomes.
To examine the interaction between UBR3/6 and Nav1.5, we performed co-IP to check whether Nav1.5 binds to UBR3/6, using HEK293T cells overexpressing Nav1.5 proteins. However, no significant binding was shown by the co-IP tests (data not shown). The reasons may lie in technical issues. The amount of Nav1.5 proteins expressed by HEK293T cells might be too small to be detected by co-IP, and the weak and transient interaction between UBR3/6 and Nav1.5 may not allow its detection by co-IP. Also, there is a possibility that the action of UBR3/6 on Nav1.5 ubiquitylation and degradation is not direct but occurs through other ligases, such as Nedd4-2 that has been described previously 1,10. More study is required to determine whether UBR3/6 directly take effect on Nav1.5 or not.
The major cardiac voltage-gated sodium channel Nav1.5 plays a key role in the generation of AP. The opening and closing of this channel can regulate the flow of Na+ and control the voltage gradient between the inside and outside of cells to initiate AP and conduct electrical impulses 31,32. Abnormalities of the Nav1.5 channel may distort AP and thereby lead to different types of arrhythmia syndromes and some congenital and acquired cardiac disorders 33–35. Based on the present evidence, the molecular mechanisms associated with Nav1.5 in these diseases are multitudinous. Nav1.5-associated proteins that can regulate its synthesis, transportation, structure, activity and degradation may also be associated with its function. In contrast to the numerous mutations found in the SCN5A gene 36, the number of related proteins that can regulate Nav1.5 expression is limited 9,37. Deletion of forkhead box protein O1 enhance the expression of cardiac Nav1.5, increase Na+ channel activity and result in a shortened QRS 38. In contrast, defects in the PDZ domain-binding motif of Nav1.5 may reduce its expression and the sodium current in the lateral myocyte membrane, leading to increased anisotropy of the ventricular conduction and Brugada syndrome 39. This finding suggests that Nav1.5 expression is a crucial step in its normal function in cardiomyocytes.
In the present study, we discovered two new regulators of Nav1.5 expression that could reduce the protein levels of Nav1.5 channels by ubiquitination. The UBR3/6-mediated degradation of Nav1.5 affected the opening of ion channels and thereby the amplitude of APs. This discovery has certain pathophysiological implications, suggesting a potential association of UBR3/6 with heart diseases such as arrhythmias. UBR3 and UBR6 might serve as potential targets for therapeutic intervention in ion channel diseases.
Acknowledgments
This work was supported by the National Key Basic Research Program of China (2013CB531100, to Yi-Han Chen), the Fund for National Innovative Research Groups of the National Natural Science Foundation of China (81221001, to Yi-Han Chen), the Major International Joint Research Program Fund of China (81120108004, to Yi-Han Chen), and the General Program of National Natural Science Foundation of China (81170224 and 81270313, to Jun Li; 31271214, to Yi-Han Chen).
Conflicts of interest
The authors declare no competing financial interests.
References
- van Bemmelen MX, Rougier JS, Gavillet B, et al. Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ Res. 2004;95:284–91. doi: 10.1161/01.RES.0000136816.05109.89. [DOI] [PubMed] [Google Scholar]
- Rook MB, Evers MM, Vos MA, et al. Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc Res. 2012;93:12–23. doi: 10.1093/cvr/cvr252. [DOI] [PubMed] [Google Scholar]
- Bongiorno D, Schuetz F, Poronnik P, et al. Regulation of voltage-gated ion channels in excitable cells by the ubiquitin ligases Nedd4 and Nedd4-2. Channels. 2011;5:79–88. doi: 10.4161/chan.5.1.13967. [DOI] [PubMed] [Google Scholar]
- Shao D, Okuse K, Djamgoz MB. Protein-protein interactions involving voltage-gated sodium channels: post-translational regulation, intracellular trafficking and functional expression. Int J Biochem Cell Biol. 2009;41:1471–81. doi: 10.1016/j.biocel.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Kang L, Zheng MQ, Morishima M, et al. Bepridil up-regulates cardiac Na+ channels as a long-term effect by blunting proteasome signals through inhibition of calmodulin activity. Brit J Pharmacol. 2009;157:404–14. doi: 10.1111/j.1476-5381.2009.00174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann J, Ciechanover A, Lerman LO, et al. The ubiquitin-proteasome system in cardiovascular diseases-a hypothesis extended. Cardiovasc Res. 2004;61:11–21. doi: 10.1016/j.cardiores.2003.09.033. [DOI] [PubMed] [Google Scholar]
- Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol. 2003;19:141–72. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- Abriel H. Cardiac sodium channel Na(v)1.5 and interacting proteins: physiology and pathophysiology. J Mol Cell Cardiol. 2010;48:2–11. doi: 10.1016/j.yjmcc.2009.08.025. [DOI] [PubMed] [Google Scholar]
- Abriel H, Kass RS. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc Med. 2005;15:35–40. doi: 10.1016/j.tcm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Rougier JS, van Bemmelen MX, Bruce MC, et al. Molecular determinants of voltage-gated sodium channel regulation by the Nedd4/Nedd4-like proteins. Am J Physiol Cell Physiol. 2005;288:C692–701. doi: 10.1152/ajpcell.00460.2004. [DOI] [PubMed] [Google Scholar]
- Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. 2008;9:679–90. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T, Zakrzewska A, Dudgeon DD, et al. The substrate recognition domains of the N-end rule pathway. J Biol Chem. 2009;284:1884–95. doi: 10.1074/jbc.M803641200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CS, Sukalo M, Batygin O, et al. Ubiquitin ligases of the N-end rule pathway: assessment of mutations in UBR1 that cause the Johanson-Blizzard syndrome. PLoS ONE. 2011;6:e24925. doi: 10.1371/journal.pone.0024925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T, Sohr R, Xia Z, et al. Biochemical and genetic studies of UBR3, a ubiquitin ligase with a function in olfactory and other sensory systems. J Biol Chem. 2007;282:18510–20. doi: 10.1074/jbc.M701894200. [DOI] [PubMed] [Google Scholar]
- An JY, Seo JW, Tasaki T, et al. Impaired neurogenesis and cardiovascular development in mice lacking the E3 ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Proc Natl Acad Sci USA. 2006;103:6212–7. doi: 10.1073/pnas.0601700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Pal K, Tasaki T, et al. Synthetic heterovalent inhibitors targeting recognition E3 components of the N-end rule pathway. Proc Natl Acad Sci USA. 2008;105:100–5. doi: 10.1073/pnas.0708465105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T, Kim ST, Zakrzewska A, et al. UBR box N-recognin-4 (UBR4), an N-recognin of the N-end rule pathway, and its role in yolk sac vascular development and autophagy. Proc Natl Acad Sci USA. 2013;110:3800–5. doi: 10.1073/pnas.1217358110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yan B, Huo Z, et al. beta2- but not beta1-adrenoceptor activation modulates intracellular oxygen availability. J Physiol. 2010;588:2987–98. doi: 10.1113/jphysiol.2010.190900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster G, Vicente R, Coma M, et al. One-step reverse transcription polymerase chain reaction for semiquantitative analysis of mRNA expression. Methods Find Exp Clin Pharmacol. 2002;24:253–9. doi: 10.1358/mf.2002.24.5.802301. [DOI] [PubMed] [Google Scholar]
- Lecona E, Rojas LA, Bonasio R, et al. Polycomb protein SCML2 regulates the cell cycle by binding and modulating CDK/CYCLIN/p21 complexes. PLoS Biol. 2013;11:e1001737. doi: 10.1371/journal.pbio.1001737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudoh A, Daikoku T, Sugaya Y, et al. Inhibition of S-phase cyclin-dependent kinase activity blocks expression of Epstein-Barr virus immediate-early and early genes, preventing viral lytic replication. J Virol. 2004;78:104–15. doi: 10.1128/JVI.78.1.104-115.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach DS, Jones J, Clawson BC, et al. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS ONE. 2013;8:e77843. doi: 10.1371/journal.pone.0077843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein ML, Musa H, Balbuena DP, et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci USA. 2012;109:E2134–43. doi: 10.1073/pnas.1109370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Azzo A, Bongiovanni A, Nastasi T. E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic. 2005;6:429–41. doi: 10.1111/j.1600-0854.2005.00294.x. [DOI] [PubMed] [Google Scholar]
- Tasaki T, Mulder LC, Iwamatsu A, et al. A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol Cell Biol. 2005;25:7120–36. doi: 10.1128/MCB.25.16.7120-7136.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Xia Z, An JY, et al. Female lethality and apoptosis of spermatocytes in mice lacking the UBR2 ubiquitin ligase of the N-end rule pathway. Mol Cell Biol. 2003;23:8255–71. doi: 10.1128/MCB.23.22.8255-8271.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehmer C, Wilhelm V, Palmada M, et al. Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel SCN5A. Cardiovasc Res. 2003;57:1079–84. doi: 10.1016/s0008-6363(02)00837-4. [DOI] [PubMed] [Google Scholar]
- Yang B, Kumar S. Nedd4 and Nedd4-2: closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ. 2010;17:68–77. doi: 10.1038/cdd.2009.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisenberg C, Tait PS, Dianova II, et al. Ubiquitin ligase UBR3 regulates cellular levels of the essential DNA repair protein APE1 and is required for genome stability. Nucleic Acids Res. 2012;40:701–11. doi: 10.1093/nar/gkr744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta-Camacho E, Kozlov G, Li FF, et al. Structural basis of substrate recognition and specificity in the N-end rule pathway. Nat Struct Mol Biol. 2010;17:1182–7. doi: 10.1038/nsmb.1894. [DOI] [PubMed] [Google Scholar]
- Despa S, Bers DM. Na(+) transport in the normal and failing heart - remember the balance. J Mol Cell Cardiol. 2013;61:2–10. doi: 10.1016/j.yjmcc.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bay J, Kohlhaas M, Maack C. Intracellular Na(+) and cardiac metabolism. J Mol Cell Cardiol. 2013;61:20–7. doi: 10.1016/j.yjmcc.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res. 2011;108:884–97. doi: 10.1161/CIRCRESAHA.110.238469. [DOI] [PubMed] [Google Scholar]
- Ge J, Sun A, Paajanen V, et al. Molecular and clinical characterization of a novel SCN5A mutation associated with atrioventricular block and dilated cardiomyopathy. Circ Arrhythm Electrophysiol. 2008;1:83–92. doi: 10.1161/CIRCEP.107.750752. [DOI] [PubMed] [Google Scholar]
- Makiyama T, Akao M, Shizuta S, et al. A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. J Am Coll Cardiol. 2008;52:1326–34. doi: 10.1016/j.jacc.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Remme CA. Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J Physiol. 2013;591:4099–116. doi: 10.1113/jphysiol.2013.256461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shy D, Gillet L, Abriel H. Cardiac sodium channel Nav1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim Biophys Acta. 2013;1833:886–94. doi: 10.1016/j.bbamcr.2012.10.026. [DOI] [PubMed] [Google Scholar]
- Cai B, Wang N, Mao W, et al. Deletion of FoxO1 leads to shortening of QRS by increasing Na(+) channel activity through enhanced expression of both cardiac Nav1.5 and beta3 subunit. J Mol Cell Cardiol. 2014;74:297–306. doi: 10.1016/j.yjmcc.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shy D, Gillet L, Ogrodnik J, et al. PDZ domain-binding motif regulates cardiomyocyte compartment-specific Nav1.5 channel expression and function. Circulation. 2014;130:147–60. doi: 10.1161/CIRCULATIONAHA.113.007852. [DOI] [PubMed] [Google Scholar]