

Introduction
The pathological self-assembly and deposition of proteins is a hallmark of a wide variety of devastating human disorders classified as protein misfolding or amyloid diseases (reviewed in [1-3]). These disorders are widespread and severe, encompassing 3 of the top 15 causes of death in the USA (Alzheimer’s disease (AD), Type II Diabetes Mellitus (T2D) and Parkinson’s disease (PD)) [4] as well as other major conditions including Chronic Traumatic Encephalopathy (CTE), Huntington’s disease and Creutzfeldt-Jakob disease. There is therefore urgent and significant clinical interest in understanding the factors that trigger pathological self-assembly, and the development of pharmacological means by which this disease-related phenomenon may be arrested or mitigated. Initial efforts to understand the molecular mechanisms of amyloid diseases focused on characterization of fibrillar aggregates. Since then, however there has been growing interest in the structures formed along the aggregation pathway (Figure 1a), investigating the hypothesis that these early structures are relevant to toxicity and thus are potential targets for therapeutic intervention.
Figure 1.

Conversion of soluble protein to amyloid aggregates. Proteins may convert from their native states or conformational ensemble (i) to an aggregation-prone form (ii), and then proceed via a population of on-pathway oligomers (iii) to amyloid fibers (iv). Proteins may also form off-pathway oligomers (v), or bind to membranes (vi) in modes which could inhibit or enhance fiber formation. Biologically relevant toxicity could result from states ii, iii, v or vi, while states i and iv are thought to be comparatively inert. b) Schematic of aggregation kinetics displayed by amyloidogenic proteins. Sigmoidal fibrillization kinetics are observed in buffer (black) that can be accelerated, in many cases, by the addition of suitable lipid compositions (blue). The lag phase is bypassed upon the addition of a seed of preformed fibrillar material, demonstrating the nucleation-dependent nature of this process.
The aggregation of amyloidogenic proteins generally display nucleation-dependent kinetics, wherein the conversion from monomer to aggregate displays a lag phase dependent on the accumulation of a nucleus – an on-pathway, high-energy intermediate that must be populated in order for subsequent self-assembly to progress. The lag phase is followed by a rapid, cooperative fibril growth phase until the reaction saturates upon depletion of monomer, and may be reduced in duration or eliminated by the addition of a small amount of ‘seed’ of preformed amyloid material (Figure 1b) [5, 6]. This behavior means that the conformation and population of specific, transiently-populated intermediates early in the self-assembly process play major roles in determining the nature and prevalence of subsequent toxic states. The dynamic and often heterogeneous nature of these transient intermediates makes them highly challenging targets for conventional structural biology approaches. Indeed, recent years have seen the development and application of a number of powerful biophysical approaches to the characterization of intermediates in the self-assembly process [7]. Significant insight has been gained into the thermodynamics driving aggregation as well as the secondary, tertiary and in some cases quarternary structures of molecular species relevant to pathological self-assembly [8-10].
Fluorescence spectroscopy has a number of advantages for the characterization of amyloidogenic proteins, including compatibility with low concentrations of protein (pM or nM in many cases) which inhibits rapid fiber formation, as well as enabling measurements under conditions (such as bound to membranes or even in cells) that are not easily accessible by other approaches. The major drawback is a lack of high resolution structural information relative to techniques such as NMR or EPR. However, in conjunction with molecular dynamics simulations and other computational modeling approaches, fluorescence methods can overcome resolution limitations and generate unique and valuable insights into the conformation and dynamics of amyloidogenic proteins in monomeric and oligomeric states [7, 11, 12]. In this review, we survey selected ensemble and single molecule fluorescence studies that demonstrate the level of residue-level structural information that can be obtained on intermediate species of interest.
Fluorescence approaches to the investigation of amyloidogenic proteins can be broadly divided into three classes. The first capitalizes on the phenomenon of Förster resonance energy transfer (FRET), wherein the efficiency of non-radiative energy transfer from a donor fluorophore to an acceptor reports on the distance between them. FRET measurements may be obtained in steady-state or time-resolved ensemble formats, or via single molecule FRET (smFRET; reviewed in [13-16]). Donor and acceptor fluorophores may be placed on the same molecule for intramolecular FRET, which allows for an assessment of the conformational ensemble and dynamics of the protein, or may be placed on different molecules for intermolecular FRET to interrogate oligomerization of the protein. The second class of measurement involves the site-specific attachment of environmentally-sensitive fluorophores to particular locations in the target protein chain, so as to probe local environment and dynamics in aggregation-prone states or through the course of the aggregation reaction. The third class relies on environment-sensitive fluorescence from histological and diagnostic dyes that bind non-covalently to fibers and/or intermediates in the assembly process, such as Thioflavin-T (ThT) and -S (ThS), Congo Red, Nile Red, and 1-anilinonaphthalene-8-sulfonic acid (ANS) [17-19]. While there has been recent progress in defining the structural modes of interaction of these probes with amyloid and non-amyloid states [20-23], and these methods have generated fundamental insights into our understanding of pathological self-assembly, structural information from environment-sensitive dyes is generally low-resolution and coarse-grained relative to site-specific approaches. The use of these dyes also raises the prospect of false positives [20] and negatives [24] with respect to the existence of amyloid structure. In this review, we therefore focus on the first two classes of measurement described above, and survey selected ensemble and single molecule fluorescence studies that demonstrate the level of residue-level structural information that can be obtained on intermediate species of interest.
We have selected three amyloidogenic proteins for further discussion: islet amyloid polypeptide (IAPP; also called amylin), α-synuclein (AS) and tau. IAPP, AS and tau are implicated in the pathologies of T2D, PD and AD respectively, and share intriguing commonalities in terms of toxic mechanism, the nucleated kinetics of self-assembly, and the importance of electrostatics and templated self-assembly to the aggregation process. Moreover, they are all examples of intrinsically disordered proteins (IDPs): in solution, under physiological conditions, they each sample an ensemble of diverse, extended conformations rather than folding into a single, well-defined structure. The highly dynamic and heterogeneous nature of the native states of this class of proteins presents its own set of challenges to biophysical characterization [7, 25, 26]. These three proteins differ in native function and size, spanning a broad range from 37 (IAPP) to 441 (full-length tau) residues, and thus illustrate the versatility of fluorescence approaches applied to important misfolding diseases. We will describe insights into the nature of aggregation-prone states populated in solution, the effects of molecular inducers of aggregation, and the structures of membrane-bound states of these proteins, followed by a discussion of how future advances in fluorescence methods might impact our understanding of aggregation-prone states and early oligomers.
IAPP
IAPP is a 37-residue, C-terminally amidated, intrinsically disordered peptide hormone secreted by pancreatic islet β-cells (Figure 2a). IAPP has proposed functional roles in the satiety response and the regulation of gastric emptying [27], and is normally packaged into secretory granules along with a ~100-fold excess of insulin. In T2D, IAPP is thought to be responsible for the loss of pancreatic β-cell mass, and is the primary constituent of the characteristic amyloid plaques often observed in T2D patients [28-30].
Figure 2.

a) Sequence of human IAPP and the rat isoform (rIAPP). The native disulfide bond is indicated with a solid line, and the approximate region of helical structure upon binding a lipid bilayer is indicated in bold. b) Representative model of a membrane-bound rIAPP dimer obtained using intermolecular smFRET measurements [31].
The in vitro conversion of IAPP from a disordered monomeric peptide to highly ordered amyloid fibrils has been extensively studied, typically using light scattering by large aggregates or environment-sensitive fluorescence of extrinsic dyes such as ThT as proxies for the formation of high-order assemblies [32, 33]. While these approaches provide useful information on the thermodynamics and kinetics of amyloid formation, they do not report on structural details of oligomeric or fibrillar species. Padrick and Miranker [34] addressed this deficiency in early work by utilizing the intrinsic fluorescence of the phenylalanine (F15, F23) and tyrosine (Y37) residues present in wild-type IAPP. While intrinsic tryptophan fluorescence is a commonly used probe of protein structure and dynamics, the less intense signals typically observed from tyrosine and (especially) phenylalanine make their analogous use comparatively rare in the literature; however, the absence of tryptophan residues in IAPP enabled their use as reporters of conformation and self-assembly in this case. Based on steady-state fluorescence measurements, Padrick and Miranker observed Förster resonance energy transfer (FRET) between one or both of the Phe residues and Tyr37 in different stages of the IAPP aggregation reaction. Padrick and Miranker measured ETeff values of 0.48 for IAPP in the lag phase and 0.31 for the non-amyloidogenic rat isoform (rIAPP), indicating mean Phe-Tyr distances of ~13 Å and ~14 Å respectively. These values imply that monomeric IAPP and rIAPP in solution are significantly compacted relative to an ideal random coil (which would display inter-residue distances of 30 Å for F23-Y37 and 40 Å for F15-Y37), as later confirmed by tryptophan triplet quenching [35] and hydrodynamic [36, 37] measurements. Based on the high energy transfer energy (ETeff) value of 0.84 observed in fibrillar IAPP, the authors estimated an upper bound of 11 Å between either Phe and Tyr37 in the mature amyloid structure. This constraint suggested that fibrillar IAPP assumes a conformation with at least one turn interrupting stretches with β-strand character, a prediction validated by subsequent solid-state NMR [38] and EPR [39] measurements of IAPP fibers. Finally, the authors used steady-state quenching and anisotropy measurements on Tyr37 to characterize the dynamics of the IAPP C-terminus, and found that this region is highly ordered in the fibrillar state, with measured anisotropy values close to the fundamental anisotropy for tyrosine (0.278). Furthermore, the self-assembly kinetic measured by changes in steady-state anisotropy closely matched that measured by ThT fluorescence, suggesting that the observed increase in order was associated with the acquisition of β-sheet-rich structure.
An elegant extension of these studies was presented by Marek et al. [40], based on the introduction of the unnatural amino acid p-cyanophenylalanine (PheCN) at selected positions in the IAPP sequence. The presence of PheCN at positions 15, 23 or 37 of IAPP did not significantly alter fibrillization kinetics as observed by ThT fluorescence, enabling meaningful site-specific fluorescence measurements at any one of these residues during the self-assembly reaction. PheCN fluorescence decreased at all three positions upon fiber formation, a change attributable to either increased FRET to the remaining aromatic residues in IAPP, a decrease in PheCN fluorescence, or both. Based on relative fluorescence intensities in fibrillar IAPP, the authors suggested that Phe15 is more solvent-exposed than either Phe23 or Tyr37. While this model is inconsistent with either the ssNMR- or EPR-derived structures [38, 39] of IAPP fibers, it is possible the discrepancy is due to variations in the experimental conditions employed for aggregation reactions in the three different studies.
Membranes play a crucial role in modulating IAPP self-assembly: IAPP binds with high affinity to anionic lipid membranes, and converts from its disordered solution state to a partially structured state with increased α-helical structure. This binding dramatically catalyzes fiber formation, both decreasing the lag time and increasing the rate of fiber growth [41]. NMR measurements have shown that regions of IAPP assume helical structure upon binding model membranes, and also transiently sample helical states in solution [37, 42-45]. If these α-helical states are aggregation intermediates, membrane binding may serve to enhance fiber formation both by increasing the local concentration of IAPP and by biasing the conformational ensemble towards aggregation-prone states (Figure 2).
Another intriguing facet of IAPP-lipid interactions is that IAPP potently disrupts membranes and causes leakage [46, 47]. Evidence exists in the literature that suggests pore formation and detergent-like interactions may serve as underlying mechanisms for membrane disruption. Both IAPP and rIAPP have been shown to cause all-or-none, pore-type leakage in vesicles in a concentration-dependent manner, with membrane disruption mediated by oligomeric species that are formed by stochastic nucleation and evolve more potent leakage activity above a critical concentration [48]. Alternative models have proposed that Ca2+ potentiates a detergent-like mechanism of membrane disruption [49]. Membrane disruption by either mechanism may mediate the cytotoxicity exhibited by IAPP and rIAPP, with significant contributions from non-amyloid or pre-amyloid oligomeric states. There is thus considerable interest in the structures of membrane-bound IAPP oligomers, but the heterogeneity exhibited by these species at the protein concentrations (> 10 µM) required for NMR, EPR or infrared spectroscopic approaches complicates structural investigations.
Single-molecule fluorescence approaches provide a powerful method of dissecting this heterogeneity, while operating at concentrations sufficiently low to arrest higher-order oligomerization. We recently applied smFRET to define the structure of a membrane-bound rIAPP dimer [31]. Initial measurements on double-labeled rIAPP indicated a monodisperse, compact ensemble in solution that underwent a conversion to lower ETeff consistent with the acquisition of helical structure upon the addition of model membranes, in excellent accord with previous work [35, 37, 44]. We then employed smFRET in an intermolecular format to selectively probe oligomeric states in a predominantly monomeric population: separate samples of rIAPP were labeled with donor and acceptor fluorophores, and mixed together at pM concentrations in the presence of a model membrane system (phospholipid bilayer Nanodiscs). A small fraction (~0.5%) of rIAPP assembled into membrane-bound dimers bearing both donor and acceptor fluorophores and thus displayed high ETeff values, while the remaining particles (rIAPP monomers either in solution or bound to Nanodiscs) bore only one fluorophore and thus displayed low apparent ETeff values. The mean ETeff of the dimeric population reports on the structure of the dimer; five independent inter-residue ETeff values were used to constrain monomer-monomer Monte Carlo docking calculations implemented in Rosetta [50] to generate low-energy dimer models fully consistent with experimental measurements, in a procedure roughly analogous to structure determination by NMR. This protocol resulted in three distinct dimer models that together represent the first set of species formed upon membrane-mediated IAPP self-association. All three dimer models differed slightly in inter-helical angle and register, but shared a topology with the N-terminal regions of both monomers forming a symmetric, antiparallel helix-helix dimer and the C-termini remaining largely disordered (Figure 2). It is important to note that, while the topology of these models is primarily determined by spFRET constraints, the details of local structure are a function of the Rosetta refinement procedure. Nevertheless, they constitute the most detailed structural insight into membrane-mediated IAPP self-assembly obtained to date, and preliminary experiments based on ligand design and screening indicate they may be viable pharmacological targets for the inhibition of IAPP-mediated cytotoxicity.
AS
AS is a 140 amino acid, abundant neuronal protein that is the primary component of Lewy bodies, the dense cytoplasmic inclusions found in the substantia nigra of persons suffering from PD. Its primary sequence consists of three domains: an N-terminal membrane-binding region that contains repeats of the highly conserved KTKEGV sequence; the hydrophobic NAC central region which contains the minimal aggregation sequence of AS and forms the core of the fibrillar aggregates; and a highly acidic C-terminal region (Figure 3a). Both point mutants of AS (A30P, E46K, and A53T) and multiplication of the AS gene are linked to familial forms of PD [51-54].
Figure 3.
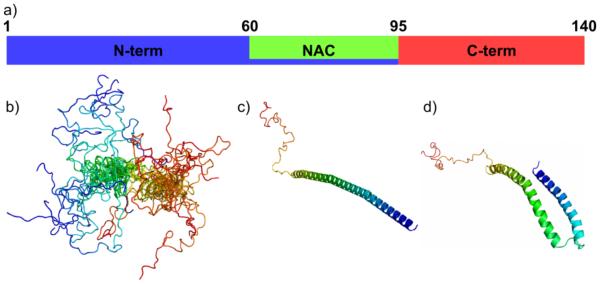
a) Schematic of AS sequence, indicating the residues demarcating the N-terminal membrane-binding region, the hydrophobic NAC region, and the C-terminal segment that is highly negatively charged at neutral pH and uncharged at low pH. Relevant structural states of AS include b) a highly dynamic ensemble of states sampled in solution, c) an extended helix formed upon binding low-curvature lipid bilayers, and d) a horseshoe-shaped helix formed upon binding high-curvature membrane mimetics.
AS has been extensively characterized using smFRET. Because the putative native function of AS in regulating synaptic vesicles is thought to involve direct interactions with cellular membranes, early studies focused on defining the structure of its membrane bound state. While circular dichroism provided evidence that AS became partially α-helical upon binding membranes [55], and NMR provided a more detailed view of that structure [56-58], there was still controversy as to the precise topology of the α-helical region, with both elongated (Figure 3c) and horseshoe-shaped (Figure 3d) structures proposed. Subramaniam and colleagues published the first smFRET study of AS using the detergent sodium dodecyl sulfate (SDS) as a membrane mimetic. Labeling AS with donor and acceptor fluorophores within the membrane binding domain, they measured an intramolecular ETeff compatible with the horseshoe-shaped model of membrane-bound AS [59]. This was followed shortly after by work from our own lab where we used both lipid vesicles and SDS as membrane mimetics. By labeling AS at three different sets of amino acids spanning various regions within the membrane-binding domain (residue pairs 9-33, 33-72, and 9-72) we, in agreement with the Subramaniam lab, measured ETeff consistent with a horseshoe-shaped helix in the presence of SDS. However, upon binding to more physiological lipid vesicles, we measured an extended helical conformation [60] which we have also observed for AS bound to lipid Nanodiscs [61]. The smFRET work confirmed the findings of two EPR studies published during the same timeframe using a variety of lipid mimetics, also found that the curvature of the mimetic system played a major role in determining the structure of AS, with the horseshoe found on highly curved, micellar mimetics, and elongated form dominating on more planar surfaces [62, 63]. A subsequent EPR study indicated that both the horseshoe and elongated conformations may coexist, in equilibrium, when bound to certain vesicle systems [64]. This body of work demonstrates that AS is able to assume different structures in response to the physical restrictions placed on it by its membrane partner. With growing evidence supporting a role for AS in membrane remodeling [65-68], such conformational flexibility may be a necessary mechanism for the generation or maintenance of high curvature lipid bilayers. The presence of multiple membrane-bound conformations also has important implications for membrane-templated aggregation of AS, which has been observed under certain conditions with high AS binding density [69-71]; it implies that like IAPP, α-helical oligomers may be important intermediates for this aggregation pathway and nucleation of aggregation may reflect the relative populations of the different conformers of AS.
Concurrent with our work, the Deniz lab published a comprehensive study mapping out the SDS-induced folding of AS [72]. One important finding of this study is that AS populates different conformations as a function of SDS concentration. In agreement with the previous SDS and vesicle studies, at SDS concentrations above the critical micelle concentration (SDS) resulted in AS forming the bent horseshoe helix, whereas the extended helix was populated at very high SDS concentrations, where the detergent forms elongated tubular structures. Strikingly, they also found that concentrations of SDS below the CMC induced populations of both horseshoe-shaped and extended helical conformations of AS. In subsequent work, the SDS folding pathways of the three PD-associated point mutants of AS were probed. While E46K and A53T displayed very similar behavior to wild-type AS, A30P was found to never populate the extended α-helical state at sub-CMC SDS concentrations [14]. The importance of misfolding or (for IDPs) partial folding in the amyloid diseases is well-recognized as evidenced by their classification as “misfolding diseases”. The A30P variant of AS binds anionic vesicles weakly compared to wild-type and the other PD mutants [73], and these data suggest that altered sampling of α-helical states in the folding pathway may be predictive of this decreased affinity for lipid bilayers..
Also of interest for AS is the characterization of aggregation intermediates, in particular those species that are thought to be directly relevant to toxicity by binding to and disrupting lipid bilayers. Building upon previous work that had established conditions for forming stable oligomers of AS that were capable of disrupting lipid vesicles composed of DOPS [74], Subramaniam and co-workers created AS variants containing a single tryptophan at one of 6 locations throughout the protein sequence [75]. Spectral measurements of the oligomers showed significant blue-shifting of the tryptophan fluorescence, indicative of a more hydrophobic environment, from tryptophans located in both the N-terminal and NAC region of the protein relative to the monomer protein. The two C-terminal tryptophans also exhibited blue-shifted fluorescence emission, but to a lesser extent. Upon binding of the oligomers to DOPS vesicles, the emission of three of the four N-terminal/NAC tryptophans shifted to even shorter wavelengths, whereas the C-terminal tryptophans were unaffected. As is well-established for the monomer protein, these results provide evidence of the importance of the N-terminus in mediating membrane-binding of oligomers as well. To compare oligomer and fiber structures, the emission of each tryptophan was measured in fully aggregated samples. The blue-shift in the NAC region of the fibers was much greater than that of the indicating a more tightly-packed, hydrophobic environment in the fibers. The general degree to which these tryptophans were buried within the core of the oligomer or the lipid bilayer was confirmed by measuring the accessibility of the each of the tryptophans to a molecular quencher, acrylamide. The oligomers are more structured than the disordered monomer protein, but still flexible enough to allow conformational rearrangement to accommodate both membrane-binding and fiber formation.
A more recent paper took a similar approach, but used the extrinsic label dansyl instead of tryptophan as the environment sensitive probe [76]. Individual dansyl fluorophores were placed at positions in the N- (residue 7) and C-termini (residue 136), as well as a positions which are expected to be involved in the fiber core (residues 26 and 100) [77], and their fluorescence emission was measured during an AS aggregation reaction. Changes in the core-fiber probes followed the kinetics of aggregation as measured by light scattering and ThT fluorescence, while the dansyl probes located at either the N- or C-termini showed evidence of a change in environment preceding fiber formation. These terminal probes are surprisingly sensitive given that they are thought to be disordered in fibers, and indicate that there are conformational changes in these distal domains of the proteins that precede the core hydrophobic interactions that drive fiber packing. Both NMR and smFRET measurements have suggested that long-range interactions between the N- and C-termini of AS may be important for modulating self-association and this work finds evidence that perturbation of these interactions occur at an early stage in the aggregation pathway [78-81].
There has also been significant effort to simply characterize the conformational ensemble of AS in solution (Figure 3b) to understand how it is altered by conditions that favor aggregation. Time-resolved intramolecular FRET measurement of AS variants, each containing a single tryptophan (donor) and a single nitro-tyrosine (acceptor) at neutral pH were compared with those at acidic pH, a condition which promotes aggregation of the protein [82]. At pH 7.4, the distance distributions calculated showed evidence of both very compact and very extended conformations. While the distributions for A53T were very similar to wild-type AS, A30P showed a loss of compact conformations in the C-terminus, and greater average separations in the intermediate and extended populations. These findings are qualitatively similar to those of the Deniz group which suggest that the conformational space sampled by A30P is most altered conformational space relative to wild-type protein [14]. Lowering the pH to 4.4 had the general effect of increasing the amplitude of the compact conformations in wild-type AS. The most notable change was in the C-terminus of the protein, which revealed a new, very compact population, as well as contraction of the most extended conformation to a more compact one. Our own single molecule FRET studies of AS at low pH found similar population of more compact states in the C-terminus of wild-type AS [78]. The enhanced aggregation of AS at low pH may be the result in part from the more compact, hydrophobic character of the C-terminus.
Modeling of the distance distributions obtained in time-resolved FRET measurements allows for the extraction of information about the dynamics of the protein chain [11]. Work from the Haas group used this approach to compare the temperature dependence of the conformational ensembles and dynamics of 8 overlapping segments in the N-terminus and NAC domain of AS [83, 84]. They found that each sub-domain of the protein behaved as a random, disordered chain, with the exception of the NAC region. Specifically, this region showed evidence of slowed intramolecular diffusion and a pronounced conformational bias. At increased temperature, a condition that accelerates aggregation of AS, the NAC region was again found to behave differently. While all of the sub-domains probed generally showed a monotonic increase in their diffusion coefficients with increasing temperature, the magnitude of this change with significantly lower for the NAC domain. These intriguing results emphasize that the potential importance of intrinsic chain dynamics in the self-association of disordered protein states – both IDPs as well as partially or fully denatured globular proteins – is worth further consideration.
Tau
The microtubule-associated protein tau is a neuronal IDP up to 441 residues in length (Figure 4: dependent on alternative splicing) that normally functions to stabilize microtubules. Tau is of major clinical relevance because of its presence in intracellular amyloid-type deposits called neurofibrillary tangles (NFTs) frequently observed in the brains of individuals affected by AD [85]. Extracellular amyloid plaques consisting of deposits of the IDP amyloid-β (Aβ) are also observed in AD brains more often than in healthy brains, and both classes of aggregate are linked to the pathology of the disease [86]. In addition, tau aggregate formation is associated with a range of other neurodegenerative disorders termed ‘tauopathies’, including CTE, Pick’s disease and Frontotemporal Dementia [87, 88].
Figure 4.

a) Schematic of tau sequence, indicating the residues that demarcate the proline-rich region (PRR) and microtubule-binding region (MTBR). b) Contour plot of ETeff from smFRET measurements between residues 244 and 354 as a function of heparin concentration, showing that this region of the protein undergoes compaction as a distinct two-state transition upon addition of a molecular inducer of aggregation. c) Charge-hydrophobicity plot of portions of the tau sequence, demonstrating that different regions of this protein are expected to vary widely in their polymer properties and response to changes in the electrostatic environment.
A major trigger of tau aggregation in vivo is thought to be hyperphosphorylation, i.e., phosphorylation at a large number of serine and threonine sites throughout the tau sequence, with a concomitant increase in negative charge [89]. The details of which specific regions or phosphorylation site are most responsible for the increased aggregation propensity are not understood. However, tau aggregation can be efficiently induced in vitro by the addition of suitable polyanions (such as the molecular inducer heparin) [90] or the presence of anionic surfaces or interfaces (including negatively charged lipids and polystyrene microspheres) [91, 92]. These diverse effects highlight the importance of electrostatic effects in modulating the self-assembly behavior of tau.
As with AS and IAPP, tau is disordered in solution but more compact than an ideal random coil [93-95], forms amphipathic helical domains upon membrane binding [96, 97], and displays accelerated aggregation upon membrane binding under some conditions [91, 92]. Full-length tau is sufficiently large that NMR experiments are highly challenging and require specialized acquisition and assignment procedures [95, 98], and so most spectroscopic work to date has been performed on shorter constructs of tau. Constructs spanning the microtubule-binding region (MTBR; residues 244-369) are potent amyloid-formers; inclusion of the remainder of the protein significantly slows aggregation kinetics, suggesting that it may be necessary to the full-length protein in order to gain accurate insight into the thermodynamics and kinetics of tau self-assembly [99]. Other significant regions of full-length tau are the proline-rich region (PRR; residues 151-243) and the N-terminal projection domain [100] (Figure 4a).
Seminal fluorescence studies on the conformation of tau in solution were performed by Mandelkow and co-workers, based on both intrinsic tryptophan fluorescence [101, 102] and FRET between tryptophan residues inserted into the tau sequence and dye (IAEDANS) moieties ligated via cysteine-maleimide chemistry [103, 104]. Notably, fluorophores were introduced at several different residue pairs in tau, including one pair spanning virtually the complete tau sequence, and two pairs within the central MTBR. Using steady state measurements, Jeganathan et al. [103] measured ETeff values for energy transfer from Trp to IAEDANS for ten residue pairs. Strikingly, they observed FRET between the N- and C-termini of tau, as well as from the C-terminus to the central MTBR. This indicates that tau forms a highly compact structure in solution relative to an ideal random coil, with the N-terminus in relatively close proximity to (but not necessarily in contact with) the C-terminus, and the latter close to both the N-terminus and the MTBR. These findings constitute the so-called ‘paperclip’ model of tau, and have been highly influential in subsequent conformational studies. One potential caveat with the quantitative interpretation of this work is that the authors converted ETeff values to inter-residue distances using the canonical form of the Förster equation instead of convolving the Förster equation with an estimate of the inter-residue distance distribution of a protein chain (as is necessary and appropriate for accurate interpretation of FRET-derived for dynamic polymers [105]); another is that ensemble FRET measurements can be highly sensitive to artifacts caused by incomplete dye labeling. Mandelkow and co-workers subsequently used similar ensemble FRET methods [106] to characterize the relative changes induced by pseudo-phosphorylation (i.e., mutations mimicking the added negative charge associated with phosphorylation) at specific locations in tau, and found that phosphorylation at the ‘AT8’ locus (residues 199, 202 and 205) increased the distance between N- and C-termini, while the ‘PHF1’ locus (residues 396 and 404) decreased the corresponding distance, and the ‘AT100’ locus (residues 212 and 214) had little effect. However, combining all three pseudo-phosphorylation modifications left the N-C distance relatively unaffected while decreasing the distances from N- and C-termini to the MTBR. This study provides a level of qualitative insight into the types of conformational changes induced by post-translational modifications of tau.
Our group more recently used smFRET to investigate in greater detail the conformational ensembles populated by tau in the presence and absence of the aggregation inducer heparin [12, 107]. Full-length tau was double-labeled at 12 distinct pairs of residues, spanning the sequence, with donor and acceptor dyes (Alexa Fluors 488 and 594, respectively) and an extensive set of intramolecular ETeff values were measured in buffer and in various concentrations of heparin. Computational methodology was also developed to generate conformational ensembles of IDPs from these spFRET constraints while rigorously accounting for polymer chain dynamics and appropriately quantifying the errors introduced at each stage of experiment and analysis. Qualitatively, our findings on the conformational ensemble of tau in solution resembled that of Mandelkow and co-workers [103], in that the protein chain was significantly more compact than an ideal random coil, and that long-range interactions brought the N- and C-termini into relatively close contact. We also detected interactions between the N-terminus and MTBR that were not reported by Jeganathan et al., and also extensively characterized conformation in the PRR and other regions of tau not studied in the earlier work. Broadly speaking, heparin induced a compaction in the MTBR and significant expansion through the rest of the protein, suggesting that the heparin-induced aggregation-prone state may be distinct from the analogous state putatively induced by the pseudophosphorylation mutants. On a quantitative level, the inter-residue distances derived from our smFRET data were almost universally larger in magnitude than the corresponding distances from the data of Jeganathan et al. This discrepancy may be due to the larger Förster radius of the dye pair we employed (~54 Å vs. ~22 Å), the greater sensitivity of single-molecule instrumentation, and/or our use of appropriate corrections to account for the dynamics of the tau protein chain.
Two details of our studies with tau highlight particularly well the unique features of amyloidogenic proteins that are also intrinsically disordered. First, the addition of heparin to tau induces a distinct two-state conformational shift (Figure 4b), indicating that heparin is stabilizing or inducing a particular conformation of tau, rather than mediating a non-specific transition analogous to a polymeric coil-globule transition that has been observed in salt titrations of other disordered proteins [108]. Second, different domains of tau responded differentially to binding of heparin. Further analysis of the various domains using a net-charge/hydrophobicity plot (also called an Uversky plot; Figure 4c) [109] revealed that domains could be classified in terms of how closely they resemble canonical globular (high hydrophobicity, low net charge) or intrinsically disordered (low hydrophobicity, high net charge) proteins: the C-terminal sequence (residues 354-433) lies well in the globular region, the MTBR lies intermediated between disordered and globular, and the N-terminal projection domain (residues 17-103) lies far into the disordered part of the map. Each of these domains, then, is predicted to respond differentially to electrostatic perturbations – a prediction validated by their response to increasing salt concentrations [107]. Importantly, in contrast to globular proteins whose unfolding can be described by a two-state transition, the types of continuous conformational changes displayed by the various regions of tau are better modeled using polymer physics paradigms [110]. This study highlights the importance of characterizing multiple domains of the protein in detail in order to develop a complete picture of the conformational changes relevant to the potentiation of aggregation.
Challenges and Opportunities: The Future of Fluorescence Studies of Amyloidogenic Proteins
The examples discussed above demonstrate how the variety of fluorescence methodologies available to modern biophysicists, including ensemble, steady-state and time-resolved approaches, can be employed to generate powerful insight into the pathological self-assembly of amyloid proteins. We have also seen how these findings complement the foundational insights from other spectroscopic and biochemical methods. However, major questions about the mechanisms and pathways by which amyloid proteins, particularly those whose native form is intrinsically disordered, gain toxic function remain unanswered, and will require the development of corresponding new techniques. Here, we outline a few potential areas in which fluorescence-based methods may prove particularly useful over the next few years.
A variety of modern fluorescence approaches, including fluorescence correlation spectroscopy, fluorescence fluctuation spectroscopy and two-color coincidence detection (TCCD) have been successfully employed to assess the stoichiometry of oligomeric intermediates in amyloidogenic pathways [111-118]. However, obtaining detailed structural insight into these larger species remains challenging. The most promising recent efforts have probably been from Klenerman and co-workers, who have adapted TCCD approaches to measure what might be called small-oligomer FRET [112]. Translating these types of data into useful models could require improvements in computational techniques to constraint generation from ambiguous data as well as, potentially, in adapting concepts from three- and four-color FRET approaches.
Of emerging interest, but not nearly as well-explored as conformational studies, is a role the intrinsic dynamics of unfolded and disordered protein in predicting aggregation propensity [119]. Established probes of dynamic behavior, such as FRET-FCS [120] and contact-quenching FCS [121], could be applied to study the dynamic behavior of amyloidogenic IDPs under diverse conditions, at sufficiently low concentrations that aggregation ought not to be a concern. Complementary tools such as time-resolved FRET [11] could also serve to interrogate the distributions of distances sampled on sub-microsecond timescales.
Finally, the capacity of fluorescence approaches to rapidly characterize amyloidogenic proteins at low concentrations and in heterogeneous, complex and biologically relevant environments make them ideally suited for the screening of conformational modulators. These are drug-like small molecules, peptides or peptidomimetics that alter the conformation of a target protein in a therapeutically useful manner, either inhibiting self-assembly or inducing the formation of biologically inert oligomeric species. The sensitivity of fluorescence relative to other spectroscopic tools allows for the use of much smaller amounts of protein, which is attractive not just from an economic perspective but also because it enables the accurate determination of binding constants over a correspondingly broader dynamic range of modulator affinity. Fluorescence could thus serve not only to uncover the fundamental mechanisms and general principles behind pathological protein self-assembly, but as a route to the discovery and development of new classes of therapeutic molecules targeting this ubiquitous and pharmacologically challenging phenomenon.
Abbreviations
- AD
Alzheimer’s disease
- ANS
1-anilinonaphthalene-8-sulfonic acid
- AS
α-synuclein
- CMC
critical micellar concentration
- CTE
chronic traumatic encephalopathy
- EPR
electron paramagnetic resonance
- ETeff
energy transfer efficiency
- FRET
Förster resonance energy transfer
- IAPP
islet amyloid polypeptide
- IDP
intrinsically disordered protein
- MTBR
microtubule-binding region
- NFT
neurofibrillary tangle
- NMR
nuclear magnetic resonance
- PD
Parkinson’s disease
- PheCN
p-cyanophenylalanine
- PRR
proline-rich region
- SDS
sodium dodecyl sulfate
- T2DM
Type II Diabetes Mellitus
- TCCD
two-color coincidence detection
- ThT
thioflavin-T
References
- 1.Hebda JA, Miranker AD. The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: Insights from type II diabetes. Annu. Rev. Biophys. 2009;38:125–52. doi: 10.1146/annurev.biophys.050708.133622. [DOI] [PubMed] [Google Scholar]
- 2.Uversky VN, Oldfield CJ, Midic U, Xie H, Xue B, Vucetic S, Iakoucheva LM, Obradovic Z, Dunker AK. Unfoldomics of human diseases: Linking protein intrinsic disorder with diseases. BMC Genomics. 2009;10(Suppl 1):S7. doi: 10.1186/1471-2164-10-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turoverov KK, Kuznetsova IM, Uversky VN. The protein kingdom extended: Ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog. Biophys. Mol. Biol. 2010;102:73–84. doi: 10.1016/j.pbiomolbio.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoyert DL, Xu J. Deaths: Preliminary data for 2011. National Vital Statistics Reports. 2012;61:1–50. [PubMed] [Google Scholar]
- 5.Jarrett JT, Lansbury PT., Jr. Seeding "one-dimensional crystallization" of amyloid: A pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–8. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 6.Powers ET, Powers DL. The kinetics of nucleated polymerizations at high concentrations: Amyloid fibril formation near and above the "supercritical concentration". Biophys. J. 2006;91:122–32. doi: 10.1529/biophysj.105.073767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eliezer D. Biophysical characterization of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2009;19:23–30. doi: 10.1016/j.sbi.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abedini A, Raleigh DP. A critical assessment of the role of helical intermediates in amyloid formation by natively unfolded proteins and polypeptides. Protein Eng. Des. Sel. 2009;22:453–9. doi: 10.1093/protein/gzp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fandrich M. Oligomeric intermediates in amyloid formation: Structure determination and mechanisms of toxicity. J. Mol. Biol. 2012;421:427–40. doi: 10.1016/j.jmb.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Langkilde AE, Vestergaard B. Methods for structural characterization of prefibrillar intermediates and amyloid fibrils. FEBS Lett. 2009;583:2600–9. doi: 10.1016/j.febslet.2009.05.040. [DOI] [PubMed] [Google Scholar]
- 11.Haas E. Ensemble FRET methods in studies of intrinsically disordered proteins. Methods Mol Biol. 2012;895:467–98. doi: 10.1007/978-1-61779-927-3_28. [DOI] [PubMed] [Google Scholar]
- 12.Nath A, Sammalkorpi M, DeWitt DC, Trexler AJ, Elbaum-Garfinkle S, O'Hern CS, Rhoades E. The conformational ensembles of α-synuclein and tau: Combining single-molecule FRET and simulations. Biophys. J. 2012;103:1940–9. doi: 10.1016/j.bpj.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H, Rhoades E. Fluorescence characterization of denatured proteins. Curr. Opin. Struct. Biol. 2008;18:516–24. doi: 10.1016/j.sbi.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferreon AC, Moran CR, Ferreon JC, Deniz AA. Alteration of the α-synuclein folding landscape by a mutation related to Parkinson's disease. Angew. Chem. Int. Ed. Engl. 2010;49:3469–72. doi: 10.1002/anie.201000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haas E. The study of protein folding and dynamics by determination of intramolecular distance distributions and their fluctuations using ensemble and single-molecule FRET measurements. Chemphyschem. 2005;6:858–870. doi: 10.1002/cphc.200400617. [DOI] [PubMed] [Google Scholar]
- 16.Nettels D, Schuler B. Single-molecule FRET of protein-folding dynamics. In: Komatsuzaki T, Kawakami M, Takahashi S, Yang H, Silbey RJ, editors. Single-molecule Biophysics: Experiment and Theory. Vol. 146. 2012. pp. 23–48. [Google Scholar]
- 17.Bolognesi B, Kumita JR, Barros TP, Esbjorner EK, Luheshi LM, Crowther DC, Wilson MR, Dobson CM, Favrin G, Yerbury JJ. ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 2010;5:735–40. doi: 10.1021/cb1001203. [DOI] [PubMed] [Google Scholar]
- 18.Reinke AA, Gestwicki JE. Insight into amyloid structure using chemical probes. Chem. Biol. Drug Des. 2011;77:399–411. doi: 10.1111/j.1747-0285.2011.01110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mishra R, Sjolander D, Hammarstrom P. Spectroscopic characterization of diverse amyloid fibrils in vitro by the fluorescent dye Nile Red. Mol. Biosyst. 2011;7:1232–40. doi: 10.1039/c0mb00236d. [DOI] [PubMed] [Google Scholar]
- 20.Harel M, Sonoda LK, Silman I, Sussman JL, Rosenberry TL. Crystal structure of Thioflavin T bound to the peripheral site of torpedo californica acetylcholinesterase reveals how Thioflavin T acts as a sensitive fluorescent reporter of ligand binding to the acylation site. J. Am. Chem. Soc. 2008;130:7856–61. doi: 10.1021/ja7109822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landau M, Sawaya MR, Faull KF, Laganowsky A, Jiang L, Sievers SA, Liu J, Barrio JR, Eisenberg D. Towards a pharmacophore for amyloid. PLoS Biol. 2011;9:e1001080. doi: 10.1371/journal.pbio.1001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schutz AK, Soragni A, Hornemann S, Aguzzi A, Ernst M, Bockmann A, Meier BH. The amyloid-Congo Red interface at atomic resolution. Angew. Chem. Int. Ed. Engl. 2011;50:5956–60. doi: 10.1002/anie.201008276. [DOI] [PubMed] [Google Scholar]
- 23.Wolfe LS, Calabrese MF, Nath A, Blaho DV, Miranker AD, Xiong Y. Protein-induced photophysical changes to the amyloid indicator dye Thioflavin T. Proc. Natl. Acad. Sci. U S A. 2010;107:16863–8. doi: 10.1073/pnas.1002867107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cloe AL, Orgel JP, Sachleben JR, Tycko R, Meredith SC. The Japanese mutant Aβ (ΔE22-Aβ(1-39)) forms fibrils instantaneously, with low-Thioflavin T fluorescence: Seeding of wild-type Aβ(1-40) into atypical fibrils by ΔE22-Aβ(1-39) Biochemistry. 2011;50:2026–39. doi: 10.1021/bi1016217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 26.Wright PE, Dyson HJ. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999;293:321–31. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 27.Young A, Pittner R, Gedulin B, Vine W, Rink T. Amylin regulation of carbohydrate metabolism. Biochem. Soc. Trans. 1995;23:325–31. doi: 10.1042/bst0230325. [DOI] [PubMed] [Google Scholar]
- 28.Clark A, Nilsson MR. Islet amyloid: A complication of islet dysfunction or an aetiological factor in type 2 diabetes? Diabetologia. 2004;47:157–69. doi: 10.1007/s00125-003-1304-4. [DOI] [PubMed] [Google Scholar]
- 29.Haataja L, Gurlo T, Huang CJ, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 2008;29:303–16. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson KH, O'Brien TD, Betsholtz C, Westermark P. Islet amyloid polypeptide: Mechanisms of amyloidogenesis in the pancreatic islets and potential roles in diabetes mellitus. Lab Invest. 1992;66:522–35. [PubMed] [Google Scholar]
- 31.Nath A, Miranker AD, Rhoades E. A membrane-bound antiparallel dimer of rat islet amyloid polypeptide. Angew. Chem. Int. Ed. Engl. 2011;50:10859–62. doi: 10.1002/anie.201102887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kayed R, Bernhagen J, Greenfield N, Sweimeh K, Brunner H, Voelter W, Kapurniotu A. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J. Mol. Biol. 1999;287:781–96. doi: 10.1006/jmbi.1999.2646. [DOI] [PubMed] [Google Scholar]
- 33.Abedini A, Raleigh DP. The role of His-18 in amyloid formation by human islet amyloid polypeptide. Biochemistry. 2005;44:16284–91. doi: 10.1021/bi051432v. [DOI] [PubMed] [Google Scholar]
- 34.Padrick SB, Miranker AD. Islet amyloid polypeptide: Identification of long-range contacts and local order on the fibrillogenesis pathway. J. Mol. Biol. 2001;308:783–94. doi: 10.1006/jmbi.2001.4608. [DOI] [PubMed] [Google Scholar]
- 35.Vaiana SM, Best RB, Yau WM, Eaton WA, Hofrichter J. Evidence for a partially structured state of the amylin monomer. Biophys. J. 2009;97:2948–57. doi: 10.1016/j.bpj.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soong R, Brender JR, Macdonald PM, Ramamoorthy A. Association of highly compact type II diabetes related islet amyloid polypeptide intermediate species at physiological temperature revealed by diffusion NMR spectroscopy. J. Am. Chem. Soc. 2009;131:7079–85. doi: 10.1021/ja900285z. [DOI] [PubMed] [Google Scholar]
- 37.Williamson JA, Loria JP, Miranker AD. Helix stabilization precedes aqueous and bilayer-catalyzed fiber formation in islet amyloid polypeptide. J. Mol. Biol. 2009;393:383–96. doi: 10.1016/j.jmb.2009.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luca S, Yau WM, Leapman R, Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: Constraints from solid-state NMR. Biochemistry. 2007;46:13505–22. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bedrood S, Li Y, Isas JM, Hegde BG, Baxa U, Haworth IS, Langen R. Fibril structure of human islet amyloid polypeptide. J. Biol. Chem. 2012;287:5235–41. doi: 10.1074/jbc.M111.327817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marek P, Mukherjee S, Zanni MT, Raleigh DP. Residue-specific, real-time characterization of lag-phase species and fibril growth during amyloid formation: A combined fluorescence and IR study of p-cyanophenylalanine analogs of islet amyloid polypeptide. J. Mol. Biol. 2010;400:878–88. doi: 10.1016/j.jmb.2010.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knight JD, Miranker AD. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 2004;341:1175–87. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 42.Williamson JA, Miranker AD. Direct detection of transient α-helical states in islet amyloid polypeptide. Protein Sci. 2007;16:110–7. doi: 10.1110/ps.062486907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nanga RP, Brender JR, Vivekanandan S, Ramamoorthy A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim Biophys Acta. 2011;1808:2337–42. doi: 10.1016/j.bbamem.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nanga RP, Brender JR, Xu J, Hartman K, Subramanian V, Ramamoorthy A. Three-dimensional structure and orientation of rat islet amyloid polypeptide protein in a membrane environment by solution NMR spectroscopy. J. Am. Chem. Soc. 2009;131:8252–61. doi: 10.1021/ja9010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patil SM, Xu S, Sheftic SR, Alexandrescu AT. Dynamic α-helix structure of micelle-bound human amylin. J. Biol. Chem. 2009;284:11982–91. doi: 10.1074/jbc.M809085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sparr E, Engel MF, Sakharov DV, Sprong M, Jacobs J, de Kruijff B, Hoppener JW, Killian JA. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 2004;577:117–20. doi: 10.1016/j.febslet.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 47.Knight JD, Hebda JA, Miranker AD. Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry. 2006;45:9496–508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 48.Last NB, Rhoades E, Miranker AD. Islet amyloid polypeptide demonstrates a persistent capacity to disrupt membrane integrity. Proc. Natl. Acad. Sci. U S A. 2011;108:9460–5. doi: 10.1073/pnas.1102356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sciacca MF, Milardi D, Messina GM, Marletta G, Brender JR, Ramamoorthy A, La Rosa C. Cations as switches of amyloid-mediated membrane disruption mechanisms: Calcium and IAPP. Biophys. J. 2013;104:173–84. doi: 10.1016/j.bpj.2012.11.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaufmann KW, Lemmon GH, Deluca SL, Sheehan JH, Meiler J. Practically useful: What the ROSETTA protein modeling suite can do for you. Biochemistry. 2010;49:2987–98. doi: 10.1021/bi902153g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nature Genetics. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 52.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 53.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 54.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Annals of Neurology. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 55.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998;273:9443–9. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 56.Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC. A broken α-helix in folded α-synuclein. J Biol Chem. 2003;278:15313–8. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- 57.Eliezer D, Kutluay E, Bussell R, Browne G. Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 58.Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005;280:9595–603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 59.Veldhuis G, Segers-Nolten I, Ferlemann E, Subramaniam V. Single-molecule FRET reveals structural heterogeneity of SDS-bound α-synuclein. Chembiochem. 2009;10:436. doi: 10.1002/cbic.200800644. + [DOI] [PubMed] [Google Scholar]
- 60.Trexler AJ, Rhoades E. α-synuclein binds large unilamellar vesicles as an extended helix. Biochemistry. 2009;48:2304–6. doi: 10.1021/bi900114z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nath A, Trexler AJ, Koo P, Miranker AD, Atkins WM, Rhoades E. Single-molecule fluorescence spectroscopy using phospholipid bilayer Nanodiscs. Methods Enzymol. 2010;472:89–117. doi: 10.1016/S0076-6879(10)72014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. Membrane-bound α-synuclein forms an extended helix: Long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J. Am. Chem. Soc. 2008;130:12856. doi: 10.1021/ja804517m. + [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U. S. A. 2008;105:19666–71. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robotta M, Braun P, van Rooijen B, Subramaniam V, Huber M, Drescher M. Direct evidence of coexisting horseshoe and extended helix conformations of membrane-bound α-synuclein. Chemphyschem. 2011;12:267–9. doi: 10.1002/cphc.201000815. [DOI] [PubMed] [Google Scholar]
- 65.Mizuno N, Varkey J, Kegulian NC, Hegde BG, Cheng NQ, Langen R, Steven AC. Remodeling of lipid vesicles into cylindrical micelles by α-synuclein in an extended α-helical conformation. J. Biol. Chem. 2012;287:29301–29311. doi: 10.1074/jbc.M112.365817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Braun AR, Sevcsik E, Chin P, Rhoades E, Tristram-Nagle S, Sachs JN. Alpha-synuclein induces both positive mean curvature and negative gaussian curvature in membranes. J. Am. Chem. Soc. 2012;134:2613–20. doi: 10.1021/ja208316h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamp F, Beyer K. Binding of α-synuclein affects the lipid packing in bilayers of small vesicles. J. Biol. Chem. 2006;281:9251–9. doi: 10.1074/jbc.M512292200. [DOI] [PubMed] [Google Scholar]
- 68.Zhu M, Li J, Fink AL. The association of α-synuclein with membranes affects bilayer structure, stability, and fibril formation. J. Biol. Chem. 2003;278:40186–97. doi: 10.1074/jbc.M305326200. [DOI] [PubMed] [Google Scholar]
- 69.Jo E, Darabie AA, Han K, Tandon A, Fraser PE, McLaurin J. α-synuclein-synaptosomal membrane interactions: Implications for fibrillogenesis. Eur. J. Biochem. 2004;271:3180–9. doi: 10.1111/j.1432-1033.2004.04250.x. [DOI] [PubMed] [Google Scholar]
- 70.Lee JH, Hong CS, Lee S, Yang JE, Park YI, Lee D, Hyeon T, Jung S, Paik SR. Radiating amyloid fibril formation on the surface of lipid membranes through unit-assembly of oligomeric species of α-synuclein. PLoS One. 2012;7:e47580. doi: 10.1371/journal.pone.0047580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Necula M, Chirita CN, Kuret J. Rapid anionic micelle-mediated α-synuclein fibrillization in vitro. J. Biol. Chem. 2003;278:46674–80. doi: 10.1074/jbc.M308231200. [DOI] [PubMed] [Google Scholar]
- 72.Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. U. S. A. 2009;106:5645–50. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Middleton ER, Rhoades E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys. J. 2010;99:2279–88. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Rooijen BD, Claessens M, Subramaniam V. Lipid bilayer disruption by oligomeric α-synuclein depends on bilayer charge and accessibility of the hydrophobic core. Biochimica Et Biophysica Acta-Biomembranes. 2009;1788:1271–1278. doi: 10.1016/j.bbamem.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 75.van Rooijen BD, van Leijenhorst-Groener KA, Claessens M, Subramaniam V. Tryptophan fluorescence reveals structural features of α-synuclein oligomers. J. Mol. Biol. 2009;394:826–833. doi: 10.1016/j.jmb.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 76.Yap TL, Pfefferkorn CM, Lee JC. Residue-specific fluorescent probes of α-synuclein: Detection of early events at the N- and C-termini during fibril assembly. Biochemistry. 2011;50:1963–1965. doi: 10.1021/bi2000824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen M, Margittai M, Chen J, Langen R. Investigation of α-synuclein fibril structure by site-directed spin labeling. J. Biol. Chem. 2007;282:24970–9. doi: 10.1074/jbc.M700368200. [DOI] [PubMed] [Google Scholar]
- 78.Trexler AJ, Rhoades E. Single molecule characterization of α-synuclein in aggregation-prone states. Biophys. J. 2010;99:3048–55. doi: 10.1016/j.bpj.2010.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cho MK, Nodet G, Kim HY, Jensen MR, Bernado P, Fernandez CO, Becker S, Blackledge M, Zweckstetter M. Structural characterization of α-synuclein in an aggregation prone state. Protein Sci. 2009;18:1840–6. doi: 10.1002/pro.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu KP, Weinstock DS, Narayanan C, Levy RM, Baum J. Structural reorganization of α-synuclein at low pH observed by NMR and REMD simulations. J. Mol. Biol. 2009;391:784–96. doi: 10.1016/j.jmb.2009.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McClendon S, Rospigliosi CC, Eliezer D. Charge neutralization and collapse of the C-terminal tail of α-synuclein at low pH. Protein Sci. 2009;18:1531–40. doi: 10.1002/pro.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee JC, Langen R, Hummel PA, Gray HB, Winkler JR. Alpha-synuclein structures from fluorescence energy-transfer kinetics: Implications for the role of the protein in Parkinson's disease. Proc. Natl. Acad. Sci. U S A. 2004;101:16466–16471. doi: 10.1073/pnas.0407307101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grupi A, Haas E. Segmental conformational disorder and dynamics in the intrinsically disordered protein α-synuclein and its chain length dependence. J. Mol. Biol. 2011;405:1267–1283. doi: 10.1016/j.jmb.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 84.Grupi A, Haas E. Time-resolved FRET detection of subtle temperature-induced conformational biases in ensembles of α-synuclein molecules. J. Mol. Biol. 2011;411:234–247. doi: 10.1016/j.jmb.2011.04.056. [DOI] [PubMed] [Google Scholar]
- 85.Pritchard SM, Dolan PJ, Vitkus A, Johnson GV. The toxicity of tau in Alzheimer disease: Turnover, targets and potential therapeutics. J. Cell. Mol. Med. 2011;15:1621–35. doi: 10.1111/j.1582-4934.2011.01273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.LaFerla FM. Pathways linking Aβ and tau pathologies. Biochem. Soc. Trans. 2010;38:993–5. doi: 10.1042/BST0380993. [DOI] [PubMed] [Google Scholar]
- 87.Lee G, Leugers CJ. Tau and tauopathies. Prog. Mol. Biol. Transl. Sci. 2012;107:263–93. doi: 10.1016/B978-0-12-385883-2.00004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bouchard M, Suchowersky O. Tauopathies: One disease or many? Can. J. Neurol. Sci. 2011;38:547–56. doi: 10.1017/s0317167100012087. [DOI] [PubMed] [Google Scholar]
- 89.Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Discov. 2007;6:464–79. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- 90.Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–3. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- 91.Chirita CN, Kuret J. Evidence for an intermediate in tau filament formation. Biochemistry. 2004;43:1704–14. doi: 10.1021/bi036034b. [DOI] [PubMed] [Google Scholar]
- 92.Elbaum-Garfinkle S, Ramlall T, Rhoades E. The role of the lipid bilayer in tau aggregation. Biophys. J. 2010;98:2722–2730. doi: 10.1016/j.bpj.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.von Bergen M, Barghorn S, Jeganathan S, Mandelkow EM, Mandelkow E. Spectroscopic approaches to the conformation of tau protein in solution and in paired helical filaments. Neurodegener. Dis. 2006;3:197–206. doi: 10.1159/000095257. [DOI] [PubMed] [Google Scholar]
- 94.Mukrasch MD, Markwick P, Biernat J, Bergen M, Bernado P, Griesinger C, Mandelkow E, Zweckstetter M, Blackledge M. Highly populated turn conformations in natively unfolded tau protein identified from residual dipolar couplings and molecular simulation. J. Am. Chem. Soc. 2007;129:5235–43. doi: 10.1021/ja0690159. [DOI] [PubMed] [Google Scholar]
- 95.Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009;7:e34. doi: 10.1371/journal.pbio.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kunze G, Barre P, Scheidt HA, Thomas L, Eliezer D, Huster D. Binding of the three-repeat domain of tau to phospholipid membranes induces an aggregated-like state of the protein. Biochim. Biophys. Acta. 2012;1818:2302–13. doi: 10.1016/j.bbamem.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Barre P, Eliezer D. Folding of the repeat domain of tau upon binding to lipid surfaces. J. Mol. Biol. 2006;362:312–26. doi: 10.1016/j.jmb.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 98.Narayanan RL, Durr UH, Bibow S, Biernat J, Mandelkow E, Zweckstetter M. Automatic assignment of the intrinsically disordered protein tau with 441-residues. J. Am. Chem. Soc. 2010;132:11906–7. doi: 10.1021/ja105657f. [DOI] [PubMed] [Google Scholar]
- 99.Wille H, Drewes G, Biernat J, Mandelkow EM, Mandelkow E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J. Cell Biol. 1992;118:573–84. doi: 10.1083/jcb.118.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33:9511–22. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- 101.Li L, von Bergen M, Mandelkow EM, Mandelkow E. Structure, stability, and aggregation of paired helical filaments from tau protein and ftdp-17 mutants probed by tryptophan scanning mutagenesis. J. Biol. Chem. 2002;277:41390–400. doi: 10.1074/jbc.M206334200. [DOI] [PubMed] [Google Scholar]
- 102.von Bergen M, Li L, Mandelkow E. Intrinsic fluorescent detection of tau conformation and aggregation. Methods Mol. Biol. 2005;299:175–84. doi: 10.1385/1-59259-874-9:175. [DOI] [PubMed] [Google Scholar]
- 103.Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–93. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- 104.Jeganathan S, Chinnathambi S, Mandelkow EM, Mandelkow E. Conformations of microtubule-associated protein tau mapped by fluorescence resonance energy transfer. Methods Mol. Biol. 2012;849:85–99. doi: 10.1007/978-1-61779-551-0_7. [DOI] [PubMed] [Google Scholar]
- 105.O'Brien EP, Morrison G, Brooks BR, Thirumalai D. How accurate are polymer models in the analysis of Förster resonance energy transfer experiments on proteins? J Chem. Phys. 2009;130:124903. doi: 10.1063/1.3082151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of tau and generates a pathological (MC-1) conformation. J. Biol. Chem. 2008;283:32066–76. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- 107.Elbaum-Garfinkle S, Rhoades E. Identification of an aggregation-prone structure of tau. J. Am. Chem. Soc. 2012;134:16607–16613. doi: 10.1021/ja305206m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Muller-Spath S, Soranno A, Hirschfeld V, Hofmann H, Ruegger S, Reymond L, Nettels D, Schuler B. From the cover: Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U S A. 2010;107:14609–14. doi: 10.1073/pnas.1001743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Uversky VN, Gillespie JR, Fink AL. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins. 2000;41:415–27. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 110.Pappu RV, Wang X, Vitalis A, Crick SL. A polymer physics perspective on driving forces and mechanisms for protein aggregation. Arch. Biochem. Biophys. 2008;469:132–41. doi: 10.1016/j.abb.2007.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bader B, Nubling G, Mehle A, Nobile S, Kretzschmar H, Giese A. Single particle analysis of tau oligomer formation induced by metal ions and organic solvents. Biochem. Biophys. Res. Commun. 2011;411:190–6. doi: 10.1016/j.bbrc.2011.06.135. [DOI] [PubMed] [Google Scholar]
- 112.Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, Bertoncini CW, Wood NW, Knowles TP, Dobson CM, Klenerman D. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. 2012;149:1048–59. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ding H, Wong PT, Lee EL, Gafni A, Steel DG. Determination of the oligomer size of amyloidogenic protein β-amyloid(1-40) by single-molecule spectroscopy. Biophys. J. 2009;97:912–21. doi: 10.1016/j.bpj.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Matsumura S, Shinoda K, Yamada M, Yokojima S, Inoue M, Ohnishi T, Shimada T, Kikuchi K, Masui D, Hashimoto S, Sato M, Ito A, Akioka M, Takagi S, Nakamura Y, Nemoto K, Hasegawa Y, Takamoto H, Inoue H, Nakamura S, Nabeshima Y, Teplow DB, Kinjo M, Hoshi M. Two distinct amyloid β-protein (Aβ) assembly pathways leading to oligomers and fibrils identified by combined fluorescence correlation spectroscopy, morphology, and toxicity analyses. J. Biol. Chem. 2011;286:11555–62. doi: 10.1074/jbc.M110.181313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Narayan P, Orte A, Clarke RW, Bolognesi B, Hook S, Ganzinger KA, Meehan S, Wilson MR, Dobson CM, Klenerman D. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β(1-40) peptide. Nat. Struct. Mol. Biol. 2012;19:79–83. doi: 10.1038/nsmb.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Paredes JM, Casares S, Ruedas-Rama MJ, Fernandez E, Castello F, Varela L, Orte A. Early amyloidogenic oligomerization studied through fluorescence lifetime correlation spectroscopy. Int. J. Mol. Sci. 2012;13:9400–18. doi: 10.3390/ijms13089400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Powell LR, Dukes KD, Lammi RK. Probing the efficacy of peptide-based inhibitors against acid- and zinc-promoted oligomerization of amyloid-β peptide via single-oligomer spectroscopy. Biophys. Chem. 2012;160:12–9. doi: 10.1016/j.bpc.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zijlstra N, Blum C, Segers-Nolten IM, Claessens MM, Subramaniam V. Molecular composition of sub-stoichiometrically labeled α-synuclein oligomers determined by single-molecule photobleaching. Angew. Chem. Int. Ed. Engl. 2012;51:8821–4. doi: 10.1002/anie.201200813. [DOI] [PubMed] [Google Scholar]
- 119.Ahmad B, Chen YJ, Lapidus LJ. Aggregation of α-synuclein is kinetically controlled by intramolecular diffusion. Proc. Natl. Acad. Sci. U S A. 2012;109:2336–2341. doi: 10.1073/pnas.1109526109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Torres T, Levitus M. Measuring conformational dynamics: A new FCS-FRET approach. J Phys. Chem. B. 2007;111:7392–400. doi: 10.1021/jp070659s. [DOI] [PubMed] [Google Scholar]
- 121.Chattopadhyay K, Elson EL, Frieden C. The kinetics of conformational fluctuations in an unfolded protein measured by fluorescence methods. Proc. Natl. Acad. Sci. U S A. 2005;102:2385–9. doi: 10.1073/pnas.0500127102. [DOI] [PMC free article] [PubMed] [Google Scholar]