

Abstract
Background and Objective
There is general agreement that certain fatty acids and lipopolysaccharides (LPS) promote inflammation through toll-like receptor 4 (TLR4), and that inflammation promotes insulin resistance. We therefore hypothesized that mice with periodontitis and a TLR4 loss-of-function (LOF) mutation fed a high-fat (HF) diet would develop improved glucose homeostasis compared with wild-type (WT) animals with periodontitis fed a HF diet.
Material and Methods
Wild-type and TLR4 mutant mice fed a HF diet were divided into four groups (n = 6/group): WT; WT with periodontitis (WT/P); mutant (Mut); and mutant with periodontitis (Mut/P). Periodontitis was induced by placing LPS soaked ligatures around maxillary second molars. Fasting insulin and glucose levels were measured weekly for 10 wk. Glucose tolerance was evaluated at baseline (week 1) and at 9 wk. Insulin signaling (phosphorylation of Akt) and tumor necrosis factor-α (TNF-α) mRNA levels in liver were determined when the mice were killed at week 10.
Results
Mut/P mice developed less alveolar bone loss compared with WT/P mice (p < 0.05). Fasting glucose levels were improved after 8 wk of feeding a HF diet (weeks 9 and 10) in Mut/P mice compared with Mut, WT and WT/P mice (p < 0.05). Glucose tolerance was impaired in all groups compared with baseline (p < 0.05), except for the Mut/P group. Insulin signaling was improved (p < 0.05), and expression of TNF-α was decreased (p < 0.05) in the liver of Mut/P mice compared with the liver of WT/P mice.
Conclusion
The TLR4 LOF mutation partially protects against alveolar bone loss and improves glucose homeostasis in mice with periodontitis fed a HF diet.
Keywords: diabetes, inflammation, periodontal disease, alveolar bone
Epidemiological and cross-sectional studies indicate a strong association among periodontitis, obesity and type 2 diabetes mellitus (T2DM), with chronic inflammation as a possible common denominator (1–5). However, the precise mechanism responsible for this association has not been established because of the difficulties in investigating a cause-and-effect relationship in human subjects. Temporal, and also cause-and-effect, relationships can be more effectively controlled in animal models to study associations between periodontitis/inflammation and diabetes. To demonstrate a causal effect of periodontitis on the development of insulin resistance (IR) and T2DM, we recently used Zucker diabetic fatty (ZDF) rats as a ligature-induced periodontitis model system (6). ZDF rats are prone to obesity as a result of the presence of a mutation in the leptin receptor, and female ZDF rats develop diabetes when provided a high-fat (HF) diet (7). We reported that periodontitis influences the onset of IR and hyperglycemia in female ZDF rats fed a HF diet, but not in rats fed a low-fat (LF) diet (6). These findings indicate that periodontitis, in combination with a HF diet, promotes the development of hyperglycemia and IR in diabetic-prone animals. However, the mechanism accounting for this effect on glucose homeostasis is currently unknown.
Toll-like receptor 4 (TLR4) is a pattern recognition receptor expressed by various cell types, including macrophages, hepatocytes and pancreatic β-cells (8–14). TLR4 recognizes certain bacterial lipopolysaccharides (LPS) (15), including certain LPS originating from periodontal pathogens (16–18).
The activation of TLR4 by LPS results in increased signal transduction that upregulates the transcription of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin (IL)-6 and IL-1β (15,19–23). Interestingly, certain fatty acids (FAs) also activate TLR4 signaling, thereby increasing the production of proinflammatory cytokines (20). There are several mechanisms by which proinflammatory cytokines induce IR. For example, TNF-α can phosphorylate insulin receptor substrate-1 and -2 on a serine residue and impair normal insulin signal transduction (24).
Having demonstrated that induced periodontitis in diabetic-prone rats, in combination with a HF diet, affects plasma glucose levels (6), we now test the hypothesis that disruption of TLR4 in animals with periodontitis fed a HF diet will result in improved glucose homeostasis. We therefore induced periodontitis in mice fed a HF diet and which have a TLR4 loss-of-function (LOF) mutation rendering them resistant to stimulation with certain types of LPS and FAs via this receptor. To assess glucose homeostasis, we measured fasting plasma glucose and insulin levels and performed glucose tolerance tests. To evaluate a possible involvement of TLR4 (15) in the development of insulin resistance, and thus glucose homeostasis, we assessed insulin signaling (phosphorylation of Akt) by Western blotting (22), and measured TNF-α mRNA levels using quantitative real-time PCR in the liver, which is a major target of insulin action and plays a central role in the regulation of glucose homeostasis.
Material and methods
Animals
Twelve, 8-wk-old male control mice [C3H/HeNCrl, wild-type (WT)] (Charles River Laboratories, Wilmington, MA, USA) and 12, 8-wk-old male TLR4 mutant mice (C3H/HeJ) (Mut) (Jackson Laboratories, Bar Harbor, ME, USA) were purchased. The TLR4 mutant mice have been characterized previously by Poltorak et al. (25) and the mutation is known to confer a loss of TLR4 function. Upon arrival, the mice were placed three per cage and maintained on a LF diet (10 kcal % fat) (Research Diets, Inc., New Brunswick, NJ, USA) and autoclaved tap water ad libitum for 7 d before starting the studies. The mice were housed at constant temperature (22°C) and humidity (45–55%) in a 14-h light/10-h dark cycle. After the acclimatization period, the diet was changed to HF (60 kcal % fat) (Research Diets, Inc.) for all mice. Ligatures were placed around maxillary second molars in mice at week 1 and the animals were killed 9 wk later (week 10). The study was conducted in accordance with the University of Illinois at Chicago Institutional Animal Care guidelines.
Study design
Twenty-four mice were divided into four groups: WT mice with a healthy periodontium (WT); WT mice with periodontitis (WT/P); Mut mice with a healthy periodontium (Mut); and mutant mice with periodontitis (Mut/P). To induce periodontitis, 8-0 silk sutures (Kono Seisakujo, Ichikawa, Japan) were placed around maxillary second molars in six WT and six Mut mice at the beginning of the study (referred to as week 1) subsequent to obtaining plasma samples for baseline measurements of glucose and insulin levels. Escherichia coli LPS (2.5 ng in phosphate-buffered saline) (Sigma, St Louis, MO, USA) was soaked into the mesial and distal interproximal portion of ligatures and ligature placement was confirmed weekly from weeks 2 to 8. General anesthesia was given by intraperitoneal (ip) injection using ketamine (7.5 mg/100 g body weight) (Hospira, Inc., Lake Forest, IL, USA) and xylazine (1 mg/100 g body weight) (Lloyd, Inc., Shenandoah, IA, USA) for placing ligatures and for weekly confirmation of ligature placement. Control animals (WT and Mut) were also administered general anesthesia to control for the effects of anesthesia. Before the administration of general anesthesia, glucose levels were measured every week after fasting for 14 h. At weeks 1 and 9, glucose tolerance tests were performed as described below. Body weight was measured weekly, following overnight fasting, to assure the health of the animals. At week 10, the mice were killed by CO2 inhalation and cervical dislocation 8 min following ip injection of insulin (Novolin, Princeton, NJ, USA) at a concentration of 10 U/kg body weight. Livers were harvested and snap frozen in liquid nitrogen and then stored at −85°C for later use, and maxillas were collected for analysis as described below. Food consumption was not determined because the HF diet is very soft, crumbles easily relative to normal chow and is therefore prone to excess spillage into bedding.
Determination of plasma fasting blood-glucose and insulin levels
Fasting blood-glucose levels following a 14 h fast were determined using blood from nicked tail veins once a week using a OneTouch Glucometer (Life Scan, Milpitas, CA, USA).
Following determination of fasting blood-glucose levels, approximately 200 μL of tail blood was collected into heparinized tubes and plasma was collected. The plasma samples were used to determine fasting insulin levels using a mouse ultrasensitive insulin ELISA kit (Mercodia Inc., Winston Salem, NC, USA).
Glucose tolerance test
To further characterize glucose homeostasis, intraperitoneal glucose tolerance tests (ipGTT) were performed at baseline (week 1) and 8 wk later (week 9). Briefly, following a 14 h fast and after collection of blood samples to determine glucose and insulin levels, 50% dextrose (2 g/kg body weight) was administered intraperitoneally and glucose levels in tail blood were determined after 15, 30, 60, 90 and 120 min using a glucometer.
Alveolar bone loss
After the mice were killed (week 10), maxillas were collected and then defleshed. Alveolar bone loss per tooth was assessed using stereomicoscopy (Zeiss Stemi 2000-C; Carl Zeiss, Thornwood, NY, USA) and image analysis (NIH ImageJ; National Institutes of Health, Bethesda, MD, USA), as described by Tatakis & Guglielmoni (26).
Western blot analysis of Akt phosphorylation
Liver lysates were prepared in RIPA buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA (pH 8.0), 1% IGEPAL® (Sigma)] containing protease and phosphatase inhibitors which were then homogenized, sonicated and incubated on ice for 20 min. Following centrifugation, aliquots of the supernatant were stored at −85°C until assays were conducted. Western blotting was performed according to Sambrook et al. (27). Phosphorylated Akt (pAkt) and total Akt were detected using 0.077 μg/mL and 0.01 μg/mL, respectively, of rabbit polyclonal antibodies against mouse pAkt and Akt (Cell Signaling, Danvers, MA, USA) as primary antibodies and 0.01 μg/mL of goat anti-rabbit horseradish peroxidase-conjugated immunoglobulin G (Pierce Biotechnology, Rockford, IL, USA) as the secondary antibody. Signal was detected using an enhanced chemiluminescent substrate (SuperSignal West Dura Extended Duration Substrate; Pierce Biotechnology) followed by exposure of membrane to film. Results were quantified by densitometry, and phosphorylation of Akt was expressed relative to the levels of total Akt in each sample (i.e. the pAkt/Akt ratio).
Real-time PCR
RNA was extracted from 2–3 mg of frozen liver according to the manufacturer’s instructions (Ambion RNAqueous™-4PCR Kit; Applied Biosystems/Ambion, Austin, TX, USA). Complementary DNA (cDNA) was synthesized using an Applied Biosystems High Capacity cDNA Reverse Transcription Kit (Applied Biosystems/Ambion) and real-time PCR was performed with cDNA from 25 ng of total RNA/sample in a 20 μL final volume using a TaqMan® Gene Expression Master Mix and StepOne Plus Real Time PCR System (Applied Biosystems, Austin, TX, USA) with mouse TNF-α primers (Mm99999068m1; Applied Biosystems). Each sample was run in duplicate, and the mean value of each set of duplicates was normalized to mouse β-actin and used to calculate relative gene expression by the ΔΔCt method using StepOne Plus software (Applied Biosystems).
Statistical analysis
Statistical analysis was performed using a Mann–Whitney U-test for between-group comparisons and a Wilcoxon rank sum test for within-group repeated measures over the study period with a significance level of p < 0.05.
Results
Alveolar bone loss from periodontitis
To assess the severity of induced periodontitis, we measured alveolar bone loss as the area (mm2) bordered by the cemento–enamel junction, the crest of alveolar bone, and the mesial and distal line angles on the buccal and lingual sides of maxillary second molars (Fig. 1A). This area of bone loss includes areas of connective tissue and epithelial cell attachment that are shown in control groups (Fig. 1A, a,c). Ligature placement with LPS treatment resulted in significant bone loss in WT/P mice compared with the WT control mice (Fig. 1A, a,b). Bone loss also occurred in TLR4 Mut mice after ligature/LPS treatment (Mut/P) compared with the Mut control mice (Fig. 1A c,d), but the severity of bone loss was reduced by approximately 50% in Mut/P mice compared with WT/P mice (Fig. 1A, b,d), and this difference was statistically significant (p < 0.05), as shown in Fig. 1B.
Fig. 1.

(A) Alveolar bone loss captured by stereomicroscopy of defleshed mouse maxilla. (a) Maxilla from a control wild-type (WT) mouse without periodontitis; (b) maxilla from a WT mouse with periodontitis (WT/P); (c) maxilla from a toll-like receptor 4 (TLR4) mutant mouse without periodontitis (Mut); (d) maxilla from a Mut mouse with periodontitis (Mut/P). Red lines are drawn on the mesial and distal transition line angles. The area within the line was calculated as bone loss. The areas were measured from the buccal and palatal sides, and the total area of bone loss was calculated per tooth. (B) Average bone loss per tooth calculated using software (ImageJ). Data are presented as mean ± SEM. Statistical analysis was performed using a Mann–Whitney U-test. *p < 0.05 between direct comparison groups.
Fasting glucose and insulin levels over a 10 wk period on a HF diet
In between-group comparisons, a significant improvement/reduction in plasma fasting glucose values was observed at weeks 9 and 10 in Mut/P mice and this decrease was statistically significant compared with that for week 8 in the within-group analysis (p < 0.05) (Table 1) as well as that observed in Mut, WT and WT/P mice for both weeks 9 and 10 (p < 0.05) (Table 1). There were no statistically significant differences between WT vs. WT/P or Mut vs. Mut/P groups during the first 8-wk period (Table 1). A within-group comparison revealed that fasting glucose levels were increased significantly relative to baseline in all groups (p < 0.05) except for Mut/P mice, which developed improved/decreased glucose levels at weeks 9 and 10 (p < 0.05) (Table 1).
Table 1.
Weekly plasma fasting glucose and insulin levels
| Mouse group
|
||||
|---|---|---|---|---|
| WT/P | WT | Mut/P | Mut | |
| Fasting glucose (mg/dL) | ||||
| Week 1 | 128.2 ± 4.222 | 109.0 ± 9.438 | 121.0 ± 6.633 | 117.3 ± 5.760 |
| Week 2 | 152.2 ± 9.428 | 143.5 ± 5.408 | 170.6 ± 12.734 | 153.2 ± 4.923 |
| Week 3 | 152.7 ± 8.554 | 142.2 ± 11.068 | 155.0 ± 20.547 | 170.4 ± 6.997 |
| Week 4 | 169.0 ± 4.967 | 161.8 ± 6.509 | 146.8 ± 15.261 | 165.6 ± 9.832 |
| Week 5 | 145.5 ± 5.852 | 153.8 ± 3.942 | 173.5 ± 10.348 | 181.8 ± 6.343 |
| Week 6 | 165.5 ± 9.561 | 159.8 ± 7.645 | 173.8 ± 5.154 | 183.3 ± 3.521 |
| Week 7 | 171.3 ± 7.532 | 171.3 ± 6.033 | 180.5 ± 11.420 | 167.5 ± 8.703 |
| Week 8 | 148.5 ± 10.145 | 169.0 ± 3.512 | 167.5 ± 10.137 | 166.5 ± 2.986 |
| Week 9 | 157.8 ± 9.860 | 163.0 ± 8.185 | 135.5 ± 9.438* | 171.0 ± 8.367 |
| Week 10 | 175.3 ± 11.056 | 179.7 ± 6.691 | 135.3 ± 6.969* | 167.8 ± 2.983 |
| Fasting insulin (μg/L) | ||||
| Week 1 | 0.286 ± 0.055 | 0.193 ± 0.020 | 0.178 ± 0.017 | 0.203 ± 0.025 |
| Week 2 | 0.468 ± 0.109 | 0.433 ± 0.038 | 0.783 ± 0.048 | 0.674 ± 0.070 |
| Week 3 | 0.483 ± 0.108 | 0.463 ± 0.040 | 0.781 ± 0.087 | 0.718 ± 0.077 |
| Week 4 | 0.732 ± 0.230 | 0.609 ± 0.077 | 1.134 ± 0.205 | 0.758 ± 0.115 |
| Week 5 | 0.677 ± 0.195 | 0.587 ± 0.084 | 1.081 ± 0.081 | 0.858 ± 0.106 |
| Week 6 | 0.791 ± 0.143 | 0.754 ± 0.199 | 0.978 ± 0.082 | 1.147 ± 0.242 |
| Week 7 | 1.095 ± 0.141 | 1.303 ± 0.220 | 1.324 ± 0.230 | 1.194 ± 0.058 |
| Week 8 | 0.880 ± 0.143 | 1.334 ± 0.400 | 1.963 ± 0.367 | 1.516 ± 0.393 |
| Week 9 | 1.264 ± 0.183 | 1.291 ± 0.404 | 2.086 ± 0.824 | 1.546 ± 0.165 |
| Week 10 | 1.975 ± 0.320 | 1.539 ± 0.306 | 1.910 ± 0.541 | 1.442 ± 0.172 |
Mut, toll-like receptor 4 (TLR4) mutant mice without periodontitis; Mut/P, Mut mice with periodontitis; WT, wild-type mice without periodontitis; WT/P, WT mice with periodontitis.
Data are presented as mean ± SEM. Statistical analysis was performed using a Mann–Whitney U-test for between-group comparisons (*p < 0.05) and a Wilcoxon rank sum test for within-group repeated measures over the study period. A within-group comparison indicates that fasting glucose levels were increased significantly relative to baseline in all groups (p < 0.05) except for Mut/P mice which developed improved/decreased glucose levels at weeks 9 and 10 (p < 0.05). Statistical significance (*) is not shown for within-group comparisons because of the number of significant data points for both glucose and insulin levels.
To understand the effects of periodontitis and TLR4 LOF on insulin levels and insulin sensitivity/resistance, we also measured fasting insulin levels. In between-group comparisons, no statistical difference was found in WT vs. WT/P or Mut vs. Mut/P. The mean insulin levels reached 2 μg/L (hyperinsulinemia) in both WT/P (week 10) and Mut/P (weeks 8 and 9) mice, but not in WT or Mut mice. Comparison of baseline insulin levels with weekly values in within-group analyses revealed a statistically significant increase in insulin levels starting at week 2 in WT, Mut and Mut/P and at week 4 in WT/P groups (p < 0.05) (Table 1).
To demonstrate more clearly, the interplay between insulin and glucose levels in the four groups, the glucose and insulin data are displayed together in Fig. 2. In Mut/P mice, the development of mean insulin levels near 2.0 μg/L (hyperinsulinemia) occurred in week 8 and was associated with a significant decrease in fasting glucose levels at weeks 8–9 (Fig. 2D). By contrast, in WT/P mice, the mean insulin levels continued to rise during weeks 8–10 and the mean insulin level reached 2.0 μg/L at week 10 while the glucose levels remained elevated (Fig. 2B). In both WT and Mut mice, insulin levels never reached 2.0 μg/L and both glucose and insulin levels plateaued after week 7 (Fig. 2A,C).
Fig. 2.

Composite graph showing temporal relationships between fasting glucose (dashed line) and insulin (solid line) levels in wild-type mice without periodontitis (WT) (A), in WT mice with periodontitis (WT/P) (B), in toll-like receptor 4 (TLR4) mutant mice without periodontitis (Mut) (C) and in Mut mice with periodontitis (Mut/P) (D) over a 10 wk period. Left y-axis: fasting insulin levels (μg/L). Right y-axis: fasting glucose levels (mg/dL).
Effect of periodontitis and involvement of TLR4 on glucose tolerance
The effect of periodontitis on glucose homeostasis in TLR4 mutants and WT mice was also assessed by glucose tolerance testing at baseline and at 9 wk. There was no difference in glucose tolerance between WT and Mut mice at baseline (before ligature placement) at any time-point (Fig. 3A). At week 9, all groups exhibited impaired glucose tolerance compared with baseline (p < 0.01), except for Mut/P mice, which exhibited no statistically significant difference (Fig. 3A). Although the mean glucose levels at 90 and 120 min were > 300 mg/dL for WT/P mice at 9 wk, the difference in glucose tolerance in WT/P vs. WT mice did not reach statistical significance (Fig. 3B, e) (p = 0.064). By contrast, glucose tolerance was not impaired in Mut/P vs. Mut mice, as evidenced by significantly reduced glucose levels in Mut/P mice at 60, 90 and 120 min after glucose administration (p < 0.05) (Fig. 3B, g). In addition, glucose tolerance was significantly better in Mut/P vs. WT/P mice at 90 and 120 min after glucose administration (p < 0.05) (Fig. 3B, h).
Fig. 3.

(A) Comparison of intraperitoneal glucose tolerance (ipGTT) test results obtained at weeks 1 (baseline; dashed line) and 9 (solid line) within a group in wild-type mice without periodontitis (WT) (a), WT mice with periodontitis (WT/P) (b), toll-like receptor 4 (TLR4) mutant mice without periodontitis (Mut) (c) and Mut mice with periodontitis (Mut/P) (d). (B) Comparison of ipGTT results obtained at week 9 between the following groups: WT vs. WT/P (e), WT vs. Mut (f), Mut vs. Mut/P (g) and WT/P vs. Mut/P (h). Values for each time-point are expressed as mean ± SEM. The x-axis indicates min following intraperitoneal dextrose injection. The y-axis indicates glucose levels in mg/dL. Statistical analysis was performed using the Wilcoxon rank sum test with *p < 0.05. p = 0.064 at the 120-min time-point in Fig. 3B, e.
Effects of periodontitis and involvement of TLR4 on Akt phosphorylation in the liver
As the liver is a major target of insulin action and plays an important role in determining fasting glucose levels and glucose tolerance, we also examined the effect of periodontitis and mutation of the TLR4 receptor on insulin signaling in the liver. Insulin-stimulated phosphorylation of Akt was enhanced in TLR4 Mut/P and Mut mice compared with WT/P mice (p < 0.05) (Fig. 4A). Although the mean pAkt/Akt values appeared to differ, there was no statistically significant difference in pAkt/Akt between WT and WT/P mice or between Mut/P and Mut mice.
Fig. 4.
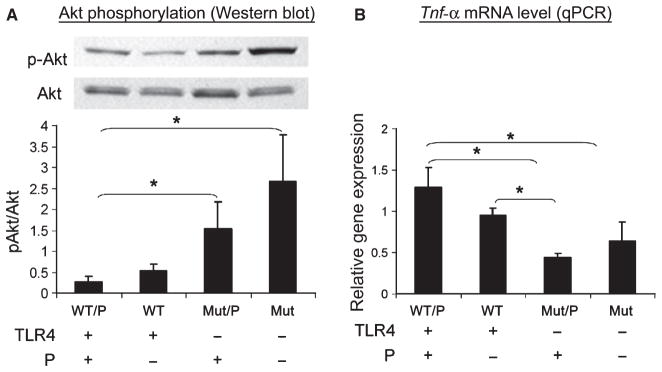
(A) Akt phosphorylation in liver as determined by Western blot analysis. Upper panel: Western blot (representative of four independent experiments). Lower panel: pAkt/Akt ratio (y-axis) determined from densitometric scanning of four independent western blot experiments. (B) Tumor necrosis factor-α (TNF-α) mRNA levels in liver, determined by real-time quantitative (q)PCR. P indicates periodontitis. (A), (B) Statistical analysis was performed using a Mann–Whitney rank sum test. *p < 0.05 between groups.
Quantitative PCR analysis of liver TNF-α
As activation of TLR4 stimulates the expression of TNF-α, and TNF-α signaling can cause insulin resistance, we also examined the expression of TNF-α in the liver using real-time PCR. As shown in Fig. 4B, the levels of TNF-α mRNA were significantly lower in both Mut and Mut/P mice compared with WT/P mice (p < 0.05). Although the mean expression levels of TNF-α also appear to be different in Mut vs. Mut/P, the difference was not statistically significant (Fig. 4B).
Discussion
There is general agreement that FAs and LPS promote inflammation, at least in part, through TLR4, and inflammation promotes IR (20,24, 28–30). We therefore hypothesized that disruption of TLR4 would improve glucose homeostasis in animals with periodontitis provided a HF diet. To test this hypothesis, we induced periodontitis by placing ligatures soaked with E. coli LPS around maxillary second molars in WT and TLR4 Mut mice. In this study, we focused on a HF diet, based on our previous study results and also on the results of other studies which indicate that TLR4 mice fed a HF diet develop IR, but very little IR develops in mice fed a LF diet (20,22,31). In general, consumption of a HF diet more closely resembles the human diet, at least in western societies. Thus, our focus in this study was to examine the effects of periodontitis and disruption of TLR4 function in the context of a HF diet.
TLR4 Mut mice have a point mutation in the Tlr4 gene, rendering these animals unresponsive to certain types of LPS, whereas the corresponding WT mice remain responsive to LPS (20,32). LPS from certain periodontal pathogens activates TLR4 to produce proinflammatory cytokines in vitro (16,17) and initiates alveolar bone resorption. The results from an in vivo study, which involved repeated gingival injections of Aggregatibacter actinomycetemcomitans LPS, showed less bone resorption in TLR4 Mut vs. WT mice using immunohistochemistry (18). Thus, it has been postulated that one pathway leading to alveolar bone loss involves the activation of TLR4 and the subsequent production of proinflammatory cytokines (33). Activation of other TLRs may also be involved in the initiation of bone loss during periodontitis. For example, Burns et al. (34) showed that TLR2−/− mice are protected from bone loss when Porphyromonas gingivalis LPS is administered orally.
We used a ligature-induced periodontitis model in mice to simulate the induction and progression of chronic periodontitis over a 10 wk period. Ligature-induced periodontitis has been used as a model system for studying periodontitis in other animals such as rats, primates and dogs (35–37). The results of our current study clearly demonstrate that the LOF mutation of TLR4 significantly reduces alveolar bone loss in ligature-induced periodontitis in Mut mice compared with WT mice by approximately 50%. Thus, the TLR4 LOF mutation partially protects against alveolar bone loss from ligature-induced periodontitis. As mentioned, the lack of full protection from alveolar bone loss may be caused by the activation of other TLRs, such as TLR2. It is well documented that subgingival plaque consists of different species and strains of periodontal pathogens (38), and products from these bacteria may activate different TLRs. Therefore, combinations of TLRs may contribute to alveolar bone loss. We chose E.coli LPS to soak the ligatures because it is a prototype LPS that binds to and activates TLR4 (39–43). Further studies are needed to investigate whether TLR2-activating products might also be present in this ligature-induced periodontitis model. To our knowledge, this is the first study to demonstrate definitively that mice with a TLR4 LOF mutation are partially protected against alveolar bone loss occurring in response to LPS-soaked ligature-induced periodontitis, which simulates chronic periodontitis over a 10 wk period. Based on the results from our current study and from other studies (44,45), ligature-induced periodontitis in mice, although technically challenging, appears to be useful for investigating the systemic effects of periodontitis.
We monitored fasting glucose and insulin levels over a 10 wk period to determine the effect of periodontitis and TLR4 on glucose homeostasis. Fasting glucose and insulin levels at week 10 were increased significantly in all groups relative to baseline; however, the glucose levels in Mut/P mice at weeks 9 and 10 became close to normoglycemic. This suggests that disruption of TLR4 improves glucose homeostasis in the presence of periodontitis in conjunction with a HF diet. The normalization of glucose levels observed in Mut/P mice was associated with the development of hyperinsulinemia at weeks 9 and 10. The mean insulin levels in Mut/P mice reached 2.0 μg/L by week 8, which was 2 wk earlier than observed in WT/P mice. The development of hyperinsulinemia associated with periodontitis is consistent with our previous study results using ZDF rats except that the onset of hyperinsulinemia is much earlier in the rats. The difference in the onset of hyperinsulinemia between our previous study and the current study may again be attributed to the fact that ZDF rats are prone to diabetes, and mice used in this study are not.
It is not clear why Mut/P mice, but not Mut mice, developed improved glucose homeostasis. In this comparison, the only difference was the presence or absence of periodontitis. In Mut/P mice, hyperinsulinemia occurred at weeks 8–10, and this may be associated with the TLR4 regulation of insulin production/secretion by pancreatic β-cells. During the development of IR and T2DM, β-cells undergo compensatory alterations to increase insulin secretion (hyperinsulinemia) by increasing β-cell mass (hyperplasia) in an early stage of IR followed by declining insulin secretion resulting from β-cell apoptosis in T2DM (46). It has been reported that LPS modulates glucose-dependent insulin secretion by inducing IL-1α, TNF-α and IL-6 expression in pancreatic β-cells (47). Therefore, it is possible that periodontitis may modulate insulin secretion via TLR4 on β-cells, and the absence of TLR4 may lead to the early development of hyperinsulinemia.
We also performed glucose tolerance tests to provide further evidence of the effect of TLR4 and periodontitis on glucose homeostasis. Following 8 wk on a HF diet, all groups developed impaired glucose tolerance compared with baseline, except for Mut/P mice. Glucose tolerance was significantly impaired in WT/P mice compared with Mut/P mice. Taken together, our data on glucose levels, insulin levels and ipGTT in Mut/P vs. WT/P mice indicated that TLR4 LOF improves glucose homeostasis in the presence of periodontitis.
In contrast to our previous study in diabetes-prone ZDF rats, we did not detect a significant effect of periodontitis on fasting glucose or ipGTT results in WT mice provided a HF diet. This difference may be attributed to differences in the animal models, and suggests that an underlying genetic predisposition may be necessary to observe the effect of periodontitis on glucose and insulin levels. It is also possible that more prolonged exposure to a HF diet and/or more prolonged duration of periodontitis may be required to demonstrate an effect on fasting glucose levels in this mouse model.
The glucose levels continued to increase in WT/P mice despite the increased mean insulin levels observed at week 10, and this suggests that insulin sensitivity may be impaired in WT/P mice during weeks 8–10 compared with earlier time-points. The same interplay between glucose and insulin levels was not observed in WT mice, as clearly shown in the composite figure presented. It is possible that the hyperinsulinemia observed in Mut/P mice would have developed in WT/P mice if the study period had been longer than 10 wk, because mean insulin levels reach 2 μg/L at week 10, which is 2 wk later than observed in Mut/P mice.
The liver plays a central role in the regulation of glucose metabolism, and it is a major target of insulin action. Therefore, in this study we also examined the role of TLR4 in mediating effects on cytokine expression and insulin signaling in the livers of mice with/without periodontitis fed a HF diet. The major cell types in the liver are hepatocytes and Kupffer cells, both of which express TLR4 (48). Circulating LPS is taken up predominantly by Kupffer cells, which constitute 80–90% of all macrophages in the body (49). LPS uptake is thought to be mediated largely through interaction with TLR4, which activates pathways that promote the expression of proinflammatory cytokines, including TNF-α (20). Because certain free fatty acids (FFAs) also interact with and activate TLR4, it is reasonable to speculate that circulating LPS and FFAs both may augment proinflammatory cytokine production in the liver via TLR4 and thereby impair insulin signaling and glucose control in hepatocytes. In the present study, insulin-stimulated phosphorylation of Akt in animals with periodontitis significantly improved when TLR4 was suppressed (Mut/P), indicating that the LOF mutation of TLR4 serves to maintain insulin action in the liver in mice with periodontitis when fed a HF diet. Insulin signaling, assessed by pAkt/Akt, was decreased in WT/P mice compared with WT mice, although this difference did not reach significance, perhaps because of the limited time-frame of the study. The pAkt/Akt data showing a significant difference between WT/P and Mut/P mice indicate that disrupting TLR4 alone results in improved insulin signaling, and therefore insulin sensitivity in the liver. Mut/P mice have suppressed insulin signaling compared with Mut mice, although the difference is not statistically significant. This relative decrease in insulin signaling may be attributed to the fact that other receptors, including TLR2, may be still functional in Mut/P mice.
As certain inflammatory cytokines impair insulin signaling and promote insulin resistance (25), we also examined the expression of TNF-α in the livers of WT and Mut mice, with or without periodontitis. The level of expression of TNF-α mRNA was significantly decreased in the liver of TLR4 mutant mice compared with WT/P mice, especially in the presence of periodontitis (Mut/P). Taken together, these findings support the concept that suppressing the function of TLR4 may help to maintain insulin sensitivity in the liver, at least in part, by decreasing the expression of TNF-α.
In conclusion, our results indicate that the TLR4 LOF mutation improves glucose homeostasis in mice with periodontitis that are fed a HF diet. Additional studies with a larger number of animals that have been fed a HF diet for a longer period of time may help to demonstrate more marked effects of periodontitis on glucose homeostasis. Further studies are required to identify the contribution of other TLRs, as well as the effect of periodontitis on proinflammatory and insulin signaling pathways in other tissues such as skeletal muscle, adipose tissue and pancreatic β-cells.
Acknowledgments
This study was supported by a College of Dentistry Research Seed Grant, College of Dentistry, University of Illinois at Chicago (K.W.), an NIH/NIDCR R21 DE 019194 (K.W.), by a College of Dentistry Urban Health Program grant (A.A.), and the Department of Veterans Affairs Merit Review System (T.U.). The authors thank Ms Grace Viana and Dr Ellen Begole at the University of Illinois at Chicago for assisting with statistical analyses, and Dr Vineela Redla at the University of Illinois for her technical assistance.
Footnotes
The authors report no conflicts of interest related to this study.
References
- 1.Genco JR, Grossi SG, Ho A, Nishimura F, Murayama Y. A proposed model linking inflammaion to obesity, diabetes, and periodontal infection. J Periodontol. 2005;76:2075–2084. doi: 10.1902/jop.2005.76.11-S.2075. [DOI] [PubMed] [Google Scholar]
- 2.Sjoholm A, Nystrom T. Inflammation and the etiology of type 2 diabetes. Diabetes metab Res Rev. 2006;22:4–10. doi: 10.1002/dmrr.568. [DOI] [PubMed] [Google Scholar]
- 3.Saito T, Shimazaki Y. Metabolic disorders related to obesity and periodontal disease. Peirodontol 2000. 2007;43:254–266. doi: 10.1111/j.1600-0757.2006.00186.x. [DOI] [PubMed] [Google Scholar]
- 4.Pischon N, Heng N, Bernimoulin J-P, Kleber B-M, Willich SN, Pischon T. Obesity, inflammation and periodontal disease. J Dent Res. 2007;86:400–409. doi: 10.1177/154405910708600503. [DOI] [PubMed] [Google Scholar]
- 5.Schoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132:2169–2180. doi: 10.1053/j.gastro.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe K, Petro BJ, Shlimon AE, Unterman TG. Effect of periodontitis on insulin resistance and the onset of Type 2 diabetes mellitus in Zucker Diabetic Fatty Rats. J Periodontol. 2008;79:1208–1216. doi: 10.1902/jop.2008.070605. [DOI] [PubMed] [Google Scholar]
- 7.Corsetti JP, Sparks JD, Peterson RG, Smith RL, Sparks CE. Effect of dietary fat on the development of non-insulin dependent diabetes mellitus in obese Zucker diabetic fatty male and female rats. Atherosclerosis. 2000;148:231–241. doi: 10.1016/s0021-9150(99)00265-8. [DOI] [PubMed] [Google Scholar]
- 8.Wang PL, Ohura K, Fujii T, et al. DNA microarray analysis of human gingival fibroblasts from healthy and inflammatory gingival tissues. Biochem Biophys Res Commun. 2003;305:970–973. doi: 10.1016/s0006-291x(03)00821-0. [DOI] [PubMed] [Google Scholar]
- 9.Migita K, Abiru S, Nakamura M, et al. Lipopolysaccharide signaling induces serum amyloid A (SAA) synthesis in human hepatocytes in vitro. FEBS Lett. 2004;569:235–239. doi: 10.1016/j.febslet.2004.05.072. [DOI] [PubMed] [Google Scholar]
- 10.Hemmi H, Akira S. TLR signaling and the function of dendritic cells. Chem Immunol Allergy. 2005;86:120–135. doi: 10.1159/000086657. [DOI] [PubMed] [Google Scholar]
- 11.Kinane DF, Shiba H, Stathopoulou PG, et al. Gingival epithelial cells heterozygous for Toll-like receptor 4 polymorphism Asp299Gly ad Thr399ile are hyporesponsive to Porphyromonas gingivalis. Genes Immun. 2006;7:190–200. doi: 10.1038/sj.gene.6364282. [DOI] [PubMed] [Google Scholar]
- 12.Bes-Houtmann S, Roche R, Hoareau L, et al. Presence of functional TLR2 and TLR4 on human adipocytes. Histochem Cell Biol. 2007;127:131–137. doi: 10.1007/s00418-006-0230-1. [DOI] [PubMed] [Google Scholar]
- 13.Reyna SM, Ghosh S, Tantiwong P, et al. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes. 2008;57:2595–2602. doi: 10.2337/db08-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Y, Shu R, Zhang MZ, Wu AP. Toll-like receptor 4 singaling plays a role in triggering periodontal infection. FEMS Immunol Med Microbiol. 2008;52:362–369. doi: 10.1111/j.1574-695X.2008.00386.x. [DOI] [PubMed] [Google Scholar]
- 15.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 16.Kikkert R, Laine ML, Aaden LA, van Winkelhoff AJ. Activation of toll-like receptors 2 and 4 by gram-negative periodontal bacteria. Oral Microbiol Immunol. 2007;22:145–154. doi: 10.1111/j.1399-302X.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- 17.Sawada N, Ogawa T, Asai Y, Makimura Y, Sugiyama A. Toll-like receptor 4-dependent recognition of structurally different forms of chemically synthesized lipid As of Porphyromonas gingivalis. Clin Exp Immunol. 2007;148:529–536. doi: 10.1111/j.1365-2249.2007.03346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura H, Fukusaki Y, Yoshimura A, et al. Lack of Toll-Like Receptro 4 decreases lipopolysaccharides-induced bone resorption in C3H/HeJ mice in vivo. Oral Microbiol Immunol. 2008;23:190–195. doi: 10.1111/j.1399-302X.2007.00410.x. [DOI] [PubMed] [Google Scholar]
- 19.Weatherill AR, Lee JY, Zhao L, Lemay DG, Youn HS, Hwang DH. Saturated and polyunsaturated fatty acids reciprocally modulate dendritic cell functions mediated through TLR4. J Immunol. 2005;174:5390–5397. doi: 10.4049/jimmunol.174.9.5390. [DOI] [PubMed] [Google Scholar]
- 20.Shi H, Kokoeva MV, LInouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen MTA, Favelyukis S, Nguyen AK, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-Like Receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–35292. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- 22.Tsukumo DM, Carvalho-Filno MA, Carvalheira JB, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- 23.Song MJ, Kim KH, Yoon JM, Kim JB. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem Biophys Res Commun. 2006;346:739–745. doi: 10.1016/j.bbrc.2006.05.170. [DOI] [PubMed] [Google Scholar]
- 24.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentarions in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 26.Tatakis DN, Guglielmoni P. HLA-B27 transgenic rats are susceptible to accelerated alveolar bone loss. J Periodontol. 2000;71:1395–1400. doi: 10.1902/jop.2000.71.9.1395. [DOI] [PubMed] [Google Scholar]
- 27.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. Plainview, New York: Cold Spring Harbor Laboratory Press; 1989. pp. 18.60–18.75. [Google Scholar]
- 28.Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem. 2000;275:15985–15991. doi: 10.1074/jbc.275.21.15985. [DOI] [PubMed] [Google Scholar]
- 29.Senn JJ, Kolver PJ, Nowak IA, Mooney RA. Inerleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes. 2002;51:3391–3399. doi: 10.2337/diabetes.51.12.3391. [DOI] [PubMed] [Google Scholar]
- 30.Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol. 2004;24:5434–5446. doi: 10.1128/MCB.24.12.5434-5446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim F, Pham M, Luttrell I, et al. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res. 2007;100:1589–1596. doi: 10.1161/CIRCRESAHA.106.142851. [DOI] [PubMed] [Google Scholar]
- 32.Hoshino K, Takeuchi O, Kawai T, et al. Cutting Edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharides: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 33.Yamaguchi R, Yoshimura A, Yoshioka H, Kaneko T, Hara Y. Ability of supragingival plaque to induce toll-like receptor 4-mediated stimulation is associated with cytokine production by peripheral blood mononuclear cells. J Periodontol. 2009;80:512–520. doi: 10.1902/jop.2009.080393. [DOI] [PubMed] [Google Scholar]
- 34.Burns E, Bachrach G, Shapira L, Nussbaum G. Cutting Edge: TLR2 is required for the innate response to Prophyromonas gingivalis activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. 2006;177:8296–8300. doi: 10.4049/jimmunol.177.12.8296. [DOI] [PubMed] [Google Scholar]
- 35.Schroeder HE, Lindhe J. Conditions and pathological features of rapidly destructive, experimental periodontitis in dogs. J Periodontol. 1980;51:6–19. doi: 10.1902/jop.1980.51.1.6. [DOI] [PubMed] [Google Scholar]
- 36.Siegrist B, Kornman KS. The effect of supragingival plaque control on the composition of the subgingival microbial flora in ligature-induced periodontitis in the monkey. J Dent Res. 1982;61:936–941. doi: 10.1177/00220345820610071001. [DOI] [PubMed] [Google Scholar]
- 37.Pontes Andersen CC, Buschard K, Flyvbjerg A, Stoltze K, Holmstup P. Periodontitis deteriorates metabolic control in Type 2 Diabetic Goto-Kakizaki rats. J Periodontol. 2006;77:350–356. doi: 10.1902/jop.2006.050184. [DOI] [PubMed] [Google Scholar]
- 38.Paster BJ, Boches SK, Galvin JL, et al. Bacterial diversity in human subgingival plaque. J Bacteriol. 2001;183:3770–3783. doi: 10.1128/JB.183.12.3770-3783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang Q, Akashi S, Miyake K, Petty HR. Lipopolysaccharide induces physical proximity between CD14 and toll-like receptor 4 (TLR4) prior to nuclear translocation of NF-kappa B. J Immunol. 2000;165:3541–3544. doi: 10.4049/jimmunol.165.7.3541. [DOI] [PubMed] [Google Scholar]
- 40.Flo TH, Ryan L, Latz E, et al. Involvement of toll-like receptor (TLR) 2 and TLR4 in cell activation by mannuronic acid polymers. J Biol Chem. 2002;277:35489–35495. doi: 10.1074/jbc.M201366200. [DOI] [PubMed] [Google Scholar]
- 41.Raffeiner B, Dejaco C, Duftner C, et al. Between adaptive and innate immunity: TLR4-mediated perforin production by CD28null T-helper cells in ankylosing spondylitis. Arthritis Res Ther. 2005;7:R1412–R1420. doi: 10.1186/ar1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang Y, Chen G, Zheng Y, et al. TLR4 signaling induces functional nerve growth factor receptor p75NTR on mouse dendritic cells via p38MAPK and NF-kappa B pathways. Mol Immunol. 2008;45:1557–1566. doi: 10.1016/j.molimm.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 43.Lee KM, Seong SY. Partial role of TLR4 as a receptor responding to damage-associated molecular pattern. Immunol Lett. 2009;125:31–39. doi: 10.1016/j.imlet.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 44.Kimura S, Nagai A, Onitsuka T, et al. Induction of experimental periodontitis in mice with Phophyromonas gingivalis-adhered ligatures. J Periodontol. 2000;71:1167–1173. doi: 10.1902/jop.2000.71.7.1167. [DOI] [PubMed] [Google Scholar]
- 45.Amar S, Zhou Q, Shaki-Dasthagirisaheb Y, Leeman S. Diet-induced obesity in mice causes changes in imuune responses and bone loss manifested by bacterial challenge. Proc Natl Acad Sci USA. 2007;104:20466–20471. doi: 10.1073/pnas.0710335105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rafacho A, Cestari TM, Taboga SR, Boschero AC, Bosquerio JR. High doses of dexamethasone induce increased β-cell proliferation in pancreatic rat islets. Am J Physiol Endocrinol Metab. 2009;21:E681–E689. doi: 10.1152/ajpendo.90931.2008. [DOI] [PubMed] [Google Scholar]
- 47.Vives-PI M, Somoza N, Fernandez-Alvares J, et al. Evidence of expression of endotoxin receptors CD14, toll-like receptors TLR4 and TLR2 and associated molecule MD-2 and of sensitivity to endotoxin (LPS) in islet beta cells. Clin Exp Immunol. 2003;133:208–218. doi: 10.1046/j.1365-2249.2003.02211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsieh YC, Frink M, Thobe BM, et al. 17β-Estradiol downregulates Kupffer cell TLR4-dependent p38MAPK pathway and normalizes inflammatory cytokine production following trauma-hemorrhage. Mol Immunol. 2007;44:2165–2172. doi: 10.1016/j.molimm.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, Perlik V, Feleder C, Tang Y, Blatteis CM. Kupffer cell-generated PGE2 triggers the febrile response of guinea pigs to intravenously injected LPS. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1262–R1270. doi: 10.1152/ajpregu.00724.2005. [DOI] [PubMed] [Google Scholar]