

Abstract
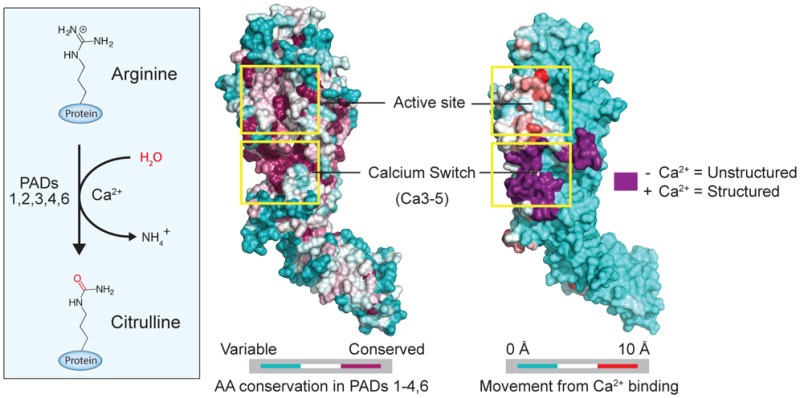
Protein arginine deiminases (PADs) are calcium-dependent histone-modifying enzymes whose activity is dysregulated in inflammatory diseases and cancer. PAD2 functions as an Estrogen Receptor (ER) coactivator in breast cancer cells via the citrullination of histone tail arginine residues at ER binding sites. Although an attractive therapeutic target, the mechanisms that regulate PAD2 activity are largely unknown, especially the detailed role of how calcium facilitates enzyme activation. To gain insights into these regulatory processes, we determined the first structures of PAD2 (27 in total), and through calcium-titrations by X-ray crystallography, determined the order of binding and affinity for the six calcium ions that bind and activate this enzyme. These structures also identified several PAD2 regulatory elements, including a calcium switch that controls proper positioning of the catalytic cysteine residue, and a novel active site shielding mechanism. Additional biochemical and mass-spectrometry-based hydrogen/deuterium exchange studies support these structural findings. The identification of multiple intermediate calcium-bound structures along the PAD2 activation pathway provides critical insights that will aid the development of allosteric inhibitors targeting the PADs.
PADs are calcium-dependent enzymes that use a nucleophilic cysteine to hydrolyze guanidinium groups on arginine residues to form citrulline (Figure 1A, Figure S1A). This reaction, termed citrullination or deimination, results in the loss of positive charge, thereby affecting protein function and altering protein–protein and protein–nucleic acid interactions.1−4 Humans encode five PADs, designated PADs 1–4 and PAD6, which are highly homologous both within and between species (44–58% identity between human PADs). While PAD4 binds five calciums per monomer, none of these metal ions directly participate in catalysis. Nevertheless, they are critical for upregulating enzymatic activity by at least 10 000-fold.5 Despite their high homology, the five enzymes in this family are nonredundant and regulate numerous cellular processes including pluripotency,3,6 myelination,7 gene transcription,8 kinase signaling,9 antigen generation,10 and neutrophil11 and macrophage12 extracellular trap (NET and MET) formation. Dysregulated PAD activity, most notably PAD2 and PAD4, is associated with multiple inflammatory diseases (e.g., rheumatoid arthritis) as well as cancer,13 and PAD inhibitors, such as Cl-amidine and BB-Cl-amidine, show efficacy in multiple preclinical animal models of disease.14−20
Figure 1.

PAD2 structural analysis. (A) Deimination (citrullination) reaction catalyzed by the PADs. (B) Wild type PAD2 structures soaked with 0 mM (apoenzyme, left, PDB: 4N20) and 10 mM CaCl2 (middle, PDB: 4N2B) and the PAD2 F221/222A mutant soaked in 10 mM CaCl2 (holoenzyme, right, PDB: 4N2C). (C) Comparison of the PAD2 active site in the apo (left), 10 mM calcium (center), and holo (right) states show that the active site rearranges upon calcium binding, including proper positioning of C647, and the movement of R347 out of the active site.
PAD2 is a particularly attractive therapeutic target for breast cancer because it is recruited by the estrogen receptor (ER) to ER target gene promoters where it citrullinates histone H3 at R26, leading to ER-target gene activation.8 PAD2 also serves as a component of an ER-related gene expression signature that predicts survival benefit from adjuvant endocrine therapy,21 and its expression is positively correlated with HER2 protein levels in breast cancer cell lines and in primary HER2+ breast tumors.22 These observations indicate that PAD2 activity plays an important role in breast cancer progression. Importantly, inhibition or knockdown of PAD2 decreases ER target-gene expression17 and the PAD inhibitor Cl-amidine decreases tumor burden in a breast cancer xenograft model of ductal carcinoma in situ.(17) Overall, these data highlight the therapeutic potential of inhibitors targeting PAD2.
Herein, we use a combination of structural, biochemical, and mass-spectrometry-based hydrogen–deuterium exchange (HDX) methods to show that PAD2 binds six calcium ions in an ordered fashion. Additionally, our data indicate that PAD2 activation is controlled by a calcium switch that unshields the active site and induces proper positioning of the catalytic cysteine, thereby activating the enzyme by >7 × 105-fold. Combining our structural data with recent data on an allosteric PAD4 selective inhibitor,23 we highlight the potential of using a similar approach to develop allosteric inhibitors targeting PAD2.
Results and Discussion
Structures of apoPAD2 and PAD2·Ca(10mM)2+
To gain insight into the regulatory processes that control PAD2 activity, we first determined the structure of the apoenzyme (PAD2·Ca(0mM)2+ or apoPAD2) to 1.66 Å resolution (Figure 1B-Left, Table S1). ApoPAD2 crystallized as a stable head-to-tail dimer (Figure S1B–C), as confirmed in solution by native spray MS (Figure S1D). The monomer is comprised of two immunoglobulin-like domains, i.e., IgG1 (residues 1–115) and IgG2 (residues 116–295), as well as a C-terminal catalytic domain (residues 296–665). The structures of the active site and the regions surrounding calcium-binding sites 2, 3, 4, and 5 (Ca2–5) are similar to apoPAD4 (RMSD = 1.42 Å; Figure S1E). Even though the Ca2–5 sites are unoccupied in apoPAD2, clear electron density is observed at both the Ca1 site and a newly discovered sixth calcium-binding site (Ca6) that is conserved among PAD2 orthologues (Figure S2A–C) but is not present in PAD4 (Figure S2D). Interestingly, electron density was not observed at the Ca1 site in apoPAD4, suggesting that PAD2 binds calcium at this site with a higher affinity.5 The Ca1 and Ca6 coordination contacts are consistent with standard calcium bond lengths (2.3–2.5 Å),24 confirming that both sites bind calcium (Figure S3). Differences in the HD exchange (HDX) kinetics for peptides comprising the Ca6 site (121–131) are also consistent with Ca6 binding calcium, as the apo state undergoes more extensive exchange than the holoenzyme upon the addition of calcium (Figure S4, Table S2). Considering the 8 μM total concentration of calcium, as determined by ICP-MS analysis, and the presence of excess EDTA (0.5 mM) in the crystallization buffer, we conclude that Ca1 and Ca6 are high-affinity calcium-binding sites with KD values of <1 μM.
We next soaked the apoPAD2 crystals in 10 mM CaCl2 and determined the structure of the PAD2·Ca(10mM)2+ complex at 1.69 Å resolution (Figure 1B-Middle). The overall fold is similar to apoPAD2, except that the residues comprising the Ca3–5 sites are now ordered and bound to calcium. Even though five of the six calcium-binding sites are now occupied, the PAD2·Ca(10mM)2+ structure does not adopt a catalytically competent conformation because the active site nucleophile, C647, is approximately 12 Å away from the catalytic center (Figure 1C-Left, Center). Despite C647 being positioned away from the active site, the remaining three key catalytic residues (i.e., D351, H471, and D473) are properly positioned to promote catalysis. Since D351, H471, and D473 adopt virtually identical conformations in apoPAD2 and the PAD2 holoenzyme (see below), the PAD2·Ca(10mM)2+ structure represents an intermediate between the apoenzyme and calcium-bound holoenzyme. This intermediate conformation is likely stabilized in part by a hydrogen bond between R347 and Q350 deep within the active site, which would also inhibit the movement of C647 into the substrate-binding pocket (Figure 1C-Left, Center, Figure S5A-Left).
Structure of the PAD2 Holoenzyme
Given that F221 and F222 bind in a hydrophobic pocket on a neighboring subunit in the crystal lattice, coupled with their proximity to the Ca3–5 sites, we hypothesized that this interaction might prevent calcium-binding at Ca2 (Figure S6). Therefore, to obtain the structure of the holoenzyme, we engineered the F221/222A double mutant and solved its calcium-bound structure at 3.02 Å resolution (Figure 1B-Right). Importantly, these mutations do not significantly alter the activity or calcium dependence of the enzyme (Table 1). Notably, however, the PAD2 F221/222A·Ca(10mM)2+ structure shows clear electron density at all six calcium-binding sites (Figure S7A,B,D), and C647, the active site cysteine, now points toward the catalytic center in a conformation that is competent for catalysis (Figure 1C-Right, Figure S5a-Right, Figure S7C, Movie S1). In addition to the effects on C647, calcium binding at the Ca2 site causes R347 to move out of the active site and W348 to move in to form one wall of the substrate-binding pocket (Figure 1C-Right). W348 is critical for catalysis because a W348A substitution decreases kcat/KM by >16 500-fold (Table 1). Q350 then rotates to coordinate Ca1 (Figure 1C-Right, Figure S7B).
Table 1. PAD2 Steady-State Kinetics and Calcium Dependence with the Substrate BAEEa.

N-α-benzoyl-l-arginine ethyl ester.
Color coding refers to residue location mapped on the holoPAD2 structure (right).
ND = Not determined.
Function is inferred from comparing PAD2 residues to corresponding residues in PAD4 complexed with N-α-benzoyl-l-arginine amide (PDB: 1WDA).
The kcat/KM for these compounds was determined using the equation v = kcat/KM([Et][S]), when possible.
R347 Shields the Active Site
Despite shielding the active site in the apoenzyme and PAD·Ca(10mM)2+, R347 does not prevent calcium binding at Ca2 because the R347A mutant shows only a small effect on the concentration of calcium required for half maximal activity (i.e., K0.5; Table 1). Rather, R347 is important for substrate binding because the KM for the R347A mutant is increased by ∼30-fold. Additionally, this shielded conformation may protect against nonspecific oxidation of the active site cysteine, which would inactivate the enzyme, or, alternatively, prevent premature docking and sequestration of substrate proteins. This potential regulatory mechanism may be a universal feature of the PADs because all five isozymes contain either an arginine (PAD1, 2, 3, 6) or a glutamine (PAD4) that could similarly shield the active site in the absence of calcium (Figure S5B). Overall, our data indicate that Ca2 is critical for generating the catalytically competent conformation. Consistent with this conclusion is the fact that a near complete loss of PAD2 activity is observed upon mutation of Ca2 binding residues E352A, D370A, and D374A (Figure 3B, Table 1).
Figure 3.

Enzymatic and structural analysis of mutations to calcium binding residues. (A) Calcium coordinating residues and their binding states seen at varying calcium concentrations in the different PAD2 structures. (B) Catalytic efficiency (kcat/KM) for wild type and calcium-binding mutants. (C) Concentration of calcium required for half-maximal activity (K0.5) for wild type and calcium-binding mutants. (D) Electron density at the Ca3–5 sites from structures soaked in 10 mM Ca2+ (WT, left, PDB: 4N2B; D169A, center, PDB: 4N2G; D177A, right, PDB: 4N2I) where density is contoured at the 5σ level from Fo–Fc omit maps after calcium was removed and simulated annealing was performed during refinement. (E) Percent occupancies of calcium in crystal structures of PAD2 mutants not soaked in CaCl2 (left column) or soaked in 10 mM CaCl2 (right column; full list of PDBs in Supporting Information Table S1).
Calcium Titrations by X-ray Crystallography Identify a “Calcium Switch” That Controls PAD2 Activity
Inspired by our ability to trap PAD2 in several different calcium bound configurations, we next titrated the apoPAD2 crystals with increasing concentrations of calcium (0–10 mM) and solved a total of 16 structures at eight different calcium concentrations to 1.66–1.97 Å (Figure 2A and Methods). These calcium concentrations bracket the calcium level required for half maximal activity, i.e., the K0.5 of 180 μM (Table 1). Using the electron density of the individual metal ions, we calculated the calcium occupancy for each of the structures (Figure 2B) and thereby showed that PAD2 binds calcium in a stepwise fashion: Ca6 shows near complete occupancy over the entire concentration range, and its KD is <1 μM, the approximate concentration of free calcium in the crystallization solution (see above). Similarly, Ca1 shows 40% occupancy in the absence of added calcium. Although occupancy rises from 40% to 100% with increasing calcium, the increased occupancy likely reflects increased rigidity in the structure, rather than a true increase in calcium binding as E412 and Q350 bind and stabilize Ca1 at higher calcium concentrations (Figure 3A). Consistent with this notion is the fact that multiple peptides, including 349–354 and 412–435, which comprise the Ca1 site, show almost no difference in HDX in the presence or absence of calcium (Figure S4, Table S2). Thus, the KD for this site is also <1 μM. In contrast to Ca1 and Ca6, the Ca3, 4, and 5 sites show no occupancy until the calcium concentration is >100 μM. The titrations yield KD(app) values of ∼250 μM and Hill coefficients ranging from 3.3 to 3.8, which indicates strong cooperativity (Figure 2B). Importantly, these results mirror the in vitro values (K0.5 = 180 μM, n = 2.1; Table 1), suggesting that calcium binding to Ca3–5 acts as a “calcium switch” to control the overall calcium dependence of the enzyme. Given the conservation of the residues that coordinate Ca3–5, this calcium switch is likely a universal feature of the PADs (Figure 2C).
Figure 2.

Calcium-titration experiments by X-ray crystallography. (A) Calcium coordination at the Ca1, 3, 4, 5, and 6 sites. Electron density for calciums 1, 3, 4, 5, and 6 were generated from Fo–Fc omit maps from structures soaked in 0, 50, 100, 250, 500, 1000, 5000, and 10 000 μM Ca2+, corresponding to PDB IDs: 4N20, 4N22, 4N24, 4N25, 4N26, 4N28, 4N2A, and 4N2B, respectively. Density was contoured at the 8σ level from Fo–Fc omit maps where calcium was removed and simulated annealing was performed during refinement. The electron density of each calcium was normalized to a water molecule that was present in all structures and whose density was not affected by soaking with calcium. (B) Calcium dissociation constants (KD(app)) were determined by calculating the percent occupancy of calcium from the e/Å3 levels for each calcium at eight different soak concentrations, and two structures were solved at each soak concentration for data acquisition (n = 2 for each data point). (C) Sequence alignment of the PADs shows the conserved calcium binding residues (*) in the calcium switch. This region is highly conserved (red) among the PADs except for PAD6, which shows no enzymatic activity.
The need to quench the electrostatic repulsions generated by the eight acidic residues that comprise the Ca3–5 sites (Figure 2A,B) explains the relatively high calcium dependence of the PADs. Calcium-binding at Ca3–5 likely promotes calcium binding at the Ca2 site by modulating the conformation of a loop (residues 369–389) that connects these two regions of the enzyme (Figure S7A). D389, a Ca4 ligand that lies at the end of the loop, likely plays a key role in this process. In total, these data define a stepwise calcium-activation pathway, wherein Ca1 and Ca6 bind the enzyme first, with high affinity, followed by Ca3–Ca5. The ensuing conformational changes promote Ca2 binding, the subsequent movement of R347 out of the substrate-binding pocket, and C647 into the active site to generate the catalytically competent state (Figure 5).
Figure 5.

Summary of PAD2 regulation and activation. Conversion of PAD2 from the apo to holo form is controlled by binding of three calcium ions at the calcium switch, which promotes the subsequent relief of active site shielding (R347), proper positioning of the catalytic cysteine (C647), and subsequent docking of substrate proteins for deimination or citrullination.
The “Calcium Switch” Exists in Solution
To support the existence of this Calcium Switch, we mutated 12 residues that are important for calcium binding and determined their kinetic parameters and solution K0.5 values (Figure 3A–C, Table 1). For a subset, we also determined their structures in the absence and presence of calcium. Ca6 site mutants show minimal effects on K0.5, and the apoD123N structure still binds calcium (Figure 3E). These results are consistent with our titration experiments and confirm that Ca6 is a high affinity calcium-binding site. Given that the Ca6 site mutants are purified with lower yields, this site is likely important for protein folding. The Ca1 site mutants also show relatively small but variable effects on K0.5 (2.6-fold to 10-fold). These data coupled with the fact that both the apoQ350A and Q350A·Ca(10mM)2+ structures bind calcium at levels comparable to WT (Figure 3E) also confirms that Ca1 is a high-affinity calcium-binding site. In contrast to Ca6, however, Ca1 is critical for activity because Ca1 site substitutions decrease kcat/KM by >2350-fold. Alignments of the PAD2·Ca(10mM)2+, Q350A·Ca(10mM)2+, and E354A·Ca(10mM)2+ structures show that the positions of four key catalytic residues (i.e, D351, H471, D473, and C647) are superimposable (RMSD = 0.13–0.21 Å, Figure S7E). Thus, the observed loss in activity upon substitution at the Ca1 site is not caused by destabilization of the active site. Rather, these data support the notion that Ca1 promotes the relief of active-site shielding by acting as a ligand for Q350.
Since we do not observe calcium binding at Ca2 in our titration experiments, due to a crystal form that precludes metal binding at this site, but do achieve full occupancy at the other five sites, our data suggest that Ca2 binds either last or independently of the other calcium ions. On the basis of our data, we favor the former possibility, and interestingly, despite our inability to titrate this site, Ca2 also appears to be a high-affinity site. This conclusion is based on the fact that the K0.5 values for the Ca2 site mutants (i.e., E352A, D370A, D374A) are virtually unchanged (0.6- to 2.2-fold) from WT (Figure 3C, Table 1). By contrast, Ca3–5 are low affinity calcium-binding sites within the calcium switch that control the overall calcium dependence of PAD2. This is evidenced by the fact that the Ca3–5 site mutants (i.e., D166A (Ca3,5), D177A (Ca3,5), D389A (Ca4), D169A (Ca5)) show smaller decreases in kcat/KM, but relatively larger increases in K0.5. For example, substitution of D177, which coordinates Ca3 and Ca5, results in a dramatic 26.7-fold increase in K0.5 (Figure 3C). Consistent with these mutagenesis data, calcium occupancy at the Ca3–5 sites is reduced (60–80%) in the D177A·Ca(10mM)2+ structure, whereas calcium binding at the other sites is unaffected (Figure 3D,E). Substitution of D169, a Ca5 ligand, shows similar trends: a 2-fold decrease in kcat/KM, a relatively larger increase in K0.5 (3.6-fold), and the D169A·Ca(10mM)2+ structure showing a complete loss of electron density for Ca5 (Figure 3D). The fact that the D169A substitution retains considerable activity, despite the loss of Ca5, indicates that the Ca3 and Ca4 sites are more critical for relaying the conformational changes from the calcium switch to Ca2. Consistent with this notion is that the Ca3 and Ca4 mutants show more dramatic effects on kcat/KM and K0.5.
Calcium Binding at Ca2 Generates the Catalytically Competent Enzyme
Fortuitously, a D169A·Ca(10mM)2+ structure showed electron density for the Ca2 site in the unliganded state; electron density for this site is absent from the PAD2·Ca(10mM)2+ structure. Here, the Ca2 loop forms a cone-like structure in which K378 occupies the Ca2 site where it forms favorable electrostatic and H-bonding interactions with D370 and S371 (Ca2 ligands; Figure S7F). To bind Ca2 at this site, R347, W348, D370, D374, R373, and K378 undergo a dramatic series of conformational changes, with several residues rotating ∼180°, to effect calcium binding at Ca2 (Figure S7D, Movie S1). As a result, R373 interacts with D346 in the holoenzyme, generating a belt-like structure at the base of the active site that stabilizes the Ca2 site, promotes D370 and D374 to coordinate Ca2, and properly positions key substrate binding residues. These changes are also coupled to the coordination of Q350 to Ca1, which enables the nucleophilic C647 to move into the space vacated by Q350 and R347, generating the catalytically competent conformation. These movements are consistent with both our mutagenesis and solution HDX data (Figure S4). Importantly, the HDX kinetics for peptides 370–377 and 370–384 (part of a loop) as well as 370–392 (loop and short helix) are significantly different between the apo and holoenzyme, indicating that Ca2 becomes structured upon calcium binding, which is consistent with our structural data. Taken together, these data support the notion that holoenzyme formation is driven by calcium binding to the Ca3–5 sites and that this region of the enzyme acts as a calcium switch to control the overall activity of PAD2, and likely the other PADs.
Structural Insights into Allosteric Inhibitor Design
The structural and biochemical studies described above provide important new insights into both PAD2 regulation and the development of inhibitors targeting this enzyme. For example, they suggest that it is possible to develop allosteric inhibitors that block PAD2 activation. Indeed, Lewis et al. recently identified an allosteric PAD4 specific inhibitor, GSK199, that preferentially binds to PAD4 in the absence of Ca2.23 In fact, the structure of the PAD4·GSK199 complex is highly reminiscent of our intermediate calcium-bound PAD2 structures in that the inhibitor binds a form of the enzyme in which all calciums save Ca2 are bound to the enzyme, thereby indicating that these intermediate structures can be exploited to identify highly selective PAD inhibitors.
Notably, the nearly 100-fold selectivity of GSK199 for PAD4 over PAD2 could be explained in part by residues 638–645, which occlude the GSK199 binding pocket in the PAD2·Ca(10mM)2+ structure (compare Figure 4A to 4B). On the basis of the structure of holoPAD2, this loop is mobile, however, and as such may not provide the complete mechanistic rationale for the high selectivity. Alternatively, the lack of affinity for PAD2 may be due to the loss of a key hydrophobic interaction between the benzimidazole of GSK199 and F634 in PAD4; the corresponding residue is a serine (S636) in PAD2 (Figure 4C). The high selectivity may also be due to the fact that GSK199 binding is partially occluded by the interaction between R347 and Q350 (Figure 4B). Regardless of the specific reason for the high selectivity of GSK199, our studies highlight the potential of exploiting the structural transitions that occur upon calcium binding to identify allosteric PAD inhibitors. Indeed, PAD2 has similar but non-identical binding pockets in the vicinity of the active site that could be targeted for inhibition, and it is noteworthy that the aminopiperidine–benzimidazole scaffold (Figure 4F) can be modeled to bind the active site of PAD2 in our intermediate structure (Figure 4D).
Figure 4.

Allosteric inhibitors targeting the PADs. (A) Crystal structure of PAD4 bound to GSK199 as seen in Lewis et al. Flexible loop that interacts with GSK199 is depicted in green. Unstructured loop containing residues 639–640 is shown as a black dashed line. (B) Alignment of the PAD4 GSK199 and PAD2·Ca(10mM)2+ crystal structures, followed by removal of the PAD4 structure to depict where GSK199 could bind in PAD2·Ca(10mM)2+. The PAD2·Ca(10mM)2+ loop (yellow) consisting of residues 638–645 would have to move to accommodate GSK199. This is probable because this loop takes on a more flexible form in the holoPAD2 structure. (C) Ligplot+ representation of GSK199 interacting with PAD4 in the crystal structure, with residues from PAD4 labeled in black and the corresponding residues from PAD2 in pink. (D) Docking of a fragment of GSK199 in the PAD2·Ca(10mM)2+ structure reveals a tight binding pocket and potential inhibitor for PAD2. (E) Surface representation and top down view of the docked GSK199 fragment. (F) Inhibitor structures found in panels a–e.
Conclusions
In addition to highlighting the potential for generating allosteric inhibitors that target structural intermediates along the calcium activation pathway, our data also provide important new insights into the mechanisms by which calcium binds and activates cellular PAD activity and addresses a confounding enigma for the field; i.e., why is the concentration of calcium required for in vitro activity at least 10 times higher than cellular calcium concentrations (i.e., 0.1–10 μM).25 Our stepwise crystallographic titration data show that the apparent dissociation constants for the Ca3–5 sites are in the ∼250 μM range, quite close to the concentration of calcium required for in vitro PAD2 activity (K0.5 = 180 μM). These data argue for the notion that PAD2 activity is controlled by these sites and that Ca3–5 constitute a calcium switch to govern the overall calcium dependence of the enzyme (Figure 5).
Since the Ca3–5 sites are highly conserved among all the PADs, this calcium switch represents a universal mechanism for controlling the calcium dependence of the PADs, and its presence suggests that other cellular proteins could bind to this region and act as chaperones to lower the calcium dependence of the enzymes in vivo. Consistent with this possibility, a recent study showed that antibodies that bind PAD4 near the Ca3–5 region significantly lower the concentration of calcium required for activity.26 Alternatively, localization of PAD2 to the vicinity of intracellular calcium channels could promote the transient activation of PAD2 upon channel opening as the localized calcium concentrations are thought to reach the millimolar levels required to activate PAD activity.27,28 To allow for the temporal activation of the enzyme, a protein activator would also be required to bind PAD2 and “lock” calcium into the structure. Upon its dissociation, calcium would be released, thereby inactivating PAD2.
In summary, our studies provide the first structural data for PAD2 and show that calcium binding occurs in an ordered fashion and that a calcium switch (calcium binding sites 3, 4, and 5) controls the overall calcium dependence of the enzyme. In addition to providing a deeper understanding of PAD regulation, these data will undoubtedly impact future efforts to develop novel PAD inhibitors for breast cancer and other human diseases.
Methods
PAD2 Cloning, Protein Expression, and Purification
A PAD2 clone in the pET16b vector (EMD Millipore) produced a protein that contained vector specific residues at the N-terminus (residues MGHHHHHHHHHHSSGHIEGRH) and C-terminus (residues SRRS) for a total of 690 residues (78 571 Da), of which 665 residues are the full human PAD2 protein. All PAD2 mutations (constructs summarized in Table 1) were created using the QuikChange Site-Directed Mutagenesis Kit, followed by sequencing of purified plasmid DNA (Genewiz) to confirm the desired mutation. PAD2 was expressed in BL21(DE3) pLysS E. coli (EMD Millipore) by growing E. coli in LB medium at 37 °C with shaking at 200 rpm to an OD600 of 0.6. The temperature was lowered to 16 °C and 200 rpm, and PAD2 expression was induced with 100 μM IPTG overnight. Cell pellets were harvested by centrifugation at 4500g for 15 min, flash frozen in liquid nitrogen, and resuspended in a lysis buffer (20 mM Tris, pH 7.6, 0.4 M NaCl, 1% Triton X-100, 0.5 mM TCEP, 0.005 M Imidazole) and lysed by sonication (Branson Sonifier 450 at full power for 8 rounds of 8 s bursts with 1 min rests, and allowed to cool for 3 min before repeating one additional cycle). Lysates were cleared by centrifugation at 20 000g for 15 min at 4 °C followed by affinity purification at 4 °C using Ni-NTA resin and washing using a stepwise gradient of 20 mM Tris, pH 7.6, 0.4 M NaCl, 10% glycerol, 0.5 mM TCEP, and 0.02–0.1 M imidazole followed by elution from the resin with 20 mM Tris, pH 7.6, 0.4 M NaCl, 10% glycerol, 0.5 mM TCEP, and 0.25 M imidazole. PAD2 was diluted 1:5 in a buffer (20 mM Tris, pH 7.6, 10% glycerol, 0.5 mM TCEP) and applied to a column packed with 5 mL of Source 15Q anion exchange resin (GE Healthcare) for purification on an ÄKTA FPLC (GE Healthcare) at 4 °C with a salt gradient elution from buffer A (20 mM Tris, pH 8.5, 0.05 M NaCl, 10% glycerol, 0.5 mM TCEP) to increasing buffer B (20 mM Tris, pH 8.5, 2 M NaCl, 10% glycerol, 0.5 mM TCEP). Samples containing PAD2 were pooled and dialyzed into a long-term storage buffer (LTSB: 20 mM Tris, pH 7.6, 0.5 M NaCl, 10% glycerol, 0.5 mM TCEP, 1 mM EDTA), tested for purity and activity, and stored at −80 °C.
Crystallization
Initial crystals were obtained by screening conditions using a Gryphon robot to set sitting drops, with The Nucleix Screen (Qiagen) condition #3 (20% MPD, 50 mM MES, pH 5.6, 0.1 M magnesium acetate) producing the best diffracting crystals. Optimized crystals of WT and all mutants except F221/222A were grown at 23 °C by using the sitting-drop vapor-diffusion technique by mixing 0.6 μL of 2 mg mL–1 protein solution in LTSB with 0.6 μL of precipitant (10–20% MPD, 50 mM MES, pH 5.6, 0.12 M sodium acetate). F221/222A crystals were grown by mixing 0.6 μL of a 2 mg mL–1 protein solution in LTSB with 0.6 μL of precipitant (10% MPD, 10% ethanol, 50 mM MES, pH 5.6, 20 mM MgCl2, 1 mM spermidine). Crystal morphology and size were consistent for all PAD2 constructs. During soaking, only one crystal (∼0.1 mm × 0.05 mm) was transferred into a 2 μL drop, which provided a significant excess volume of liquid to ensure that one crystal would soak up enough Ca2+ without significantly changing (<1%) the calcium concentration of the solution. Crystal soaking in CaCl2 was carried out in precipitant solution supplemented with the indicated CaCl2 concentration for 10 min before freezing (with no added cryoprotectant) in liquid nitrogen for data collection.
Data Collection and Processing
Data were collected at the SER-CAT (single wavelength at 1.0000 Å, 100 K, on beamline 22-ID) and LS-CAT (single wavelength at 0.97875 Å, 100 K, on beamlines 21-ID-D, 21-ID-F, and 21-ID-G) sectors at Argonne National Laboratories, Argonne, IL, and at the Stanford Synchrotron Radiation Lightsource (single wavelength at 1.2836 Å, 100 K, on beamline BL7-1, Stanford, CA). Data were processed and scaled using HKL-2000.29
Model Building and Refinement
All PAD2 structures were solved by using scaled data from HKL-2000 for molecular replacement in the Phenix Suite by initially using apo PAD4 (PDB: 1WD8) to solve the apoPAD2·Ca(0mM)2+ structure, followed by holo PAD4 (PDB: 1WD9) being used to solve the F221/222A·Ca(10mM)2+ structure.30 Subsequent molecular replacement for the remaining PAD2 structures used either apoPAD2·Ca(0mM)2+ or PAD2·Ca(10mM)2+ as the model protein. The Phenix Suite was used to identify water molecules, followed by alternating rounds of manual refinement in COOT31 to fit protein density and identify MPD, acetate, and calcium ions. Multiple rounds of refinement in Phenix were used to achieve the statistics in Table S1.
Accession Numbers for Structures Deposited in the Protein Data Bank (PDB)
Coordinates and structure factors for the PAD2 X-ray structures have been deposited in the Protein Data Bank under accession codes: PAD2 0 mM Ca2+ (4N20), PAD2 10 mM Ca2+ (4N2B), PAD2 F221/222A 10 mM Ca2+ (4N2C), PAD2 50 μM Ca2+ (4N22), PAD2 100 μM Ca2+ (4N24), PAD2 250 μM Ca2+ (4N25), PAD2 500 μM Ca2+ (4N26), PAD2 1 mM Ca2+ (4N28), PAD2 5 mM Ca2+ (4N2A), PAD2 D123N 0 mM Ca2+ (4N2D), PAD2 D123N 10 mM Ca2+ (4N2E), PAD2 D169A 0 mM Ca2+ (4N2F), PAD2 D169A 10 mM Ca2+ (4N2G), PAD2 D177A 0 mM Ca2+ (4N2H), PAD2 D177A 10 mM Ca2+ (4N2I), PAD2 Q350A 0 mM Ca2+ (4N2K), PAD2 Q350A 10 mM Ca2+ (4N2L), PAD2 E354A 0 mM Ca2+ (4N2M), and PAD2 E354A 10 mM Ca2+ (4N2N).
Calcium ions 1–5 in all PAD2 structures were identified in the same location as Ca1–5 in holoPAD4, which were confirmed in holoPAD4 as calcium by generating anomalous Fourier map peaks (λ = 1.5418) corresponding to the position of each metal ion. In apoPAD2, in which crystals were not soaked in CaCl2, the newly discovered calcium 6 (Ca6) and calcium 1 (Ca)1 ions were verified as a Ca2+ by analyzing bond lengths to amino acids (Ca1 average distance = 2.48 Å, Ca6 average distance = 2.28 Å, theoretical calcium residue distance = 2.36 Å) and water (Ca1 average distance = 2.47 Å, Ca6 distance = 2.54 Å, theoretical calcium water distance = 2.39 Å), with the bond length to water being significantly too long for the presence of a magnesium ion (theoretical distance = 2.09 Å) in the Ca1 or Ca6 site.32 All structure figures in the manuscript were created using PyMOL, with the exception being Figure S1C, which was created using Chimera33,34 and Figure S3, which was created using Ligplot+.35
Enzymatic Assays
A discontinuous activity assay using benzoylarginine ethylester (BAEE) as a substrate was used to analyze citrulline production, using previously established methods.36,37 Briefly, a reaction buffer (60 μL total volume; 10 mM CaCl2, 50 mM NaCl, 100 mM Tris-HCl, pH 7.6, 2 mM DTT) containing 0–10 mM BAEE was preincubated at 37 °C before PAD2 (0.2 μM final) was added to initiate the reaction. Following the addition of an enzyme, the reaction was allowed to proceed at 37 °C for 6 min and then flash frozen in liquid nitrogen. The steady-state kinetic parameters were obtained using GraFit version 5.0.11 by fitting the initial rate data (n = 2) to eq 1:38
| 1 |
to obtain the values reported in Supporting Information Table 3.
The same assay format was used to determine the concentration of calcium required for half maximal PAD activity. Briefly, a reaction buffer (60 μL total volume; 10 mM BAEE, 50 mM NaCl, 100 mM Tris-HCl, pH 7.6, 2 mM DTT) containing various concentrations of calcium chloride was preincubated at 37 °C before PAD2 (0.2 μM final) was added to initiate the reaction. The reactions were allowed to proceed for 6 min and then flash frozen in liquid nitrogen. The kinetic parameters were obtained using GraFit version 5.0.11 by fitting the initial rate data to eq 2:38
| 2 |
where K0.5 is the dissociation constant and n is the Hill coefficient. K0.5 refers to the concentration of calcium in micromolarity that is necessary to achieve 50% of maximal enzymatic activity. Note that for these assays PAD2 was dialyzed into an EDTA-free long-term storage buffer (0.02 M Tris, pH 7.6, 0.5 M NaCl, 10% glycerol, 0.5 mM TCEP). Reactions were performed in duplicate (n = 2) to obtain the values reported in Supporting Information Table 3.
Calculation of Calcium-Binding Affinities by Crystallography
To determine the order of calcium-binding and apparent KD values for each of the individual calcium-binding sites, we employed a crystallographic approach. Briefly, WT PAD2 crystals were soaked in varying concentrations of calcium (0, 50, 100, 250, 500, 1000, 5000, and 10 000 μM) for 10 min and then flash frozen in liquid nitrogen. Duplicate data sets from crystals soaked in separate drops were collected as described above, and structures were solved for each soak, giving eight snapshots (16 structures total, n = 2 for each data point represented by a structure) of PAD2 during the conversion from the apo to holo states. To determine the relative affinities of the individual sites, calcium ions were manually removed from the model in Coot, and each data set was subjected to five rounds of simulated annealing refinement from 2.10 to 30.0 Å in the Phenix suite to create Fo-Fc omit maps. The omit maps were analyzed in Coot by visualizing the peak density (e/Å3) at which the electron density from the absent calcium was no longer visible. To further normalize the 16 data sets that comprise each eight-point dose response curve, a conserved water was used to normalize the e/Å3 values (Figure 3). The water-normalized e/Å3 values for each calcium were then used to calculate the percent occupancy for each soak concentration by setting the 10 mM calcium e/Å3 value to the 100% occupancy point. KD(app) values for Ca3–5 were determined using eq 3,
| 3 |
where KD(app) is the apparent dissociation constant for calcium binding and n is the Hill coefficient. For the Ca1 site, the data were fit to eq 4:
| 4 |
which accounts for background calcium binding.
To validate our ability to calculate partial occupancies of calcium ions by e/Å3 levels in Coot, refinement of metal ion occupancy was performed in Phenix on calcium ions and gave percent occupancies of 65% and 71% for Ca1 and Ca6, respectively, in the apoPAD2 structure. As further evidence, metal ion occupancy refinement in Phenix on the PAD2·Ca2+(250 μM) structure gave calculated occupancies of 62%, 58%, and 66% for Ca3, Ca4, and Ca5, respectively, where the e/Å3 calculations were similar at 50%, 52%, and 54%. We also relaxed the occupancies and B factors of all calcium ions in eight PAD2 data sets (0, 50 100, 250 500, 1000, 5000, and 10 000 μM CaCl2) and corefined them with all PAD2 atom coordinates, and these data still trend toward increasing ion occupancy as CaCl2 soak concentrations increase. Notably, these data correlate with our simulated annealing results (Figure S8). Also worth noting is the fact that the B-factors of the calcium ions, as well as the surrounding atom clusters (oxygen atoms from residues that coordinate calcium), in our simulated annealing data sets substantially decrease with increasing CaCl2 soaking concentrations, which again correlates positively with increased calcium occupancy. While radiation damage was not explicitly accounted for in the calculations and refinement, data sets were collected with similar exposure time and normalized for resolution, with none of the data sets being more than 2.10 Å for Ca2+ occupancy calculations. Since none of the eight data sets vary significantly in resolution (1.66–1.97 Å; Figure S8), this adds validity to the observation that the decreasing B-factors of calcium ions and their coordinating residues during higher calcium soaking concentrations are due to increased calcium occupancy, and therefore a more stable protein conformation. Overall, these data help validate our use of e/Å3 calculations from simulated annealing to determine calcium occupancy in the PAD2 crystal structures.
Inductively Coupled Plasma Mass Spectroscopy (ICP-MS) Analysis of Buffers
Inductively coupled plasma mass spectrometry (ICP-MS) analysis was performed at the Oregon Health and Science University using an Agilent 7700x system equipped with an ASX-520 Autosampler. The system was operated at a radio frequency power of 1550 W, an argon flow rate of 15 L/min, carrier gas flow rate of 1.04 L/min, and helium (He) gas flow rate of 4.3 mL/min. Data were quantified using a nine-point (0, 0.5, 1, 2, 5, 10, 50, 100, and 1000 pbb (ng/g)) calibration curve with external standards for Ca, Mn, Fe, Cu, and Zn. All data were acquired in He mode to remove interference from oxides, argides, and chlorides. For each sample, run data were acquired in triplicate and averaged. Internal standards (Sc, Ge, and Bi) introduced with the sample were used to correct for plasma instabilities. The National Institute of Standards and Technology standard reference material was used to ensure elemental recovery of >90%. Sample solutions were diluted in 1% HNO3 (Fisher, Trace Metals grade).
Native Electrospray Ionization (ESI) Mass Spectrometry
WT PAD2 as well as the F221/222A mutant stored in 20 mM Tris pH 7.6, 0.5 M NaCl, 0.5 mM TCEP, and 10% glycerol were buffer exchanged into 0.2 M ammonium acetate at pH 7.0 using a Vivaspin 500 centrifugal concentrator (30 000 MWCO) to a final protein concentration of 10 μM. PAD2 and the F221/222A mutant were analyzed with a Bruker Solarix 12 T FTICR instrument in positive ion mode, with external calibration by using cesium perfluoroheptanoic acetate up to m/z 8500.39 The protein sample was delivered by nanoelectrospray ionization with a custom-pulled tip at a flow rate of 20–50 nL/min, utilizing a capillary voltage of 1.2–1.4 kV, and a drying-gas temperature of 30 °C at a flow rate of 2.5 L/min. Ions were accumulated for 500 ms in the RF-hexapole ion trap before being transferred to the ICR trap. The pressure was ∼2.3 mbar (source), 4 × 10–6 mbar (quadrupole), and 1 × 10–9 mbar (trap). Hundreds of scans were averaged for each spectrum, and a smooth peak profile for charge deconvolution was obtained by only processing the first 32 000 of the 1 million data points with Bruker FTMS Processing and Data Analysis 4.0 software.39 Results show that both constructs exist as stable dimers as shown by a single peak in the deconvoluted spectrum at ∼157 kDa and an inability to dissociate PAD2 into a monomeric state upon applying collisional energy during MS analysis.
Hydrogen–Deuterium Exchange (HDX)
We examined the effects of calcium-binding to PAD2 at the peptide level by using HDX. All HDX experiments were carried out at 4 °C in 80% in D2O (20 mM Tris (in D2O, pD 7.6), 0.5 M NaCl, 0.5 mM TCEP) where 4 μL of protein sample (14 μM PAD2 in 20 mM Tris pH 7.6, 0.5 M NaCl, 0.5 mM TCEP, 10% glycerol) that was incubated for 60 min with either 0.5 mM EDTA or 2.5 mM CaCl2 was mixed with 16 μL D2O buffer, and HDX was analyzed at eight time points (0, 0.17, 0.5, 1, 2, 15, 60, 240 min). HDX was quenched by adding 30 μL of 3 M urea and 1% TFA at 4 °C. A total of 50 μL of quenched protein sample was injected for proteolysis with online desalting and digestion (immobilized pepsin) and analysis with fast gradient HPLC (5 min) coupled to an LTQ-Orbitrap. The protein digestion gave >96% coverage (∼280 peptic peptides were observed at sufficient sensitivity to give kinetic plots). The high sequence coverage enabled an examination of the dynamics of nearly all regions of PAD2, and significant changes in the extent of HDX were observed in the regions surrounding Ca2–6 in the presence and absence of calcium.
Docking of the Aminopiperidine-Benzimidazole Fragment of GSK199 into PAD2
Docking was performed using AutoDock version 4.240 using the PAD2·Ca(10mM)2+ structure and a truncated GSK199 structure that consisted of the aminopiperidine–benzimidazole scaffold. A random conformation of the ligand was generated to allow an unbiased docking result. In addition, three rotational bonds were assigned to the ligand before docking onto the structure of PAD2. The docking grid was centered on the active site cavity. The estimated calculated free energy of binding of the aminopiperidine–benzimidazole derivative to PAD2 is −12 kcal/mol.
Acknowledgments
We thank the staff at the Southeast Regional Collaborative Access Team (SER-CAT, Argonne National Laboratories), the Life Sciences Collaborative Access Team (LS-CAT, Argonne National Laboratories), and the Stanford Synchrotron Radiation Lightsource (SSRL) for their help with data collection. We thank Hajeung Park and Jeremy R. Lohman for crystallographic data processing recommendations. We thank Daniel M. Lewallen, Eranthie Weerapana, and Philip Cole for critical analysis of the manuscript and Weidong Cui (WU) for help with native spray MS.
Supporting Information Available
Supplemental figures, tables, and movies. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported in part by NIH grants GM079357 (P.R.T.) and 8 P41 GM103422 (M.L.G.), as well as an Era of Hope Scholar Award (W871XWH-07-0372) to S.A.C.
The authors declare the following competing financial interests: PRT is a cofounder and consultant to Padlock Therapeutics. HDL and BDB are employees of GSK.
This paper was published January 26, 2015. There has been a correction to the spelling of contributing author, Jakob Fuhrmann, and an update has been made to Reference 23. The new version re-posted on January 28, 2015.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Jones J. E.; Causey C. P.; Knuckley B.; Slack-Noyes J. L.; Thompson P. R. (2009) Protein arginine deiminase 4 (PAD4): Current understanding and future therapeutic potential. Curr. Opin. Drug Discovery Dev. 12, 616–627. [PMC free article] [PubMed] [Google Scholar]
- Vossenaar E. R.; Zendman A. J.; van Venrooij W. J.; Pruijn G. J. (2003) PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays 25, 1106–18. [DOI] [PubMed] [Google Scholar]
- Christophorou M. A.; Castelo-Branco G.; Halley-Stott R. P.; Oliveira C. S.; Loos R.; Radzisheuskaya A.; Mowen K. A.; Bertone P.; Silva J. C.; Zernicka-Goetz M.; Nielsen M. L.; Gurdon J. B.; Kouzarides T. (2014) Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 507, 104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade D. J.; Subramanian V.; Thompson P. R. (2014) Citrullination unravels stem cells. Nat. Chem. Biol. 10, 327–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita K.; Hashimoto H.; Shimizu T.; Nakashima K.; Yamada M.; Sato M. (2004) Structural basis for Ca(2+)-induced activation of human PAD4. Nat. Struct Mol. Biol. 11, 777–83. [DOI] [PubMed] [Google Scholar]
- Dwivedi N.; Neeli I.; Schall N.; Wan H.; Desiderio D. M.; Csernok E.; Thompson P. R.; Dali H.; Briand J. P.; Muller S.; Radic M. (2014) Deimination of linker histones links neutrophil extracellular trap release with autoantibodies in systemic autoimmunity. FASEB J. 28, 2840–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscarello M. A.; Pritzker L.; Mastronardi F. G.; Wood D. D. (2002) Peptidylarginine deiminase: a candidate factor in demyelinating disease. J. Neurochem 81, 335–43. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Bolt M.; Guertin M. J.; Chen W.; Zhang S.; Cherrington B. D.; Slade D. J.; Dreyton C. J.; Subramanian V.; Bicker K. L.; Thompson P. R.; Mancini M. A.; Lis J. T.; Coonrod S. A. (2012) Peptidylarginine deiminase 2-catalyzed histone H3 arginine 26 citrullination facilitates estrogen receptor alpha target gene activation. Proc. Natl. Acad. Sci. U. S. A. 109, 13331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler S. C.; Vincent C. T.; Fedorov V. D.; Patsialou A.; Cherrington B. D.; Wakshlag J. J.; Mohanan S.; Zee B. M.; Zhang X.; Garcia B. A.; Condeelis J. S.; Brown A. M.; Coonrod S. A.; Allis C. D. (2013) Dysregulation of PAD4-mediated citrullination of nuclear GSK3beta activates TGF-beta signaling and induces epithelial-to-mesenchymal transition in breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 110, 11851–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellekens G. A.; de Jong B. A.; van den Hoogen F. H.; van de Putte L. B.; van Venrooij W. J. (1998) Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J. Clin. Invest. 101, 273–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Li M.; Stadler S.; Correll S.; Li P.; Wang D.; Hayama R.; Leonelli L.; Han H.; Grigoryev S. A.; Allis C. D.; Coonrod S. A. (2009) Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 184, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanan S.; Horibata S.; McElwee J. L.; Dannenberg A. J.; Coonrod S. A. (2013) Identification of macrophage extracellular trap-like structures in mammary gland adipose tissue: a preliminary study. Front. Immunol. 4, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicker K. L.; Thompson P. R. (2013) The protein arginine deiminases: Structure, function, inhibition, and disease. Biopolymers 99, 155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumanevich A. A.; Causey C. P.; Knuckley B. A.; Jones J. E.; Poudyal D.; Chumanevich A. P.; Davis T.; Matesic L. E.; Thompson P. R.; Hofseth L. J. (2011) Suppression of colitis in mice by Cl-amidine: a novel peptidylarginine deiminase inhibitor. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G929–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight J. S.; Zhao W.; Luo W.; Subramanian V.; O’Dell A. A.; Yalavarthi S.; Hodgin J. B.; Eitzman D. T.; Thompson P. R.; Kaplan M. J. (2013) Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J. Clin. Invest. 123, 2981–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange S.; Gogel S.; Leung K. Y.; Vernay B.; Nicholas A. P.; Causey C. P.; Thompson P. R.; Greene N. D.; Ferretti P. (2011) Protein deiminases: New players in the developmentally regulated loss of neural regenerative ability. Dev. Biol. 355, 205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElwee J. L.; Mohanan S.; Griffith O. L.; Breuer H. C.; Anguish L. J.; Cherrington B. D.; Palmer A. M.; Howe L. R.; Subramanian V.; Causey C. P.; Thompson P. R.; Gray J. W.; Coonrod S. A. (2012) Identification of PADI2 as a potential breast cancer biomarker and therapeutic target. BMC Cancer 12, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis V. C.; Gizinski A. M.; Banda N. K.; Causey C. P.; Knuckley B.; Cordova K. N.; Luo Y.; Levitt B.; Glogowska M.; Chandra P.; Kulik L.; Robinson W. H.; Arend W. P.; Thompson P. R.; Holers V. M. (2011) N-alpha-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J. Immunol. 186, 4396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight J. S.; Luo W.; O’Dell A. A.; Yalavarthi S.; Zhao W.; Subramanian V.; Guo C.; Grenn R. C.; Thompson P. R.; Eitzman D. T.; Kaplan M. J. (2014) Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 114, 947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight J. S.; Subramanian V.; O’Dell A. A.; Yalavarthi S.; Zhao W.; Smith C. K.; Hodgin J. B.; Thompson P. R.; Kaplan M. J. (2014) Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum Dis. 10.1136/annrheumdis-2014-205365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmans W. F.; Hatzis C.; Sotiriou C.; Andre F.; Peintinger F.; Regitnig P.; Daxenbichler G.; Desmedt C.; Domont J.; Marth C.; Delaloge S.; Bauernhofer T.; Valero V.; Booser D. J.; Hortobagyi G. N.; Pusztai L. (2010) Genomic index of sensitivity to endocrine therapy for breast cancer. J. Clin Oncol 28, 4111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertucci F.; Borie N.; Ginestier C.; Groulet A.; Charafe-Jauffret E.; Adelaide J.; Geneix J.; Bachelart L.; Finetti P.; Koki A.; Hermitte F.; Hassoun J.; Debono S.; Viens P.; Fert V.; Jacquemier J.; Birnbaum D. (2004) Identification and validation of an ERBB2 gene expression signature in breast cancers. Oncogene 23, 2564–75. [DOI] [PubMed] [Google Scholar]
- Lewis H. D.; Liddle J.; Coote J. E.; Atkinson S. J.; Barker M. D.; Bax B. D.; Bicker K. L.; Bingham R. P.; Campbell M.; Chen Y. H.; Chung C.-W.; Craggs P. D.; Davis R. P.; Eberhard D.; Joberty G.; Lind K. E.; Locke K.; Maller C.; Martinod K.; Patten C.; Polyakova O.; Rise C. E.; Rüdiger M.; Sheppard R. J.; Slade D. J.; Thomas P.; Thorpe J.; Yao G.; Drewes G.; Wagner D. D.; Thompson P. R.; Prinjha R. K.; Wilson D. M. (2015) Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 10.1038/nchembio.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H.; Chruszcz M.; Lasota P.; Lebioda L.; Minor W. (2008) Data mining of metal ion environments present in protein structures. J. Inorg. Biochem. 102, 1765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn H. D.; Sun L.; Prywes R.; Liu J. O. (1999) Apoptosis of T cells mediated by Ca2+-induced release of the transcription factor MEF2. Science 286, 790–793. [DOI] [PubMed] [Google Scholar]
- Darrah E.; Giles J. T.; Ols M. L.; Bull H. G.; Andrade F.; Rosen A. (2013) Erosive Rheumatoid Arthritis Is Associated with Antibodies That Activate PAD4 by Increasing Calcium Sensitivity. Sci. Transl. Med. 5, 186ra65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R.; Sugimori M.; Silver R. B. (1992) Microdomains of high calcium concentration in a presynaptic terminal. Science 256, 677–9. [DOI] [PubMed] [Google Scholar]
- Tour O.; Adams S. R.; Kerr R. A.; Meijer R. M.; Sejnowski T. J.; Tsien R. W.; Tsien R. Y. (2007) Calcium Green FlAsH as a genetically targeted small-molecule calcium indicator. Nat. Chem. Biol. 3, 423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. M. W. (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–32. [DOI] [PubMed] [Google Scholar]
- Harding M. M. (2000) The geometry of metal-ligand interactions relevant to proteins. II. Angles at the metal atom, additional weak metal-donor interactions. Acta Crystallogr., Sect. D: Biol. Crystallogr. 56, 857–67. [DOI] [PubMed] [Google Scholar]
- Menard H. A.; Lapointe E.; Rochdi M. D.; Zhou Z. J. (2000) Insights into rheumatoid arthritis derived from the Sa immune system. Arthritis Res. 2, 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–12. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–86. [DOI] [PubMed] [Google Scholar]
- Kearney P. L.; Bhatia M.; Jones N. G.; Yuan L.; Glascock M. C.; Catchings K. L.; Yamada M.; Thompson P. R. (2005) Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 44, 10570–82. [DOI] [PubMed] [Google Scholar]
- Knipp M.; Vasak M. (2000) A colorimetric 96-well microtiter plate assay for the determination of enzymatically formed citrulline. Anal. Biochem. 286, 257–64. [DOI] [PubMed] [Google Scholar]
- Leatherbarrow R. J.Grafit, version 5; Erithacus Software Ltd.: Horley, U.K., 2001. [Google Scholar]
- Zhang H.; Cui W.; Wen J.; Blankenship R. E.; Gross M. L. (2011) Native electrospray and electron-capture dissociation FTICR mass spectrometry for top-down studies of protein assemblies. Anal. Chem. 83, 5598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.