

Abstract
Objective
To characterize the metabolic phenotype of 2 cases of normal weight young women who developed type 2 diabetes (T2D), severe insulin resistance (insulin requirement >200 units/day), marked hypertriglyceridemia (>2000 mg/dL), and hepatic steatosis beginning 9 years after undergoing total body irradiation (TBI) and bone marrow transplantation for childhood cancer.
Methods
Fasting plasma glucose, insulin, free fatty acids (FFAs), leptin, adiponectin, resistin, TNFα, and IL-6 were measured in each case and in 8 healthy women; Case 1 was also assessed after initiating pioglitazone. Coding regions and splice junctions of PPARG, LMNA, and AKT2 were sequenced in Case 1 and of PPARG in Case 2 to evaluate for familial partial lipodystrophies. Genotyping of APOE was performed in Case 1 to rule out type III hyperlipoproteinemia.
Results
Both cases had elevated plasma levels of insulin, leptin, resistin, and IL-6, high-normal to elevated TNFα, and low to low-normal adiponectin in keeping with post-receptor insulin resistance and adipose tissue inflammation. Case 1 experienced a biochemical response to pioglitazone. No causative mutations for partial lipodystrophies or type III hyperlipoproteinemia were identified.
Conclusion
Though metabolic derangements have previously been reported in association with TBI, few cases have described insulin resistance and hypertriglyceridemia as severe as that seen in our patients. We speculate that early childhood TBI may impede adipose tissue development leading to metabolic complications from an attenuated ability of adipose tissue to accommodate caloric excess, and propose that this extreme metabolic syndrome be evaluated for as a late complication of TBI.
INTRODUCTION
Survivors of childhood cancer are at increased risk for developing impaired glucose tolerance or type 2 diabetes (T2D), insulin resistance, and dyslipidemia (1–8). Such patients typically have a normal body mass index (BMI), but increased whole body adiposity, with preferential deposition in truncal depots (9). In some patients, endocrine sequelae to cancer and its treatment, including growth hormone deficiency, hypogonadism, and hypothyroidism, may contribute to these metabolic abnormalities (2,6,9,10); however, the most consistently demonstrated correlate of insulin resistance and dyslipidemia in childhood cancer survivors is prior total body irradiation (TBI), which is an effect independent from that of chemotherapy (1,4,6,8). In keeping with this, the risk for later development of overt T2D is particularly strong with TBI (8), with presentation a median of 9 years following exposure (4). Nevertheless, the reported insulin resistance and dyslipidemia in such patients with T2D are usually not severe.
Four patients have previously been described with insulin resistant T2D and moderate to severe hypertriglyceridemia 9 to 10 years after undergoing TBI as part of conditioning prior to bone marrow transplantation (BMT) for relapsed acute lymphocytic leukemia (ALL) (11–13). We now detail 2 cases of severe insulin resistance with T2D and severe hypertriglyceridemia presenting 9 years after exposure to TBI, prior to autologous BMT for neuroblastoma in one case, and an allogeneic BMT for relapsed ALL in the second case. Adipokine and molecular genetic studies were undertaken in an attempt to elucidate the potential etiology of the severe insulin resistance and hypertriglyceridemia, and treatment with the peroxisome proliferator-activated receptor gamma (PPARγ) agonist pioglitazone was administered in the first case. We propose that this metabolic phenotype may be accounted for by TBI-induced attenuation of the ability of adipose tissue to accommodate caloric excess.
METHODS
Two adult survivors of childhood cancer who were previously treated with TBI and later developed a constellation of metabolic abnormalities which include T2D, severe insulin resistance, hypertriglyceridemia, and hepatic steatosis were referred to our diabetes clinic for evaluation. In both cases, fasting plasma adipokine analysis was performed to determine whether the severe insulin resistance was related to adipose tissue dysfunction or inflammation. Molecular genetic testing was done to exclude forms of familial partial lipodystrophies that also manifest severe insulin resistance and hypertriglyceridemia (14) and to evaluate for apoE variants that could predispose severe hypertriglyceridemia (15,16).
Adipokine Analysis
Fasting plasma was analyzed in Case 1 prior to and after 6 and 8 months of pioglitazone therapy, and in Case 2 at 5 years after presentation. Glucose was measured by the glucose oxidase method using an automated glucose analyzer (YSI 2300; Yellow Springs Instruments, Yellow Springs, OH); free fatty acids (FFAs) by enzymatic colorimetric assay (Wako Chemicals, Richmond, VA); insulin, leptin, and adiponectin by double antibody radioimmunoassay (Millipore, Billerica, MA); and resistin, TNFα, and IL-6 using enzyme-linked immunosorbent assays (ELISA; Millipore for resistin and R&D Systems, Minneapolis, MN, for TNFα and IL-6). All assays were performed in duplicate with results compared to those obtained from 8 healthy women who participated as control subjects in previous studies (17,18).
Molecular Genetic Analysis
Genetic testing was performed after obtaining written informed consent according to procedures approved by the research ethics committee in Cambridge, UK. In both cases lymphocyte DNA was extracted from whole blood, and in Case 2, who had undergone allogeneic BMT, DNA was also extracted from cheek epithelial cells. APOE and the familial partial lipodystrophy genes PPARG, LMNA, and AKT2 were sequenced in Case 1, and PPARG in Case 2. Primer pairs were designed to cover all coding exons and intron-exon boundaries of PPARG, AKT2, exons 8 to 12 of LMNA and exon 4 of APOE using Primer3 software (sequences available on request). PCR was performed using Taq polymerase (Abgene), and PCR products were sequenced using Big Dye Terminator 3.1 DNA sequencing kit (Applied Biosystems).
RESULTS
Case Presentations
Case 1
An 18-year-old woman was referred with uncontrolled insulin resistant T2D and severe hypertriglyceridemia. She survived childhood neuroblastoma treated by partial resection and focal irradiation (1000 cGy), chemotherapy, TBI (1000 cGy), and autologous BMT between 3 and 4 years of age. Complications have included slipped femoral epiphyses, bilateral cataracts, short stature, and secondary oligomenorrhea. T2D was diagnosed at age 12, with ensuing poor control (HbA1c>11%) despite increasing doses of insulin. At 17 years of age albuminuria was noted, and therapy was initiated with an angiotensin converting enzyme inhibitor. After initiating therapy with 1.25 mg of daily oral conjugated estrogens, triglyceride levels were noted to be elevated (1537 and 4077 mg/dL) with concomitant elevation of hepatic transaminases and ultrasonographic evidence of hepatic steatosis. Despite introduction of fish oil (up to 4 g daily), acute pancreatitis developed with elevated serum amylase (302 U/L; normal 30 to 100 U/L) and lipase (6035 U/L; normal 25 to 110 U/L) at a time when serum triglycerides were 3893 mg/dL.
On examination, height was 147 cm, and weight 44.4 kg (BMI 20.5 kg/m2). Although no frank lipoatrophy was apparent, adipose deposition was more pronounced centripetally (Fig. 1 A). There was flexural acanthosis nigricans and multiple acrochordons, most pronounced in the nuchal region (Fig. 1 B) and axillae. Eruptive xanthomata were present on the dorsal surface of the forearms and upper arms, and the liver was palpably enlarged at 18 cm in the mid-axillary line.
Fig. 1.
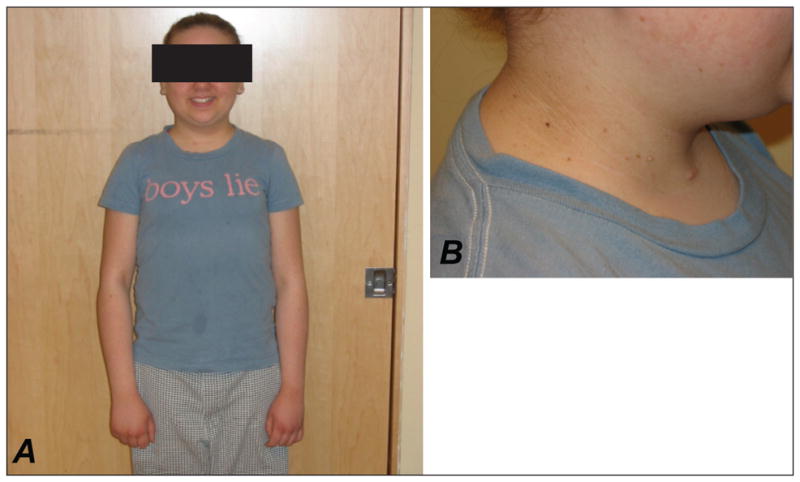
A. Slight predominance of facial and truncal adiposity without lipoatrophy. B. Flexural acanthosis nigricans and acrochordons (skin tags).
Salient chemistries from the onset of pancreatitis are summarized in Table 1. Serum creatinine, electrolytes, complete blood count, and thyroid function tests were normal. The serum C-peptide while receiving exogenous insulin was 5.7 ng/mL (normal limit: 0.8 to 3.5 ng/mL). Anti-nuclear antibodies and antibodies directed against glutamic acid decarboxylase, islet cell antigen, and insulin were negative.
Table 1.
Case 1 Chemistries and Treatment From the Episode of Pancreatitis In Months
| Months | 0 | 2 | 6 | 10 | 12 | 14 | 19 | 24 | 26 | 28 | 30 | 32 | 34 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HbA1c (%) | 11.7 (nl<6.0) | 11.8 | 10.0 | 12.8 | 11.5 | 11.6 | 9.9 | 10.2 | 10.0 | 7.8 | 8.9 | 10.2 | 10.1 |
| Triglycerides (mg/dL) | 3893–4077 (nl<150) | 1500 | 680 | 1164 | 1216 | 1318 | 859 | 881 | 676 | 220 | 537 | 855 | 325 |
| Total cholesterol (mg/dL) | 467–563 (nl<200) | 325 | 316 | 378 | 302 | 387 | 319 | 324 | 335 | 210 | 258 | 287 | 233 |
| HDL cholesterol (mg/dL) | 3–14 (nl>40) | 38 | 35 | 41 | 37 | 41 | 38 | 41 | 40 | 46 | 36 | 35 | 39 |
| LDL cholesterol (mg/dL) | N.A. (nl<130) | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | 120 | N.A. | N.A. | 129 |
| AST (U/L) | 58–68 (nl<36) | 24 | 35 | 46 | 80 | 32 | 83 | 62 | 58 | 36 | 37 | 31 | 47 |
| ALT (U/L) | 57–92 (nl<52) | 51 | 62 | 75 | 88 | 52 | 109 | 69 | 68 | 36 | 54 | 32 | 37 |
| Fish oil (g/d) | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Fenofibrate (mg/d) (mg/d) | — | 145 | 145 | 145 | 145 | 145 | 145 | 145 | 145 | 145 | — | — | 145 |
| Glargine insulin (U/d) | 45 | 50 | 50 | 55 | 60 | — | — | — | — | — | — | — | — |
| Aspart insulin (U/d) | 180 | 180 | 180 | 180 | 180 | — | — | — | — | — | — | — | — |
| U-500 insulin (U/d; U/kg/d) | — | — | — | — | — | 250; 5 | 300; 6 | 300; 6 | 300; 6 | 300; 6 | 250; 5 | 250; 5 | 250; 5 |
| Pioglitazone (mg/d) | — | — | — | — | — | — | — | — | 15 | 30 | 30 | 30 | 30 |
Abbreviations: ALT = alanine transaminase; AST = aspartate aminotransferase; g/d = grams per day; HbA1c = hemoglobin A1c; HDL = high-density lipoprotein; LDL = low-density lipoprotein. N.A. = not applicable; mg/d = milligrams per day; nl = normal limit; U/d = units per day; U/kg/d = units per kilogram per day.
A low-fat diet was initiated, oral estrogens were discontinued, and fenofibrate 145 mg daily was added; however, the patient’s triglyceride levels continued to exceed 1000 mg/dL. Glycemic control was attempted in the hospital by increasing doses of glargine insulin to 50 units at bedtime and aspart insulin to 60 units with meals. Several months later, triglyceride levels remained under 1500 mg/dL, but levels below 1000 mg/dL were seen only when the HbA1c was 10% or below, with a nadir of 680 mg/dL. The total cholesterol remained above 300 mg/dL.
Approximately 14 months after the episode of pancreatitis, insulin therapy was changed to regular U-500 insulin injected 2–4 times daily (19), and HbA1c improved from >11% to around 10%. Because glycemic and lipemic control remained inadequate, at 24 months after pancreatitis pioglitazone was initiated at 15 mg daily for 2 months, and then increased to 30 mg daily. Two months later, the HbA1c had decreased to 7.8%, serum triglycerides to 220 mg/dL, and total cholesterol to 210 mg/dL, and serum transaminases had normalized. The patient decreased the number of insulin doses to 2 to 3 times daily, in part due to hypoglycemia.
Case 2
A 22-year-old woman was referred with uncontrolled insulin resistant T2D and severe hypertriglyceridemia. She survived childhood ALL treated with chemotherapy between 3 and 6 years of age. At 7 years, she experienced a central nervous system relapse treated with focal irradiation and chemotherapy, TBI (unknown dose), and allogeneic BMT from an HLA identical sibling. The transplant was complicated by cutaneous graft-versus-host disease (GVHD) that resolved at age 11. Serum levels of hepatic transaminases were elevated, and a liver biopsy documented the presence of hepatic steatosis. Additional complications included bilateral cataracts, short stature, and secondary oligomenorrhea. At 14 years of age, albuminuria was noted, and a kidney biopsy showed chronic glomerulonephritis. At age 16, T2D was diagnosed, and was poorly controlled (HbA1c >11%) despite increasing doses of insulin therapy to glargine 60 units at bedtime and aspart 55 units with meals. At age 19, therapy with an angiotensin converting enzyme inhibitor was initiated to control albuminuria. Primary hypothyroidism was corrected with 125 μg of levothyroxine daily. Oligomenorrhea was treated with 0.625 mg daily oral conjugated estrogens (days 1 through 21 each month) and 10 mg daily oral medroxyprogesterone (days 17 through 21 each month).
On examination, height was 160 cm, and weight 57.3 kg (BMI: 22.4 kg/m2). There was a preponderance of central adiposity, but no regions of frank lipoatrophy. There was acanthosis nigricans and numerous acrochordons over the chest, abdomen, shoulders, and back. There was no evidence of thyroid or liver enlargement.
Salient chemistries are summarized in Table 2. Serum creatinine, electrolytes, thyroid function tests, and urinary free cortisol were normal, while blood count and evaluation of iron status revealed iron deficiency anemia, attributed to menorrhagia caused by a uterine fibroid, which was surgically removed. There were no detectable anti-insulin antibodies.
Table 2.
Case 2 Chemistries and Treatment From Presentation To the Diabetes Clinic
| Months | 0 | 3 | 10 | 24 | 44 | 48 | 54 | 60 | 63 |
|---|---|---|---|---|---|---|---|---|---|
| HbA1c (%) | 12.2 (nl<6.0) | 10.8 | 8.5 | 7.4 | 7.3 | 7.4 | 7.7 | 6.4 | 7.0 |
| Triglycerides (mg/dL) | 2133 (nl<150) | 956 | 507 | 732 | 465 | 591 | 427 | N.D. | 613 |
| Total cholesterol (mg/dL) | 512 (nl<200) | N.D. | 233 | 307 | 235 | 283 | 282 | N.D. | 327 |
| HDL cholesterol (mg/ dL) | 32 (nl>40) | 40 | 51 | 58 | 51 | 53 | 47 | N.D. | 60 |
| LDL cholesterol (mg/ dL) | N.A. (nl<130) | N.A. | N.A. | N.A. | 91 | N.A. | 125 | N.D. | N.A. |
| AST (U/L) | 57–92 (nl<36) | 54 | 53 | 77 | 40 | 75 | N.D. | N.D. | 43 |
| ALT (U/L) | 75–120 (nl<52) | 75 | 44 | 56 | 37 | 42 | N.D. | N.D. | 53 |
| Ezetimibe (mg/d) | — | 10 | 10 | 10 | 10 | — | — | — | — |
| Fenofibrate (mg/d) | — | — | — | — | — | 48 | 145 | 145 | 145 |
| Fish oil (g/d) | — | — | — | — | — | 4 | 4 | 4 | 4 |
| Glargine insulin (U/d) | 60 | — | — | — | — | — | — | — | — |
| Aspart insulin (U/d) | 165 | — | — | — | — | — | — | — | — |
| U-500 insulin (U/d; U/kg/d) | — | 200; 3.4 | 280; 4.7 | 280; 4.7 | 500; 8.5 | 500; 8.5 | 500; 8.5 | 515; 8.7 | 515; 8.7 |
Abbreviations: ALT = alanine transaminase; AST = aspartate aminotransferase; g/d = grams per day; HbA1c = hemoglobin A1c; HDL = high-density lipoprotein; LDL = low-density lipoprotein. N.A. = not applicable; mg/d = milligrams per day; nl = normal limit; U/d = units per day; U/kg/d = units per kilogram per day.
Oral estrogen and progesterone were changed to transdermal ethinyl estradiol 20 μg and norelgestromin 150 μg daily (days 1 through 21 each month), ezetimibe 10 mg daily was added by another physician provider and subsequently changed to fenofibrate and fish oil, and insulin was converted to multiple daily injections of U-500 insulin (19), with subsequent improvement in serum triglycerides to around 500 mg/dL, and HbA1c to around 7%. Despite this, pancreatitis developed with elevated serum levels of amylase (525 U/L; normal: 30 to 110 U/L) and lipase (>4000 U/L; normal: 10 to 230 U/L), and was attributed to relapsed hypertriglyceridemia. The dose of fenofibrate was increased from 48 to 145 mg daily.
Adipokine Analysis
The patients studied were similar in age and BMI to the female control subjects (Table 3). In both cases, insulin levels were dramatically elevated, indicating adherence to the prescribed insulin regimen. FFAs were in the normal range in Case 1 and markedly elevated in Case 2 despite hyperinsulinemia. In both cases, leptin was elevated and adiponectin was low to low-normal. Both patients also exhibited elevated resistin, high-normal to elevated TNFα, and elevated IL-6 levels. In Case 1, following initiation of pioglitizone therapy plasma levels of leptin and adiponectin increased, resistin was unchanged, and TNFα and IL-6 decreased.
Table 3.
Plasma Glucose and Hormone Levels In the Cases and Female Control Subjects (n = 8)
| Factor | Case 1 BLa | Case 1 6 moa | Case 1 8 moa | Case 2 | Control Mean (SD) | Control 95% Confidence Interval |
|---|---|---|---|---|---|---|
| Age (y) | 20 | 20 | 21 | 29 | 31 (11) | 21–41 |
| BMI (kg/m2) | 23.1 | 22.7 | 24.0 | 22.7 | 22.5 (2.1) | 20.8–24.3 |
| Glucose (mg/dL)b | 101 | 138 | 210 | 280 | 81 (6) | 76–86 |
| Insulin (μU/mL)c | 216 | 236 | 118 | 125 | 7 (3) | 5–9 |
| FFAs (μmol/L) | 358 | 473 | 246 | 1,333 | 410 (96) | 320–499 |
| Leptin (ng/mL) | 18.9 | 24.3 | 30.4 | 38.2 | 6.4 (4.3) | 2.8–10.1 |
| Adiponectin (μg/mL) | 3.1 | 4.2 | 10.2 | 8.5 | 13.0 (6.5) | 7.5–18.5 |
| Resistin (ng/mL) | 30.7 | 30.1 | 34.9 | 54.5 | 18.1 (4.4)d | 11.1–25.0d |
| TNFα (pg/mL) | 1.73 | 1.56 | 1.41 | 2.3 | 1.36 (0.35)d | 0.81–1.91d |
| IL-6 (pg/mL) | 1.18 | 0.28 | 0.25 | 1.21 | < 0.156d | <.156d |
Abbreviations: BL = baseline; FFA = free fatty acids; TNFα = tumor necrosis factor alpha; IL-6 = interleukin-6.
Time relative to the initiation of pioglitazone therapy (30 mg daily) in Case 1.
To convert glucose to mmol/L, multiply by 0.05551.
To convert insulin to pmol/L, multiply by 7.1750.
Control samples available (n = 4).
Molecular Genetic Analysis
Coding regions and splice junctions of PPARG, LMNA, and AKT2, were wild-type in Case 1; however, she was heterozygous for the PPARG Pro12Ala polymorphism. This polymorphism was not detected in Case 2, who was wild-type for PPARG in DNA extracted from both whole blood and cheek epithelial cells. APOE genotyping confirmed that Case 1 expressed the wild-type isoform, apolipoprotein E3 ruling out type III hyperlipoproteinaemia.
DISCUSSION
Our 2 patients presented with severe insulin resistance and hypertriglyceridemia 9 years after undergoing TBI prior to either autologous BMT for neuroblastoma or allogeneic BMT for relapsed ALL. Previous studies have established an association between treatment of childhood cancers with TBI (as part of conditioning prior to BMT) and the development of impaired glucose tolerance and dyslipidemia (4,6,8); however, few cases have been described where insulin resistance and hypertriglyceridemia is as severe as that seen in our patients (11–13). In the 4 previous patients described with overt T2D and marked hypertriglyceridemia, the presentation occurred around 9 years after therapy, in keeping with this report. The elevated C-peptide levels reported here and by others (12,13) offer support for the dominant metabolic problem in this setting being insulin resistance rather than β-cell failure, which has been reported after abdominal radiation therapy (20). In fact, the case reported by Tahrani and colleagues (12) exhibited a supranormal β-cell secretory response to intravenous glucagon, and an HbA1c of approximately 7% was attained with sulfonylurea therapy alone. In the case described by Rooney and Ryan (13), there was sclerodermatous GVHD resulting in marked loss of fat from the extremities. Other reports, however, have not associated GVHD with insulin resistance in childhood cancer survivors (6), and in fact our Case 1 is the first reported to develop severe insulin resistance and hypertriglyceridemia in the context of a previous autologous BMT, precluding GVHD. Thus, conditioning with TBI remains implicated as the most important risk factor for the severe insulin resistance and hypertriglyceridemia we describe.
We must acknowledge that the degree of hypertriglyceridemia initially seen in both of our cases was exacerbated by uncontrolled diabetes and oral estrogen use. Nonetheless, hypertriglyceridemia remained poorly controlled in both cases despite the discontinuation of oral estrogen, institution of high-dose fibrate therapy, and in Case 2, reasonably well-controlled glycemia. In Case 1 it was only after the introduction of pioglitazone that both reasonable lipemic and glycemic control was achieved. In the 2 cases reported by Amin and colleagues (11), the first was a male patient whose measured triglycerides were in excess of what we have ourselves reported, and while simultaneous measurement of the HbA1c was not done when the triglycerides exceeded 5000 mg/dL, the second was a female patient whose triglycerides were over 1000 mg/dL when the HbA1c was 7.0%. Furthermore, the wild-type apolipoprotein E in our Case 1 offers additional evidence against estrogen therapy or hyperglycemia exacerbating type III hyperlipoproteinemia as a sufficient explanation for these patients’ severe hypertriglyceridemia.
The pathogenesis of severe insulin resistance and hypertriglyceridemia following childhood TBI has not been definitively established. However, the metabolic phenotype including marked hepatic steatosis we have reported bears close similarities to that seen in severe obesity or partial lipodystrophies (21), and is quite at odds to the metabolic phenotype caused by congenital or acquired loss of insulin receptor function (22,23). In both these situations it has been argued that failure of adipose tissue to store dietary calories, either through manifest lack of adipose tissue in the case of lipodystrophy, or overload of adipose tissue, as in some severe obesity, leads to ectopic lipid accumulation with consequent severe hepatic steatosis, hypertriglyceridemia, and insulin resistance. Neither of our patients had frank lipodystrophy; moreover, we excluded genetic defects described in familial partial lipodystrophies in Case 1, and although the common PPARγ Pro12Ala polymorphism was present, it was not detected in Case 2. While it remains possible that an unidentified genetic factor accounted for the severity of our patients’ insulin resistance and hypertriglyceridemia, we believe this disorder is best explained as an acquired consequence of exposure to TBI in childhood.
Recent elegant use of radiocarbon dating, based on opportunistic exploitation of the defined historical window of atmospheric atomic bomb testing, has demonstrated that adipocyte cell number is set during childhood and adolescence and stays essentially static throughout adult life, although around 8% of cells turn over each year (24). This is important because lower numbers of adipocytes correlate with adipocyte hypertrophy (25), which, in turn, has been shown in many studies to correlate well with insulin resistance and to predict future diabetes (26,27). It follows from these observations that exposure to a cytotoxic therapy in childhood might deplete the population of preadipocytes available for later recruitment to the adipocyte pool, and thus program a high risk of later insulin resistance. Direct evidence for this has been provided by animal studies. Exposure of female ob/ob mice to 8 Gy of TBI followed by transplantation with syngeneic or wild-type bone marrow (28) did not reduce hyperphagia; however, reduced weight consequent to impaired accumulation of fat, but not lean, body mass was seen. Despite this reduction in body fat, treated mice had more severe insulin resistance and hepatic steatosis than untreated control ob/ob animals. Morphometric analysis of adipocyte size indicated a reduced proportion of small adipocytes in irradiated animals, and gene expression in adipose tissue revealed reduction in the preadipocyte marker MCP-1. Collectively these data support the model of impaired preadipocyte differentiation in irradiated animals leading to constrained adipose tissue expansion and function (28).
We provide some supportive evidence that adipose tissue dysfunction is at play in our patients. Reduced adiponectin and elevated leptin, resistin, TNFα, and IL6 are in keeping with post-receptor insulin resistance and adipose tissue inflammation, which is now well recognized to occur in metabolically unhealthy obesity and partial lipodystrophy (21). Histologically, this is usually correlated in cases of severe adipose tissue stress by enlarged adipocytes and the presence of macrophages in adipose tissue, often surrounding dying, perilipin-negative adipocytes to form so-called “crown-like structures” (29). We suggest that future studies concentrate on assessing adipocyte size and adipose histology in femorogluteal depots in patients with insulin resistance following TBI. This growing evidence that childhood TBI, and perhaps other cytotoxic therapies, may program high risk for later adipose insufficiency, insulin resistance, and dyslipidemia, has relevance to clinical practice. We suggest that assessment targeted at the evolving metabolic phenotype, especially around puberty and beyond, should be an important facet of “late effects” endocrine practice.
Finally, our limited experience suggests that pioglitazone may be effective in this setting. After Case 1 initiated treatment with pioglitazone, both glycemic and lipemic control improved dramatically. While the near normalization of serum triglycerides may be attributed in part to the improved glycemic control, benefit may also have been derived from direct effects of pioglitazone on adipocyte function. Pioglitazone activates PPARγ, the master regulator of preadipocyte differentiation to adipocytes, and thus is a very rational treatment in conditions where attenuated preadipocyte recruitment is the root problem. PPARγ agonists also increase expression of adiponectin and decrease expression of pro-inflammatory cytokines such as TNFα and IL-6, effects that likely contribute to improving systemic insulin sensitivity and reducing hepatic steatosis (30). In Case 1, clinical improvement on pioglitazone was associated with increases in leptin and adiponectin and decreases in TNFα and IL-6. These results are similar to those reported for familial partial lipodystrophy (31,32) where PPARγ agonists may produce marked benefits. Future comparative trials are warranted to determine optimal therapy for patients with T2D and severe insulin resistance and hypertriglyceridemia following TBI.
CONCLUSION
In conclusion, this study found that exposure to TBI in early childhood may impede adipose tissue development, leading to metabolic complications from an attenuated ability of adipose tissue to accommodate caloric excess. Therefore, this extreme metabolic syndrome should be evaluated for as a late complication of TBI later in adulthood.
Acknowledgments
We thank Dr Heather Collins of the University of Pennsylvania Diabetes Endocrinology Research Center (DERC) for performance of the immunoassays. We also thank Keli Phillips of the Institute of Metabolic Sciences for providing technical support.
This work was supported in part by Public Health Services Research Grant P30-DK19525 (Penn DERC) from the National Institutes of Health, and by the Institute for Diabetes, Obesity & Metabolism of the University of Pennsylvania. RKS is supported by the Wellcome Trust (Fellowship 080952/Z/06/Z), by the UK Medical Research Council Centre for Obesity and Related Metabolic Disease, and by the UK NIHR Cambridge Biomedical Research Center.
Abbreviations
- ALL
acute lymphocytic leukemia
- BMI
body mass index
- BMT
bone marrow transplantation
- ELISA
enzyme-linked immunosorbent assay
- FFAs
free fatty acids
- GVHD
graft-versus-host disease
- HbA1c
hemoglobin A1c
- PPARγ
peroxisome proliferator-activated receptor gamma
- TBI
total body irradiation
- T2D
type 2 diabetes
Footnotes
DISCLOSURE
The authors have no multiplicity of interest to disclose.
References
- 1.Lorini R, Cortona L, Scaramuzza A, et al. Hyperinsulinemia in children and adolescents after bone-marrow transplantation. Bone Marrow Transplant. 1995;15:873–877. [PubMed] [Google Scholar]
- 2.Talvensaari KK, Lanning M, Tapanainen P, Knip M. Long-term survivors of childhood cancer have an increased risk of manifesting the metabolic syndrome. J Clin Endocrinol Metab. 1996;81:3051–3055. doi: 10.1210/jcem.81.8.8768873. [DOI] [PubMed] [Google Scholar]
- 3.Taskinen M, Saarinen-Pihkala UM, Hovi L, Lipsanen-Nyman M. Impaired glucose tolerance and dyslipidaemia as late effects after bone-marrow transplantation in childhood. Lancet. 2000;356:993–997. doi: 10.1016/S0140-6736(00)02717-3. [DOI] [PubMed] [Google Scholar]
- 4.Traggiai C, Stanhope R, Nussey S, Leiper AD. Diabetes mellitus after bone marrow transplantation during childhood. Med Ped Oncol. 2003;40:128–129. doi: 10.1002/mpo.10098. [DOI] [PubMed] [Google Scholar]
- 5.Hoffmeister PA, Storer BE, Sanders JE. Diabetes mellitus in long-term survivors of pediatric hematopoietic cell transplantation. J Ped Hematol Oncol. 2004;26:81–90. doi: 10.1097/00043426-200402000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Neville KA, Cohn RJ, Steinbeck KS, Johnston K, Walker JL. Hyperinsulinemia, impaired glucose tolerance, and diabetes mellitus in survivors of childhood cancer: prevalence and risk factors. J Clin Endocrinol Metab. 2006;91:4401–4407. doi: 10.1210/jc.2006-0128. [DOI] [PubMed] [Google Scholar]
- 7.Shalitin S, Phillip M, Stein J, Goshen Y, Carmi D, Yaniv I. Endocrine dysfunction and parameters of the metabolic syndrome after bone marrow transplantation during childhood and adolescence. Bone Marrow Transplant. 2006;37:1109–1117. doi: 10.1038/sj.bmt.1705374. [DOI] [PubMed] [Google Scholar]
- 8.Baker KS, Ness KK, Steinberger J, et al. Diabetes, hypertension, and cardiovascular events in survivors of hematopoietic cell transplantation: a report from the bone marrow transplantation survivor study. Blood. 2007;109:1765–1772. doi: 10.1182/blood-2006-05-022335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jarfelt M, Lannering B, Bosaeus I, Johannsson G, Bjarnason R. Body composition in young adult survivors of childhood acute lymphoblastic leukaemia. Eur J Endocrinol. 2005;153:81–89. doi: 10.1530/eje.1.01931. [DOI] [PubMed] [Google Scholar]
- 10.Chemaitilly W, Boulad F, Oeffinger KC, Sklar CA. Disorders of glucose homeostasis in young adults treated with total body irradiation during childhood: a pilot study. Bone Marrow Transplant. 2009;44:339–343. doi: 10.1038/bmt.2009.40. [DOI] [PubMed] [Google Scholar]
- 11.Amin P, Shah S, Walker D, Page SR. Adverse metabolic and cardiovascular risk following treatment of acute lymphoblastic leukaemia in childhood; two case reports and a literature review. Diabetic Med. 2001;18:849–853. doi: 10.1046/j.1464-5491.2001.00591.x. [DOI] [PubMed] [Google Scholar]
- 12.Tahrani AA, Cramp C, Moulik P. The development of non-insulin-dependent diabetes after total body irradiation and bone marrow transplantation in adolescence: A case report and literature review. Ped Diabetes. 2006;7:173–175. doi: 10.1111/j.1399-543X.2006.00160.x. [DOI] [PubMed] [Google Scholar]
- 13.Rooney DP, Ryan MF. Diabetes with partial lipodystrophy following sclerodermatous chronic graft vs. host disease. Diabetic Med. 2006;23:436–440. doi: 10.1111/j.1464-5491.2006.01855.x. [DOI] [PubMed] [Google Scholar]
- 14.Semple RK, Savage DB, Cochran EK, Gorden P, O’Rahilly S. Genetic syndromes of severe insulin resistance. Endocrine Rev. 2011;32:498–514. doi: 10.1210/er.2010-0020. [DOI] [PubMed] [Google Scholar]
- 15.Weisgraber KH, Rall SC, Jr, Mahley RW. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J Biol Chem. 1981;256:9077–9083. [PubMed] [Google Scholar]
- 16.Ivanova R, Puerta S, Garrido A, et al. Triglyceride levels and apolipoprotein E polymorphism in patients with acute pancreatitis. Hepatobiliary Pancreat Dis Int. 2012;11:96–101. doi: 10.1016/s1499-3872(11)60131-8. [DOI] [PubMed] [Google Scholar]
- 17.Petrova M, Townsend R, Teff KL. Prolonged (48-hour) modest hyperinsulinemia decreases nocturnal heart rate variability and attenuates the nocturnal decrease in blood pressure in lean, normotensive humans. J Clin Endocrinol Metab. 2006;91:851–859. doi: 10.1210/jc.2005-1752. [DOI] [PubMed] [Google Scholar]
- 18.Rickels MR, Mueller R, Teff KL, Naji A. β-cell secretory capacity and demand in recipients of islet, pancreas, and kidney transplants. J Clin Endocrinol Metab. 2010;95:1238–1246. doi: 10.1210/jc.2009-2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cochran E, Musso C, Gorden P. The use of U-500 in patients with extreme insulin resistance. Diabetes Care. 2005;28:1240–1244. doi: 10.2337/diacare.28.5.1240. [DOI] [PubMed] [Google Scholar]
- 20.Teinturier C, Tournade MF, Caillat-Zucman S, et al. Diabetes-mellitus after abdominal radiation-therapy. Lancet. 1995;346:633–634. doi: 10.1016/s0140-6736(95)91461-7. [DOI] [PubMed] [Google Scholar]
- 21.Semple RK, Sleigh A, Murgatroyd PR, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest. 2009;119:315–322. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Semple RK, Soos MA, Luan J, et al. Elevated plasma adiponectin in humans with genetically defective insulin receptors. J Clin Endocrinol Metab. 2006;91:3219–3223. doi: 10.1210/jc.2006-0166. [DOI] [PubMed] [Google Scholar]
- 23.Semple RK, Halberg NH, Burling K, et al. Paradoxical elevation of high-molecular weight adiponectin in acquired extreme insulin resistance due to insulin receptor antibodies. Diabetes. 2007;56:1712–1717. doi: 10.2337/db06-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spalding KL, Arner E, Westermark PO, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453:783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- 25.Arner E, Westermark PO, Spalding KL, et al. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes. 2010;59:105–109. doi: 10.2337/db09-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weyer C, Foley JE, Bogardus C, Tataranni PA, Pratley RE. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts Type II diabetes independent of insulin resistance. Diabetologia. 2000;43:1498–1506. doi: 10.1007/s001250051560. [DOI] [PubMed] [Google Scholar]
- 27.Lonn M, Mehlig K, Bengtsson C, Lissner L. Adipocyte size predicts incidence of type 2 diabetes in women. FASEB J. 2010;24:326–331. doi: 10.1096/fj.09-133058. [DOI] [PubMed] [Google Scholar]
- 28.Ablamunits V, Weisberg SP, Lemieux JE, Combs TP, Klebanov S. Reduced adiposity in ob/ob mice following total body irradiation and bone marrow transplantation. Obesity. 2007;15:1419–1429. doi: 10.1038/oby.2007.170. [DOI] [PubMed] [Google Scholar]
- 29.Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Juurinen L, Kotronen A, Graner M, Yki-Järvinen H. Rosiglitazone reduces liver fat and insulin requirements and improves hepatic insulin sensitivity and glycemic control in patients with type 2 diabetes requiring high insulin doses. J Clin Endocrinol Metab. 2008;93:118–124. doi: 10.1210/jc.2007-1825. [DOI] [PubMed] [Google Scholar]
- 31.Moreau F, Boullu-Sanchis S, Vigouroux C, et al. Efficacy of pioglitazone in familial partial lipodystrophy of the Dunnigan type: a case report. Diabetes Metab. 2007;33:385–389. doi: 10.1016/j.diabet.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 32.Collet-Gaudillat C, Billon-Bancel A, Beressi JP. Long-term improvement of metabolic control with pioglitazone in a woman with diabetes mellitus related to Dunnigan syndrome: A case report. Diabetes Metab. 2009;35:151–154. doi: 10.1016/j.diabet.2009.01.001. [DOI] [PubMed] [Google Scholar]