

Abstract
Human adenovirus 40 (Ad40) is an interesting candidate for vector construction because of its tropism for the gastrointestinal tract. Although effective preparation of the vector is necessary for its in vivo application, the amplification of Ad40 has been very difficult. Ad40 E1 deletion mutants were detected by PCR in the viral DNA from Ad40 Dugan amplified by Ad5 E1-expressing human embryonic kidney (293) cells and in Ad40 Dugan plaques observed in Ad5 E1-expressing human retinoblastic cells. For the purpose of generating a single clone wild-type Ad40, the entirety of Ad40 DNA was cloned into a plasmid by homologous recombination. A pure Ad40 was successfully generated by the plasmid transfection and subsequent amplification with Ad5 E4orf6-inducible 293 (2V6.11) cells. 2V6.11 is an apposite cell line for effective Ad40 amplification and for future vector construction because Ad40 genetic integrity was maintained with this Ad5 E1 and E4orf6 transcomplementing cell line.
Keywords: adenovirus 40, in vitro amplification, genetic integrity
Introduction
The human adenovirus serotype 40 (Ad40) is a member of the subgroup F adenovirus (AdF). Ad40 is not only an important etiologic pathogen of infantile gastroenteritis but also an interesting candidate for vector construction targeting the gastrointestinal (GI) tract. Although an efficient method for Ad40 amplification is necessary to study Ad40 virology and vectorology, it is extremely difficult to amplify Ad40 in vitro compared to other adenoviruses (Ads) [1].
Ad40 accounts for diarrhea mainly in children below 2 years of age [2, 3], and Ad5 neutralizing antibody titers in this age group are low [4, 5, 6]. Moreover, Ad40 rarely causes diarrhea or disseminated disease even in immunocompromised adult patients, such as hematopoietic stem cell transplant recipients and HIV-positive individuals [7, 8, 9]. These data indicate that Ad40 has difficulty spreading in a human body with an immune function preventing systemic Ad5 replication, especially in the presence of anti-Ad5 neutralizing antibody.
The GI tract is a common internal organ affected by human Ads, including Ad40 and Ad5 [10]. The growth of Ad40 was reported to be complimented by the E1b55K protein of other Ads, including Ad5 [11, 12, 13]. Further, Goodrum et al. have shown that Ad5 E4orf6, in addition to Ad5 E1b55K, may cooperate to promote cell cycle-independent Ad5 growth [14]. Mautner and Mackay reported that Ad40 infection induced Ad40 E4 activity and Ad40 E4 expression preceded DNA replication [15]. These reports suggested that Ad5 E1b55K and Ad5 E4orf6 may support the replication of Ad40.
Ad40 Dugan strain [16] from the American Type Culture Collection (ATCC, Manassas, VA), which was amplified by Ad5 E1-expressing human embryonic kidney (293) cells, is readily available and has been used world wide. In the midst of the effort to amplify Ad40, Ad40 E1 deletion mutants were detected by PCR of the encapsidated viral DNA extracted from Ad40 Dugan. An Ad40 E1A- and E1b19K-deletion mutant was also observed in Ad40 Dugan plaques isolated with Ad5 E1-expressing human retinoblastic (911) cells. The lack of Ad40 E1 genetic integrity after Ad40 infection might be a major obstacle for in vitro culture of Ad40. These findings as well as the aforementioned reports led to a hypothesis that Ad5 E1b55K and Ad5 E4orf6 double-expressing cells might be suitable for Ad40 amplification, although Ad5 E1 complementation alone might be detrimental to Ad40 E1 genetic integrity.
As the first step for future Ad40-based vector construction, a single clone wild-type Ad40 (pure Ad40wt) was generated by cloning the entire Ad40 genome into a plasmid. The plasmid transfection and effective virus amplification was carried out in Ad5 E4orf6-inducible 293 (2V6.11) cells expressing both Ad5 E1 and Ad5 E4orf6. With this pure virus, the genetic integrity of Ad40 E1 and the differences in viral replication efficiency were evaluated in various propagating cell lines. These studies establish that Ad40 genetic integrity upon amplification may be supported by Ad5 early gene products.
Materials and methods
Viruses and cells
Ad40 (Dugan strain, VR-931) was obtained from ATCC. Ad5wt (Ad300wt) was used as a positive control. The 293 (CRL-1573, ATCC), 2V6.11 (CRL-2784, ATCC), 911 (a kind gift from Dr. Alex J. van der Eb, Leiden University, Leiden, Netherlands) [17], human lung epithelial adenocarcinoma (A549, CCL-185, ATCC), human cervical epithelial adenocarcinoma (HeLa; CCL-2, ATCC) and Ad5 E4-expressing African green monkey kidney (W162; CRL-2783, ATCC) cells were maintained as adherent cultures in Dulbecco modified Eagle medium (DMEM, Mediatech, Herndon, VA) supplemented with 5–10% fetal bovine serum (FBS, HyClone, Logan, UT), 2 mM L-glutamine (Mediatech), and 100 IU/ml penicillin and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA). The 2V6.11 cells were maintained with 600 μg/ml Zeocin and 100 μg/ml Hygromycin B (Invitrogen) every 3 days. Cells were grown in a dedicated virus free incubator at 37°C in a humidified atmosphere containing 5% CO2.
Construction of inducible Ad5 E4orf6-expressing 911 cells
Inducible Ad5 E4orf6-expressing 911 (911orf6) cell lines were generated based on the ecdysone system [18]. First, 911 cells were transfected with pVgRXR (a kind gift from Dr. Daniel J. Donoghue) [19] using SuperFect transfection reagent (Qiagen, Valencia, CA) and selected for 14 days with 600 μg/ml Zeocin to obtain 911VgRXR clones. Next, the 911VgRXR was transfected with pEKORF6 (a kind gift from Dr. Gary Ketner) [20], which was a derivative of the plasmid pIndHygro (Invitrogen) that contained Ad5 E4orf6 sequence under the control of the ecdysone-inducible promoter in the plasmid. The double transduced clones were obtained after 14 days with both 400 μg/ml Zeocin and 50 μg/ml Hygromycin. Individual clones were characterized for levels of expression of the Ad5 E4orf6 protein by western blotting with an anti-Ad5 Eorf6, 6/7 antiserum (M45; a kind gift from Dr. Patrick Hearing) [21]. Before transfection or infection onto 2V6.11 or 911orf6 cells, cultures were treated with 0.1% DMSO (uninduced control), or 1 or 5 μg/ml ponasterone A (Invitrogen). After additional 24 hour (h) incubation, 2V6.11 or 911orf6 cells were transfected with plasmid or infected with fresh Ad-containing medium.
Pure Ad40wt construction, titration and sequence
Total Ad40-genomic DNA was extracted from Ad40 Dugan by a Blood & Cell Culture DNA Mini Kit (Qiagen) following a DNase treatment step to eliminate nonencapsi dated viral DNA [22]. The plasmid encoding pure Ad40wt was built by the homologous recombination between the entirety of Ad40 DNA and the rescue plasmids, which contained homology regions of Ad40-left and right hand-ends (left 1849 bp between 1 and 1849 of GenBank accession no. L19443; right 494 bp between 33721 and 34214), in E.coli BJ5183 (Stratagene, La Jolla, CA). The resultant plasmid was amplified with DH5alpha (Invitrogen).
Pure Ad40wt was generated from the plasmid DNA by transfection of 2V6.11 cells using Superfect after linearization with AsiSI. Every 72 h after transfection or infection, a series of 1:3 dilutions of the resulting crude viral liquids (CVLs), which were prepared by three freeze-thaw cycles and centrifugation (3000 rpm, 10 min), were placed onto fresh 2V6.11 cells with 1 μg/ml ponasterone A in a dedicated Ad40 work incubator isolated from other viruses, especially Ad5. At every passage, encapsidated viral DNA from the 200 μl CVLs was purified by a QIAamp DNA blood mini kit (Qiagen) after a DNase treatment step and checked by PCR for Ad40 E1, fiber, and E4orf2. The PCR (QIAGEN fast cycling PCR kit, Qiagen) was performed with one fiftieth of total encapsidated viral DNA on a 9700 thermocycler (Applied Biosystems, Foster City, CA) with specific primers for Ad40 E1, fiber, and E4orf2 (Supplemental Table 1) at 35 cycles. Template-negative samples served as controls for PCR and were always undetectable. The CsCl-banded viruses were diluted into an equal amount of phosphate-buffered saline (PBS, Mediatech)-based lysis solution containing 1% SDS and 2 mM EDTA, and titered with the optical density at 260 nm (OD260; 1 OD260 = 2 × 1012 virus particle, VP/ml).
Subsequently, the PCR fragments were cloned and sequenced. PCR products were purified by a QIAquick gel extraction kit (Qiagen) and cloned into a pCR4-TOPO or pCR-XL-TOPO cloning vector using a TOPO TA or XL PCR cloning kit (Invitrogen). E.coli TOP10 (Invitrogen) was used for plasmid DNA amplification. DNA sequences of the plasmids were performed using M13-forward and M13-reverse primers independently at the DNA sequencing facility in the University of Minnesota (St. Paul, MN) and identified by using Nucleotide Blast Search against the GenBank data-base (National Center for Biotechnology Information; NCBI, Bethesda, MD).
Quantification of pure Ad40wt genomes by real-time PCR
The DNA level of Ad40 was evaluated using primers for Ad40 fiber (Supplemental Table 1) in real-time PCR analysis by SYBR green method with a QuantiTect SYBR green PCR kit (Qiagen) in an ABI sequence detection system (ABI PRISM 7400, Applied Biosystems). The PCR-cloned Ad40 fiber DNA was used as copy number standards. Encapsidated viral DNA was purified from a 20 μl viral solution of Ad40 Dugan and pure Ad40wt. The real-time PCR assay was performed with one fiftieth of the isolated viral DNAs. The lower limit of detection was 107 copies/ml of PCR-cloned Ad40 fiber DNA, and the lower limit of quantification was 108 copies/ml. Template-negative samples served as controls for real-time PCR and were always sub-detectable.
Plaque assay and isolation
Plaque assays were performed as previously described [23]. Cell monolayers of 293, 911, A549, HeLa and W162 cells were infected with 10-fold serial dilutions of pure Ad40wt virus solution. The aforementioned cells were incubated for 2 h with virus dilutions ranging from 10−2 to 10−7. Additionally, A549 was treated in a similar fashion with dilutions ranging from 10−1 to 10−6. After the 2 h incubation, the infected cells were overlaid with 0.5% agarose in DMEM supplemented with HEPES Buffer (Mediatech) and FBS. The cells were overlaid with additional growth medium on the infection day and the seventh day post-infection (pi), to avoid gel drying. Plaques were visualized in an agarose overlay and data were collected from 6 dilutions on the 10th day pi. Visualized plaques were isolated by sterile point-cutting 200 μl pipet tips, diluted in 1ml DMEM, and collected for CVLs by centrifugation (3000 rpm, 10 min) after three freeze-thaw cycles. Encapsidated viral DNA was extracted from the 200 μl CVLs. The resultant DNA was tested by PCR (QIAGEN fast cycling PCR kit) with specific primers for Ad40 E1 (by hemi-nested PCR, Supplemental Table 1), fiber, and E4orf2, and subsequently sequenced.
Detection of viral transcription in cells
To evaluate viral transcription from pure Ad40wt, 8 × 105 cells in a 60-mm dish were infected with 200 VP/cell of pure Ad40wt. After a 2 h incubation with 1 ml of medium containing virus, the virus/medium solution was replaced with fresh medium. After a total 72 h of incubation, the cell culture media were removed, and the remaining cells were collected by scraping into PBS. The suspension was centrifuged (1000 rpm, 5 min) and the supernatant was removed. The cells were lysed with RLT reagent (Qiagen) with 1% β-mercaptoethanol and the total RNA was isolated using a RNeasy mini kit (Qiagen). Contaminating DNA was removed by RNase-free DNase (Qiagen). In order to make complementary DNA (cDNA) for reverse-transcription (RT)-PCR assay, 1.5 micrograms of DNA-free total RNA was reverse transcribed using an Omniscript RT kit (Qiagen) with Oligo d(T)16 (Applied Biosystems). The detection of the Ad40 mRNA was performed with one twentieth of each cDNA using primers for Ad40 fiber in realtime RT-PCR by SYBR green method. The lower limit of real-time RT-PCR detection for Ad40 fiber mRNA was 10−4 % of total RNA, and the lower limit of quantification was 10−2 % of total RNA. For detecting the transcription of Ad40 E1b55K mRNA, RT-PCR was performed with one twentieth of each cDNA using specific primers for Ad40 E1b55K at 30 cycles (Supplemental Table 1) [24]. Template-negative, uninfected and RT-negative samples served as controls for real-time RT-PCR or RT-PCR, and were always sub-detectable.
Detection of viral replication in cells
To measure replication of pure Ad40wt, 8 × 105 cells in a 60-mm dish were incubated with 200 VP/cell of pure Ad40wt. After 2 h incubation in a total volume of 1 ml, Ad40-containing medium was replaced with fresh medium. After a total 72 h of incubation, encapsidated viral DNA was purified from the 200 μl Ad40-infected cell-free culture supernatants after centrifugation (3000 rpm, 10 min), or the 200 μl CVLs. The DNA level of Ad40 was evaluated using primers for Ad40 fiber in real-time PCR analysis by SYBR green method. The real-time PCR assay was performed with one fiftieth of the isolated viral DNAs. For detecting the intact Ad40 E1 DNA, PCR was performed with one fiftieth of total encapsidated viral DNA using specific primers for Ad40 E1 at 35 cycles, and subsequent sequencing.
Analyses of nonhomologous end joining-related cellular proteins
Cells were seeded at 1.2 × 106 cells per well in a 60-mm dish. After 24 h, cultures were treated with 0.1% DMSO, 1 or 5 μg/ml ponasterone A, or 200 VP/cell Ad5wt. After additional 24 h incubation, cells were infected with virus-free culture medium (control), 200 or 2000 VP/cell pure Ad40wt, or 200 VP/cell Ad5wt. The cells were scraped off after additional 24 h incubation and washed four times in cold PBS. The cell pellets were then lysed with sample buffer (0.125 M Tris-HCl, pH 6.8, 2% SDS, 5% β-mercaptoethanol, 10% glycerol, 0.1% bromophenol blue) plus protease inhibitor (Complete protease inhibitor cocktail EDTA-free mini tablet, Roche Applied Science, Mannheim, Germany) for 30 min on ice with gentle vortexing, followed by three freeze-thaw cycles in an ethanol-dry ice bath. After homogenization on ice by multiple passages through a 26-gauge needle, cellular debris was removed by centrifugation (14000 rpm, 10 min) at 4°C. After heating for 5 min at 95°C (except for the detection of Ad5 E4orf6), the protein extracts were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes (Bio-Rad, Hercules, CA). The membranes were blocked for 1 h at 4 °C with 5% nonfat dry milk in 0.05% Tween 20-PBS, and probed with primary antibody (mouse monoclonal anti-E1B antibody, 1:1000 dilution, 2A6, a kind gift from Dr. Thomas Shenk; anti-orf6, 6/7 antibody, 1:100, M45; anti-Mre11 antibody, 1:200, 12D7, Novus Biologicals, Littleton, CO; anti-β actin antibody, 1:10000, AC-15, Sigma-Aldrich, St Louis, MO), which was diluted in 0.05% Tween 20-PBS overnight at 4 °C. Membranes were then incubated with a horseradish peroxidase conjugated sheep anti-mouse antibody (GE Health, Piscataway, NJ) diluted in 0.05% Tween20-PBS for 1 h at 4 °C, developed by chemiluminescence (ECL plus, GE Health), and exposed to KODAK BioMax MR Film (Carestream Health, INC. Rochester, NY). β-actin was used as an internal control to adjust for differences in the amount of protein loaded in each lane.
Results
Detection of Ad40 E1 deletion mutants derived from Ad40 Dugan
PCR products of viral DNA extracted from Ad40 Dugan reproducibly showed shorter DNA fragments than that of intact Ad40 E1 (2732 bp, Fig. 1a). An Ad40 E1A-and E1b19K-deletion mutant (1251 bp deletion between 442 and 1692 of GenBank accession no. L19443; Fig. 1a) was also observed in Ad40 Dugan plaques isolated with 911 cells. After cloning and sequence analysis of those low-molecular-weight PCR products, 7 different Ad40 E1 deletion mutants were detected (Fig. 1b, c).
Fig. 1.
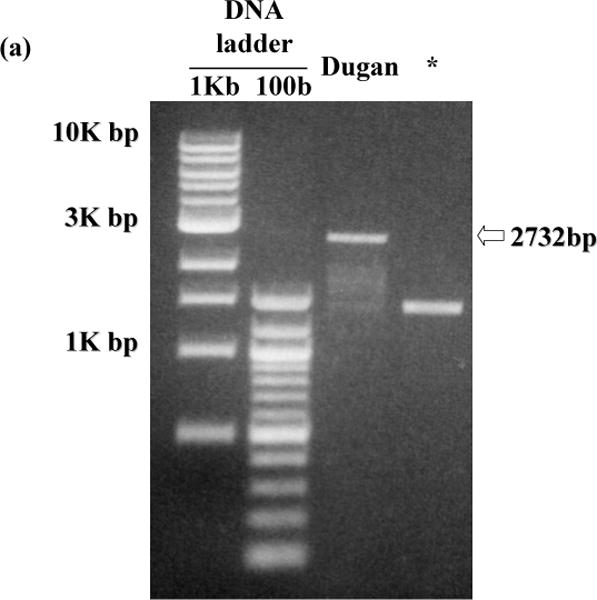

E1 region of Ad40 Dugan-derived mutants.
(a) PCR detection of E1 deletion mutants derived from Ad40 Dugan. The picture shown is representative of more than three independent experiments on the encapsidated viral DNA extracted from Ad40 Dugan obtained from ATCC and from a Ad40 Dugan plaque isolated with 911 cells. The expected DNA size with intact Ad40 E1 is shown on the right of the picture. (b) Schematic representation of the internal tandem repeat (ITR), TATA box, E1A, E1b19K and E1b55K region on the left-hand end of Ad40. The nucleotide number shown was based on GenBank accession no. L19443. (c) Schematic view of Ad40 Dugan-derived mutants. The deleted segments were indicated by dotted lines. The nucleotide number shown was the left- and right-hand breakpoints of deletions (deletion length, bp). * An Ad40 E1A- and E1b55K-deletion mutant in Ad40 Dugan plaques isolated with 911 cells; mRNA, messenger RNA; CDS, coding sequencing.
Effective amplification of pure Ad40wt on Ad5 E1 and Ad5 E4orf6 double-expressing cells
2V6.11 were transfected with linearized pure Ad40wt plasmids, harvested every 72 h after transfection or infection. Until apparent cytopathic effect (CPE, grape-like clusters and >90% cell detachment) was observed in a 60-mm dish, the same amplification was repeated using the 1:3 dilutions of CVLs (1.5 ml into 3 ml fresh medium), which were prepared by centrifugation (1500 rpm, 10 min), three freeze-thawed of 1.7 ml cell suspension, and centrifuged (3000 rpm, 10 min). After observing obvious CPE observed (9th passage) and confirming the existence of Ad40 E1, fiber, and E4orf2 by PCR, the virus was amplified subsequently with the cells (4 × 104 cells/cm2) in a 100-mm dish, a 75-cm2 flask, or 1, 2, 6 (twice), and 12 175-cm2 flasks for subsequent CsCl centrifugation (16th passage) according to standard methodology as referenced [23]. Upon effective amplification with 2V6.11 cells, the Ad40 fiber DNA levels of pure Ad40wt (5.90 × 1011 VP/ml) were approximately 100 times higher than those of Ad40 Dugan (Fig. 2). During the amplification of pure Ad40wt, obvious CPE and the Ad40 fiber DNA level were maintained until the 16th passage and longer.
Fig. 2.
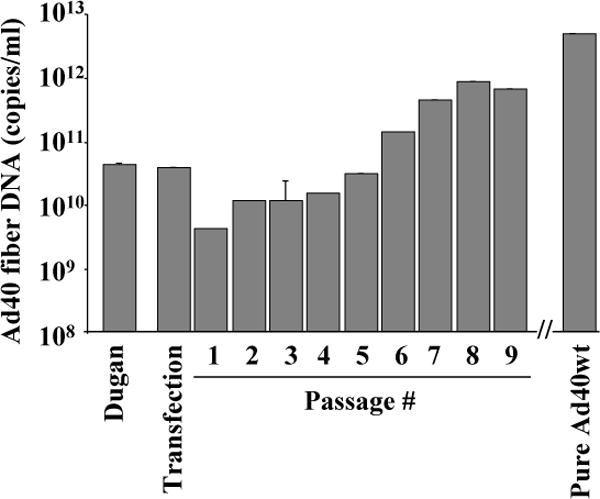
Encapsidated viral DNA levels of pure Ad40wt during propagation.
Pure Ad40wt was generated from plasmid transfection and amplified onto 2V6.11 cells with 1 μg/ml ponasterone A. In a 60-mm dish every 72 h until the appearance of obvious CPE (9th passage), the same amplification was repeated using the 1:3 dilutions of CVLs (1.5 ml into 3 ml fresh medium), which were prepared by centrifugation (1500 rpm, 10 min), reduced the volumes from 4.5 ml to 1.7 ml, three freeze-thawed and centrifuged (3000 rpm, 10 min). After observing CPE and confirming the existence of Ad40 E1, fiber, and E4orf2 by PCR, the virus was amplified continuously for CsCl centrifugation. The Ad40 fiber DNA levels were evaluated by real-time PCR on the encapsidated viral DNA from the 200 μl CVLs at each passage (in triplicate). The results shown are mean values of triplicates. Error bars indicate the 95% confidence interval.
More than 50% of pure Ad40wt (57.6%, 19738 of 34214bp) sequencing was covered by shotgun sequencing with TOPO shotgun sequencing kit (Invitrogen) and was identical to the GenBank sequencing of Ad40 (accession no. L19443). The remaining portion of the virus genome was sequenced through amplification by PCR with primers based on the known sequence. In the end, the pure Ad40wt genome sequence was identical to the GenBank sequence and the restriction analyses of pure Ad40wt with ClaI, HindIII, and SwaI complied with the GenBank sequence.
Lack of Ad40 E1 genetic integrity isolated plaques
Plaque formation of Ad40 Dugan and pure Ad40wt was observed at day 10 in 911 cells. The median (interquartile range) PFU of Ad40 Dugan and pure Ad40wt was 2.2 (1.7–2.7) × 104 PFU/ml and 9.0 (8.6–9.3) × 106 PFU/ml, respectively (in three independent experiments). In all plaques of Ad40 Dugan and pure Ad40wt (40 plaques each), both Ad40 fiber and E4orf2 DNA were detected by PCR. In plaques with pure Ad40wt, 45% (18/40) showed intact Ad40 E1 region in PCR analysis. On the other hand, only 10% (4 out of 40) of the plaques with Ad40 Dugan showed intact Ad40 E1 PCR fragments.
Maintaining Ad40 E1b55K mRNA expression and Ad40 E1 genetic integrity in Ad5 E1 and Ad5 E4orf6 double-expressing cells
Quantification of Ad40 fiber mRNA and DNA levels, respectively, were performed by real-time RT-PCR and PCR on DNA-free total RNA and encapsidated viral DNA extracted from 7 cell lines infected with pure Ad40wt: 293, 2V6.11, 911, 911orf6, A549, HeLa and W162 cells (in three independent experiments, Table 1 and Fig. 3a). Ad40 fiber mRNA and DNA in both the supernatants and CVLs were detected in 200 VP/cell pure Ad40wt infected 293, 2V6.11, 911, 911orf6, A549 and HeLa cells (Table 1). Ad40 E1b55K mRNA expression and E1 DNA was tested by RT-PCR and PCR, respectively. RT-PCR analysis for Ad40 E1b55K showed that intact Ad40 E1b55K mRNA was reproducibly detected in 200 VP/cell pure Ad40wt infected 293 and 2V6.11 cells. The result indicated that pure Ad40wt with intact Ad40 E1 was reproducibly amplified only in 200 VP/cell pure Ad40wt infected 2V6.11 cells with 1 μg/ml ponasterone A. The highest intensity of Ad40 E1 PCR fragments was observed in 200 VP/cell pure Ad40wt infected 2V6.11 cells with 1 μg/ml ponasterone A (Fig. 3b).
Table 1.
E1b55K mRNA expression and fiber mRNA levels of pure Ad40wt
| Cell lines | Ad40 E1b55K | Ad40 fiber median (IQR) | |
|---|---|---|---|
| 293 | +a | 3.20 (2.62–3.77)b | |
| 2V6.11 |

|
+ | 0.28 (0.01–0.75) |
| + | 0.44 (0.01–0.98) | ||
| + | 0.74 (0.01–1.54) | ||
| 911 | − | 3.92 (2.72–5.11) | |
| 911orf6 |
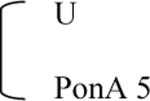
|
± | 3.86 (2.32–5.39) |
| − | 0.26 (0.01–0.66) | ||
| A549 | − | 1.67 (0.80–2.49) | |
| HeLa | − | 1.77 (0.92–2.61) | |
| W162 | − | 0.01 (0–0.01) |
The results of three independent experiments are shown.
Pure Ad40wt fiber mRNA levels (% of total RNA) were measured by real-time RT-PCR.
Plus (+) means reproducible PCR-positive, minus (−) means reproducible PCR-negative, and plus-minus (±) means unreproducible PCR-positive.
IQR, interquartile range; U, uninduced control (0.1% DMSO); PonA 1 or 5, 1 or 5 μg/ml PonA.
Fig. 3.
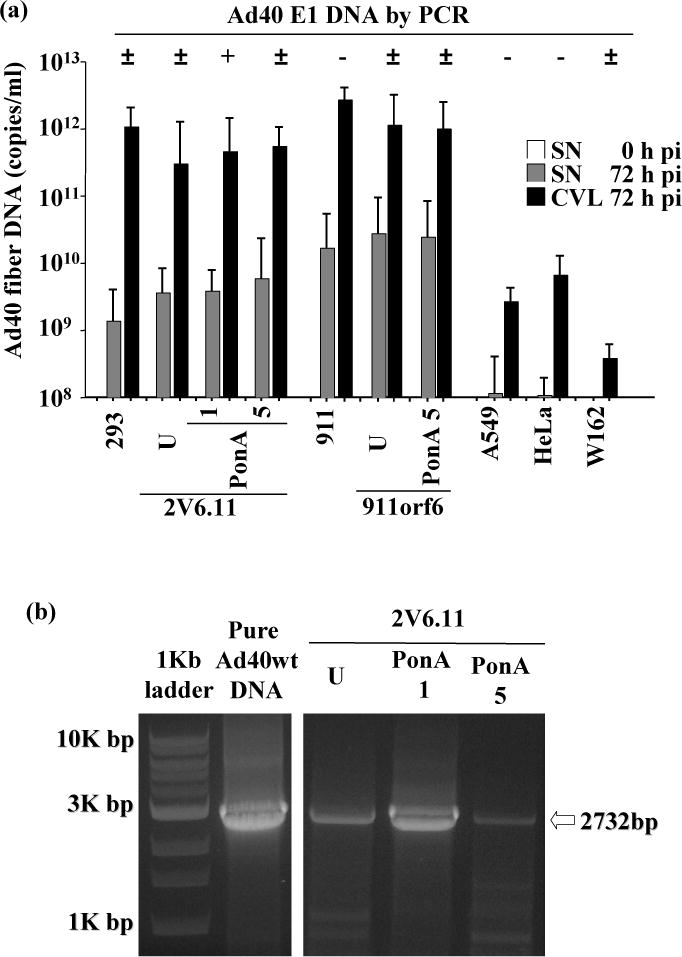
E1 and fiber DNA of pure Ad40wt.
Cells were infected with 200 VP/cell of pure Ad40wt. At 72 h pi, encapsidated viral DNA from the the 200 μl supernatants (SNs) and CVLs was purified and Ad40 E1 DNA was tested by PCR on encapsidated viral DNA from the 200 μl CVLs. The results of three independent experiments are shown on the top of figures (a). Plus (+) means reproducible PCR-positive, minus (−) means reproducible PCR-negative, and plus-minus (±) means unreproducible PCR-positive. The Ad40 fiber DNA levels (copy numbers per ml) in the SNs (0 h pi; on the left hand, empty bar graph and 72 h pi; in the middle, shaded bar graph) and CVLs (72 h pi; on the right, black bar graph) from pure Ad40wt were performed by real-time PCR (a). The results shown are mean values of three independent experiments. Error bars indicate the 95% confidence interval. The picture shown is representative of three independent experiments on pure Ad40wt infected 2V6.11 cells (b). The expected DNA size with intact Ad40 E1 is shown on the right of the picture. U, uninduced control (0.1% DMSO). PonA 1 or 5, 1 or 5 μg/ml ponasterone A.
Ad40 degraded Mre11-expression
The analysis of Ad5 E1b55K and Ad5 E4orf6 proteins showed that the expression of Ad5 E4orf6 induced with ponasterone A (1 and 5 μg/ml in 2V6.11, and 5 μg/ml in 911orf6 cells) increased the level of Ad5 E1b55K in these cells (Fig. 4a). Ponasterone A induced the Mre 11-degradation in 2V6.11 cells in a dose-dependent manner (Fig. 4b).
Fig. 4.
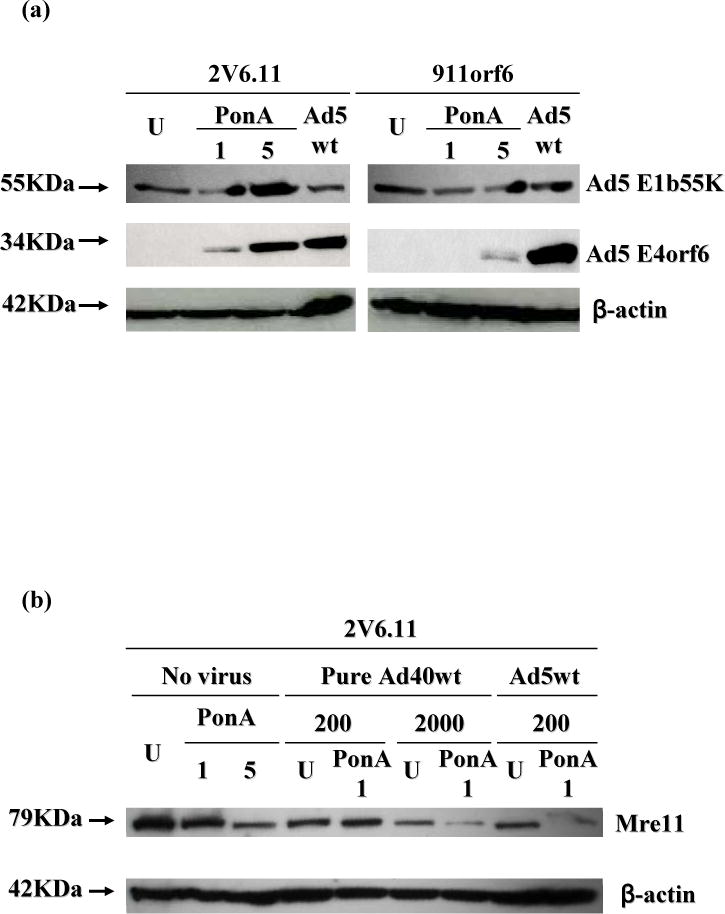
Expression of Ad5 E1b55K or Ad5 E4orf6 in 2V6.11 or 911orf6 cells, and Mre11 expression in 2V6.11 cells.
Cells were treated with 0.1% DMSO (U, uninduced control), 1 or 5 μg/ml ponasterone A (PonA 1 or PonA 5), or 200 VP/cell Ad5wt. At 24 h pi, cells were harvested (a) or cells were infected with virus-free culture medium, 200 or 2000 VP/cell pure Ad40wt, or 200 VP/cell Ad5wt (b). Cell extracts were analyzed by SDS-PAGE followed by Western blotting using approximate antibodies as indicated to the right. Expected protein sizes are shown on the left of pictures.
In order to determine whether infection of pure Ad40wt in 2V6.11 cells affected Mre11 level, the expression levels of Mre11 were assessed (Fig. 4b). Decrease of Mre11 was observed in 2V6.11 cells at 24 h pi of 2000 VP/cell pure Ad40wt, but not at 200 VP/cell. The difference of Mre11 expression between induced and uninduced cells was detected only in 2000 VP/cell pure Ad40wt infected 2V6.11 cells.
Discussion
In this study, pure Ad40wt was successfully amplified at large scale in 2V6.11 cells with ponasterone A-based Ad5 E4orf6 induction. The lack of Ad40 E1 genetic integrity after Ad40wt infection might be a primary reason why Ad40 had previously been extremely difficult to amplify in vitro. The data presented in this paper demonstrate that a coexpression of Ad5 E1 and Ad5 E4orf6 proteins overcomes the restrictions that were impeding the process of optimal Ad40wt vector amplification.
The proper transcription and effective replication of Ad40 required Ad5 E4orf6-expression in addition to Ad5 E1. Ad5 E4orf6 was reported to direct the nuclear localization of Ad5 E1b55K [25, 26], which is a multifunctional phosphoprotein that regulates viral DNA replication and nucleocytoplasmic RNA transport in lytically infected cells [27]. Ad5 E4orf6 was also known to direct both nuclear import and export [28]. Thus, ponasterone A-induced Ad5 E4orf6-expression might support the transcription of Ad40 late-onset mRNA and the subsequent release of Ad40 viruses from cells. This may explain why pure Ad40wt with intact Ad40 E1 was successfully amplified in 2V6.11 cells with ponasterone A based Ad5 E4orf6 induction. The transcription and replication of Ad40 was reported in a 72 h culture on Ad2 E1A- and E1B-expressing human epidermoid-carcinoma, 293, and A549 cell lines [12, 29, 30]. 2V6.11 cells could not be maintained more than 5 days after addition of ponasterone A (1 μg/ml), most likely due to the degradation of cellular substrates (e.g. Mre1 1) by Ad5 E1b55K and A5 E4orf6. As a result, the plaque assay withf 2V6.11 cells was not feasible. Saez et al. reported that the luciferase protein expressed with the ecdysone system was detectable within 4 h of exposure to ponasterone A, reached approximately 600-fold between 24 and 36 h, and then decreased to background levels at 20 h after ponasterone A withdrawal [31]. In 2V6.11 cells used for pure Ad40wt amplification, ponasterone A was removed upon viral infection and pure Ad40wt was amplified for 72 h. Here, the expression of Ad5 E4orf6 should have disappeared by 20 h after ponasterone A withdrawal and thus the viability of 2V6.11 cells might have been maintained for 72 h permitting pure Ad40wt amplification. The amount of ponasterone A was also important for successful amplification of pure Ad40wt because in 2V6.11 cells with 5 μg/ml ponasterone A, the intensity of Ad40 E1 PCR fragments was lower and the integrity of Ad40 E1 DNA was less reproducible compared to those with 1 μg/ml ponasterone A. This means that a 72 h passage and 1 μg/ml ponasterone A induction level is the most appropriate timing of culture and dose of ponasterone A for Ad40 amplification.
Regarding Ad40 Dugan, a majority of the clones were Ad40 E1 A and/or E1b19K mutants, and only few presented intact E1 region by PCR in plaques isolated with 911 cells. The current batch of Ad40 Dugan that was amplified in 293 cells may be contaminated with Ad40 E1 deletion mutants, which may also yield plaque formation in 911 cells. Among 5 Ad5-propagating cells, the plaque formation of Ad40 Dugan was observed in 911 cells. The different origins, which may underline the differences in morphology and growth, may be the reason why 911 cells exhibit better plaque formation than 293 cells as previously reported in Ad5 [17]. Regarding pure Ad40wt, a lack of genetic integrity in the E1 region was detected in the encapsidated viral DNA extracted from the plaques on 911 cells and from the CVLs after 72 h incubation with 911 or 911orf6 cells. The replication in 911 or 911orf6 cells may be detrimental to Ad40 E1 genetic integrity. One possible explanation of the Ad40 E1 genetic integrity after Ad5 E1 and E4orf6 transcomplementation is that the E1B promoter activity of Ad40 E1B was lower than that of Ad5 E1B and that the weak activity of Ad40 E1B could have been compensated by Ad5 E1B [32, 33]. Pure Ad40wt replicated in 2V6.11 cells with 5 μg/ml ponasterone A, where the expression of Ad5 E1B protein was higher than that in 2V6.11 cells with 1 μg/ml ponasterone A, but genetic integrity was reduced under 5 μg/ml ponasterone A. Lack of Ad40 E1 genetic integrity may explain why Ad40 was difficult to culture in vitro.
Infection with 2000 VP/cell pure Ad40wt induced the degradation of Mre11 in 2V6.11 cells with 1 μg/ml ponasterone A, but not at 200 VP/cell. This degradation of Mre11 may indicate that pure Ad40wt with Ad5 E1b55K and Ad5 E4orf6 prevents nonhomologous end joining (NHEJ) after a DNA double-strand break because of the degradation of the Mre1 1-RAD50-Nbs1 (MRN) complex which is in change of NHEJ. Moreover, pure Ad40wt infection might result in both reorganization and degradation of members of the MRN complex mediated by Ad5 E1b55K and Ad5 E4orf6, which prevents concatemerization previously reported [34]. Amplification of Ad40 may need a high level of viral particles for inducing Mre11-degradation.
For future clinical applications, Ad40-based gene transfer vectors look suitable for the GI tract because AdF possesses specific structural features and has a unique mucosal tropism for the GI tract, which may possibly be mediated by two fibers on the virion [35, 36] or lacking an RGD αv-integrin binding motif on a penton base [37]. Moreover, AdF presents significant tropism for intestinal mucosa as previously reported [16, 38]. The amplification method for effective Ad40wt without losing whole sequence integrity was established. For in vivo gene delivery, a method for effective vectors amplification is necessary. The method we established will be useful for developing an Ad40-based gene transfer vector for intestinal mucosa aiming at oral vaccination.
In summary, a method for efficient amplification of Ad40wt was established. Moreover, possible reasons for difficulty to culture Ad40 in vitro, such as lack of Ad40 E1 genetic integrity after Ad40 infection and the necessity for a high level of viral particles induced Mre11-degradation, were identified. These findings may contribute not only to the study of Ad40 virology but to the future development of gene delivery systems using this virus.
Supplementary Material
Acknowledgments
We thank Dr. Ashok K. Saluja and Dr. Selwyn M. Vickers for helpful discussions. This work was supported by NIH grants R01DK63615 and R01CA94084 (to Masato Yamamoto).
References
- 1.Tiemessen CT, Kidd AH. Adenovirus type 40 and 41 growth in vitro: host range diversity reflected by differences in patterns of DNA replication. J Virol. 1994;68:1239–1244. doi: 10.1128/jvi.68.2.1239-1244.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uhnoo I, Wadell G, Svensson L, Johansson ME. Importance of enteric adenoviruses 40 and 41 in acute gastroenteritis in infants and young children. J Clin Microbiol. 1984;20:365–372. doi: 10.1128/jcm.20.3.365-372.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carter MJ. Enterically infecting viruses: pathogenicity, transmission and significance for food and waterborne infection. J Appl Microbiol. 2005;98:1354–1380. doi: 10.1111/j.1365-2672.2005.02635.x. [DOI] [PubMed] [Google Scholar]
- 4.D’Ambrosio E, Del Grosso N, Chicca A, Midulla M. Neutralizing antibodies against 33 human adenoviruses in normal children in Rome. J Hyg (Lond) 1982;89:155–161. doi: 10.1017/s0022172400070650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thorner AR, Vogels R, Kaspers J, Weverling GJ, Holterman L, Lemckert AA, Dilraj A, McNally LM, Jeena PM, Jepsen S, Abbink P, Nanda A, Swanson PE, Bates AT, O’Brien KL, Havenga MJ, Goudsmit J, Barouch DH. Age dependence of adenovirus-specific neutralizing antibody titers in individuals from sub-Saharan Africa. J Clin Microbiol. 2006;44:3781–3783. doi: 10.1128/JCM.01249-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Appaiahgari MB, Pandey RM, Vrati S. Seroprevalence of neutralizing antibodies to adenovirus type 5 among children in India: implications for recombinant adenovirus-based vaccines. Clin Vaccine Immunol. 2007;14:1053–1055. doi: 10.1128/CVI.00173-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dionisio D, Arista S, Vizzi E, Manneschi LI, Di Lollo S, Trotta M, Sterrantino G, Mininni S, Leoncini F. Chronic intestinal infection due to subgenus F type 40 adenovirus in a patient with AIDS. Scand J Infect Dis. 1997;29:305–307. doi: 10.3109/00365549709019048. [DOI] [PubMed] [Google Scholar]
- 8.Kojaoghlanian T, Flomenberg P, Horwitz MS. The impact of adenovirus infection on the immunocompromised host. Rev Med Virol. 2003;13:155–171. doi: 10.1002/rmv.386. [DOI] [PubMed] [Google Scholar]
- 9.Echavarria M. Adenoviruses in immunocompromised hosts. Clin Microbiol Rev. 2008;21:704–715. doi: 10.1128/CMR.00052-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown M. Laboratory identification of adenoviruses associated with gastroenteritis in Canada from 1983 to 1986. J Clin Microbiol. 1990;28:1525–1529. doi: 10.1128/jcm.28.7.1525-1529.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiemessen CT, Kidd AH. Helper function of adenovirus 2 for adenovirus 41 antigen synthesis in semi-permissive and non-permissive cells. Arch Virol. 1988;103:207–218. doi: 10.1007/BF01311093. [DOI] [PubMed] [Google Scholar]
- 12.Mautner V, Mackay N, Steinthorsdottir V. Complementation of enteric adenovirus type 40 for lytic growth in tissue culture by E1B 55K function of adenovirus types 5 and 12. Virology. 1989;171:619–622. doi: 10.1016/0042-6822(89)90634-x. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto S, Sakakibara N, Kumai H, Nakai M, Sakuma S, Chiba S, Fujinaga K. Fastidious human adenovirus type 40 can propagate efficiently and produce plaques on a human cell line, A549, derived from lung carcinoma. J Virol. 1991;65:2429–2435. doi: 10.1128/jvi.65.5.2429-2435.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodrum FD, Ornelles DA. Roles for the E4 orf6, orf3, and E1B 55-kilodalton proteins in cell cycle-independent adenovirus replication. J Virol. 1999;73:7474–7488. doi: 10.1128/jvi.73.9.7474-7488.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mautner V, Mackay N. Enteric adenovirus type 40: complementation of the E4 defect in Ad2 dl808. Virology. 1991;183:433–436. doi: 10.1016/0042-6822(91)90161-4. [DOI] [PubMed] [Google Scholar]
- 16.de Jong JC, Wigand R, Kidd AH, Wadell G, Kapsenberg JG, Muzerie CJ, Wermenbol AG, Firtzlaff RG. Candidate adenoviruses 40 and 41: fastidious adenoviruses from human infant stool. J Med Virol. 1983;11:215–231. doi: 10.1002/jmv.1890110305. [DOI] [PubMed] [Google Scholar]
- 17.Fallaux FJ, Kranenburg O, Cramer SJ, Houweling A, Van Ormondt H, Hoeben RC, Van Der Eb AJ. Characterization of 911: a new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum Gene Ther. 1996;7:215–222. doi: 10.1089/hum.1996.7.2-215. [DOI] [PubMed] [Google Scholar]
- 18.No D, Yao TP, Evans RM. Ecdysone-inducible gene expression in mammalian cells and transgenic mice. Proc Natl Acad Sci USA. 1996;93:3346–3351. doi: 10.1073/pnas.93.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gastwirt RF, Slavin DA, McAndrew CW, Donoghue DJ. Spy1 expression prevents normal cellular responses to DNA damage: inhibition of apoptosis and checkpoint activation. J Biol Chem. 2006;281:35425–35435. doi: 10.1074/jbc.M604720200. [DOI] [PubMed] [Google Scholar]
- 20.Mohammadi ES, Ketner EA, Johns DC, Ketner G. Expression of the adenovirus E4 34k oncoprotein inhibits repair of double strand breaks in the cellular genome of a 293-based inducible cell line. Nucleic Acids Res. 2004;32:2652–2659. doi: 10.1093/nar/gkh593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obert S, O’Connor RJ, Schmid S, Hearing P. The adenovirus E4–6/7 protein transactivates the E2 promoter by inducing dimerization of a heteromeric E2F complex. Mol Cell Biol. 1994;14:1333–1346. doi: 10.1128/mcb.14.2.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas MA, Lichtenstein DL, Krajcsi P, Wold WS. A real-time PCR method to rapidly titer adenovirus stocks. Methods Mol Med. 2007;130:185–192. doi: 10.1385/1-59745-166-5:185. [DOI] [PubMed] [Google Scholar]
- 23.Davydova J, Le LP, Gavrikova T, Wang M, Krasnykh V, Yamamoto M. Infectivity-enhanced cyclooxygenase-2-based conditionally replicative adenoviruses for esophageal adenocarcinoma treatment. Cancer Res. 2004;64:4319–4327. doi: 10.1158/0008-5472.CAN-04-0064. [DOI] [PubMed] [Google Scholar]
- 24.Steinthorsdottir V, Mautner V. Enteric adenovirus type 40:E1B transcription map and identification of novel E1A-E1B cotranscripts in lytically infected cells. Virology. 1991;181:139–149. doi: 10.1016/0042-6822(91)90478-t. [DOI] [PubMed] [Google Scholar]
- 25.Goodrum FD, Shenk T, Ornelles DA. Adenovirus early region 4 34-kilodalton protein directs the nuclear localization of the early region 1B 55-kilodalton protein in primate cells. J Virol. 1996;70:6323–6335. doi: 10.1128/jvi.70.9.6323-6335.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marshall LJ, Moore AC, Ohki M, Kitabayashi I, Patterson D, Ornelles DA. RUNX1 permits E4orf6-directed nuclear localization of the adenovirus E1B-55K protein and associates with centers of viral DNA and RNA synthesis. J Virol. 2008;82:6395–6408. doi: 10.1128/JVI.00043-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gabler S, Schütt H, Groitl P, Wolf H, Shenk T, Dobner T. E1B 55-kilodalton-associated protein: a cellular protein with RNA-binding activity Implicated in nucleocytoplasmic transport of adenovirus and cellular mRNAs. J Virol. 1998;72:7960–7971. doi: 10.1128/jvi.72.10.7960-7971.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobbelstein M, Roth J, Kimberly WT, Levine AJ, Shenk T. Nuclear export of theE1B 55-kDa and E4 34-kDa adenoviral oncoproteins mediated by a revlike signal sequence. EMBO J. 1997;16:4276–4284. doi: 10.1093/emboj/16.14.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown M, Wilson-Friesen HL, Doane F. A block in release of progeny virus and a high particle-to-infectious unit ratio contribute to poor growth of enteric adenovirus types 40 and 41 in cell culture. J Virol. 1992;66:3198–3205. doi: 10.1128/jvi.66.5.3198-3205.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishida S, Fujinaga Y, Fujinaga K, Sakamoto N, Hashimoto S. Unusual splice sites in the E1A-E1B cotranscripts synthesized in adenovirus type 40-infected A549 cells. Arch Virol. 1994;139:389–402. doi: 10.1007/BF01310800. [DOI] [PubMed] [Google Scholar]
- 31.Saez E, Nelson MC, Eshelman B, Banayo E, Koder A, Cho GJ, Evans RM. Identification of ligands and coligands for the ecdysone-regulated gene switch. Proc Natl Acad Sci USA. 2000;97:14512–14517. doi: 10.1073/pnas.260499497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bailey A, Ullah R, Mautner V. Cell type specific regulation of expression from the Ad40 E1b promoter in recombinant Ad5/Ad40 viruses. Virology. 1994;202:695–706. doi: 10.1006/viro.1994.1391. [DOI] [PubMed] [Google Scholar]
- 33.Mautner V, Bailey A, Steinthorsdottir V, Ullah R, Rinaldi A. Properties of the adenovirus type 40 E1B promoter that contribute to its low transcriptional activity. Virology. 1999;265:10–19. doi: 10.1006/viro.1999.0014. [DOI] [PubMed] [Google Scholar]
- 34.Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 35.Kidd AH, Chroboczek J, Cusack S, Ruigrok RW. Adenovirus type 40 virions contain two distinct fibers. Virology. 1993;192:73–84. doi: 10.1006/viro.1993.1009. [DOI] [PubMed] [Google Scholar]
- 36.Favier AL, Schoehn G, Jaquinod M, Harsi C, Chroboczek J. Structural studies of human enteric adenovirus type 41. Virology. 2002;293:75–85. doi: 10.1006/viro.2001.1235. [DOI] [PubMed] [Google Scholar]
- 37.Albinsson B, Kidd AH. Adenovirus type 41 lacks an RGD alpha(v)-integrin binding motif on the penton base and undergoes delayed uptake in A549 cells. Virus Res. 1999;64:125–136. doi: 10.1016/s0168-1702(99)00087-8. [DOI] [PubMed] [Google Scholar]
- 38.Croyle MA, Stone M, Amidon GL, Roessler BJ. In vitro and in vivo assessment of adenovirus 41 as a vector for gene delivery to the intestine. Gene Ther. 1998;5:645–654. doi: 10.1038/sj.gt.3300645. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.