

Abstract
Due to devastating prognosis, novel therapies are needed for pancreatic cancer. We are in preparation for a human clinical trial of a conditionally replicative adenovirus (CRAd) we developed. While most patients in the target population are receiving either gemcitabine or 5‐fluorouracil chemotherapy, the combination with CRAd has not yet been studied. This study was designed to evaluate combination therapies with CRAd and current standard chemotherapies in pancreatic cancer. When the combination therapy was tested in vitro, gemcitabine pretreatment showed a synergistic effect in two out of four cell lines whereas CRAd followed by gemcitabine exhibited a synergistic effect in one cell line. With 5‐fluorouracil, pretreatment with 5‐fluorouracil produced a synergistic effect in three cell lines whereas post‐treatment was synergistic in only one cell line. These effects were not fully explained by either induction of cyclooxygenase (Cox) 2 activity or adenoviral receptors with chemotherapeutics. In in vivo analyses with Hs766T xenograft, 5‐fluorouracil slightly improved the CRAd antitumor effect but it was not significant. Pretreatment with gemcitabine embodied a significant tumor reduction compared with single therapy with gemcitabine. The most significant antitumor effect occurred when tumors were treated with 5/3COX2CRAdF and subsequent gemcitabine (P = 0.001 vs gemcitabine alone, P = 0.012 vs 5/3COX2CRAdF alone) at day 12. In MIA Paca‐2, pretreatments with either 5‐fluorouracil or gemcitabine improved the CRAd therapeutic effect when administered before CRAd injection (P = 0.03 and P = 0.01, respectively). These experiments indicate the possible benefit of combination therapies, and thus it is not necessary to interrupt chemotherapeutics when receiving CRAd therapy. (Cancer Sci 2009); 00: 000–000)
Cancer is the leading cause of death among women aged 40–79 years and among men aged 60–79 years.( 1 ) Pancreatic cancer is a particularly devastating disease, ranked as the fourth leading cause of cancer‐related death.( 1 ) The life expectancy upon diagnosis is usually no more than 1 year; each year, nearly 30 000 individuals die of this disease, largely because the cancer is unresectable at the time of diagnosis.( 2 ) These facts clearly indicate the need to develop new clinical interventions for improving the clinical outcome of this disease.( 3 , 4 )
Adenoviral cancer gene therapy is known for its potential as an effective therapeutic modality in treatment of refractory cancers. In clinical cancer trials, non‐replicating adenoviral vectors have been remarkably safe, with hundreds of cancer patients treated without treatment‐related mortality.( 5 ) In addition, recently conducted clinical trials have shown the impressive therapeutic potential even with early generation viruses.( 6 , 7 ) Among them, virotherapy based on conditionally replicative adenoviruses (CRAd), which are designed to replicate and spread within the tumor to overcome the issue of incomplete in vivo tumor transduction, has a unique advantage compared to the therapy based on a non‐replicative system.( 4 ) We have been developing this kind of therapeutic for pancreatic cancer.( 8 , 9 ) In particular, CRAd under the control of a cyclooxygenase (COX) 2 promoter has a strong antitumor effect in a subcutaneous xenograft model of human pancreatic cancer. Therefore, we are planning a human clinical trial with this vector.( 9 )
Currently, the standard therapies for patients with unresectable pancreatic cancer are systemic chemotherapies with either gemcitabine (Gem) or 5‐fluorouracil (FU).( 10 ) Thus, most patients in the target population of CRAd clinical trials are expected to be receiving one of these drugs, but the combination with CRAd has not been tested to date. Combination treatment presents two primary concerns. The first is that chemotherapy and CRAd therapy may impede the therapeutic function of each other. The other concern is that nucleotide analogs, which can be incorporated into DNA, may hamper replication of DNA virus and subsequent tumor lysis. In case either of these is observed, chemotherapy treatment will need to be discontinued for the duration of the CRAd clinical trial. Thus, the experimental result of combination therapy of CRAd and standard chemotherapies will significantly affect the design of the clinical trials. The combination with CRAd has not yet been studied, though several studies have examined the possibility of combination therapy including these nucleotide analogues and in many cases the benefit of effect therapy is sequence‐dependent so far.( 11 , 12 , 13 , 14 )
Currently, several CRAd are under investigation for their potential therapeutic effect for pancreatic cancer.( 4 ) Among them, fiber‐modified CRAd are a promising treatment approach (e.g. RGDCOX2F, 5/3COX2CRAdF( 8 , 9 , 15 )) that could soon go to human trials. Therefore, the study described here was carried out to determine the effect of Gem or 5‐FU with CRAd for pancreatic cancer. This analysis will help to determine the benefits and risks of the combination therapies to be applied in clinical settings.
Materials and Methods
Cell lines and animals. Pancreatic cancer cell lines (MIA Paca‐2, Hs766T, PANC‐1, and Capan‐1) were obtained from American Type Culture Collection (Manassas, VA, USA). They were maintained with the recommended medium at 37°C and 5% CO2.
Female athymic NCr‐nu nude mice (Frederick Cancer Research, Frederick, MD, USA) (6–8 weeks of age) were used for the in vivo experiments. All animals received humane care based on the American Veterinary Association guidelines. All of the animal experiment protocols were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Chemotherapeutics. Doses of Gem (AAPin Chemicals, Oxon, UK) ranged from 0.002 to 0.250 μM in vitro and 40 mg/kg in vivo. Doses of 5‐FU (Sigma, St. Louis, MO, USA) ranged from 1 to 256 μM in vitro and 25 mg/kg in vivo.
Virus. The primary virus used in this project is 5/3COX2CRAdF. The structure and function of the vector was reported previously.( 8 , 9 , 15 ) COX2 and cytomegalovirus immediate early (CMV) promoter‐driven non‐replicative adenoviral vectors (AdCOX2Luc and AdCMVLuc, respectively)( 16 ) were used for in vitro studies.
Cell viability assay.
Virus first group. Pancreatic cancer cells at 2500 cells/well were plated in a 96‐well plate. Virus was administered at doses ranging from 0.001 to 10 vp/cell and was allowed to incubate for 4 h. The media was then replaced with the new media containing Gem or 5‐FU at concentrations of 0.025–0.250 μM or 1–256 μM, respectively. Media without phenol red were used to minimize the effect on optical density reading.
Chemotherapeutic first group. Chemotherapeutics were added to the cells first and remained on the cells for 48 h. Afterwards, the chemotherapeutic was removed and virus was added for 4 h. Finally, either media or media containing chemotherapeutic was added.
After 7 days, the plates were stained using Promega Cell Titer 96 AQueous Non‐Radioactive Cell Proliferation Assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Data were analyzed using MacSynergy II (kindly provided by Marc Prichard, University of Alabama at Birmingham (UAB)).( 17 , 18 , 19 )
Effect of chemotherapeutics on Cox2 promoter activity. To assess the effect of chemotherapeutics on the COX2 promoter, which controls CRAd replication, the target pancreatic cancer cell lines were infected with the non‐replicative COX2 promoter‐driven luciferase expression vector (AdCOX2Luc)( 16 ) at 100 virus particle (vp)/cell in combination with either Gem or 5‐FU. The infection procedure was the same as those for CRAd, which is described above. Two days after infection with vector, the cells were lysed with Cell Culture Lysis Buffer (Promega), and luciferase activity was measured as described by the manufacturer.
Effect of chemotherapeutics on cell infectivity. To analyze the effect of chemotherapeutics on adenoviral infectivity, the target pancreatic cancer cell lines were infected with the non‐replicative CMV promoter‐driven luciferase expression vector (AdCMVLuc) at 100 vp/cell in combination with either Gem or 5‐FU. The infection procedure was the same as that for CRAd, which is described above. Two days after vector infection, the cells were lysed, and luciferase activity was measured.
FACS analysis of CAR, αvβ5, and α5β1. Pancreatic cancer cells treated with Gem and 5‐FU for 48 h were analyzed by flowcytometry as described previously.( 8 ) Briefly, cells (106) were stained with the primary antibodies mouse anti‐coxsackie‐adenovirus receptor (CAR) antibody RmcB (1 : 1000) (American Type Culture Collection), anti‐α5β1 antibody (1 : 100) (Chemicon, Temecula, CA, USA), and anti‐αvβ5 integrin (Chemicon) diluted 1 : 1000, then incubated with FITC‐conjugated antimouse IgG antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) diluted 1 : 50. The samples were washed and analyzed by the UAB FACS Analysis Core Facility. The data was presented as the percentile of surface marker‐positive cells.
In vivo assay. Hs766T and MIA Paca‐2 subcutaneous tumors were established on the flanks of NCR‐nu nude mice. Upon tumor establishment, the mice were divided into eight groups (untreated, Gem only, 5‐FU only, CRAd→Gem, CRAd→ 5‐FU, CRAd only, Gem→CRAd, 5‐FU→CRAd). CRAd were injected intratumorally at a dose of 1 × 1010 vp before or after administration of chemotherapeutics. Gem and 5‐FU were injected intraperitoneally at a dose of 40 and 25 mg/kg respectively.( 20 , 21 ) Tumor volumes were calculated using the formula: length × width2 × 0.52. Tumor volumes were then calculated as a percentage of tumor size just prior to the first treatment.
Statistical analysis. MTS was analyzed statistically using MacSynergy II software to 95% confidence.( 18 , 19 ) The manufacturer’s guidelines were used to determine significance. All other data were analyzed using Student’s t‐test and ANOVA.
Results
In vitro cell viability after combination therapy. After treating the pancreatic cancer cell lines with several combinations of therapies, cell viability was assessed by MTS assay. The effect of combination was then analyzed with MacSynergy II. Interestingly, the beneficial order of administration was different depending on the cell lines. These analyses were carried out in four pancreatic cancer cell lines but only the data with MIA Paca‐2 and Hs766T were shown in the figures.
When cells were treated with CRAd followed by Gem, there was an antagonistic effect in MIA Paca‐2 (Fig. 1A) and PANC‐1 (data not shown) and a synergistic effect in Hs766T (Fig. 2A). When cells were treated with Gem prior to CRAd, there was a synergistic relationship in MIA Paca‐2 (Fig. 1B), Capan‐1, and PANC‐1 (data not shown), but an antagonistic effect was observed in Hs766T (Fig. 2B).
Figure 1.

In vitro combination effect in MAI Paca‐2. Target cells were treated with various concentrations of conditionally replicative adenovirus (CRAd) (0.001–10 vp/cell) combined with gemcitabine (Gem) or 5‐fluorouracil (FU) (0.025–0.250 μM or 1–256 μM). Seven days later, the cell viability was assessed. The result was analyzed with MacSynergy II with 95% confidence. (A) CRAd→Gem: antagonistic. (B) Gem→CRAd: synergistic. (C) CRAd→ 5‐FU: antagonistic. (D) 5‐FU→CRAd: synergistic.
Figure 2.

In vitro combination effect in Hs766T cells. Target cells were treated with various concentrations of conditionally replicative adenovirus (CRAd) combined with gemcitabine (Gem) or 5‐fluorouracil (FU). The result was analyzed with MacSynergy II with 95% confidence. (A) CRAd→Gem: synergistic. (B) Gem→CRAd: antagonistic. (C) CRAd →5‐FU: antagonistic. (D) 5‐FU→CRAd: antagonistic.
In the case of 5‐FU, when cells were treated with CRAd first, there was a weak synergistic effect in PANC‐1 (data not shown) and an antagonistic effect in MIA Paca‐2 and Hs766T (1, 2). When cells were treated with 5‐FU and then virus, there was a synergistic relationship in MIA Paca‐2 (Fig. 1D), Capan‐1 (data not shown), and PANC‐1(data not shown) and an antagonistic effect in Hs766T (Fig. 2D); other cases were insignificant.
Effect of chemotherapeutics on Cox2 promoter activity. When AdCox2Luc was first added to MIA Paca‐2 (Fig. 3A), Capan‐1 (data not shown), or PANC‐1 (data not shown), followed by treatment with Gem, there was a significant increase in Cox2 activity. When AdCox2Luc was added to cells prior to 5‐FU, there was a significant increase in Cox2 activity in Capan‐1, and a significant decrease in PANC‐1 (data not shown). In all other cell lines and setups, there was no significant change in Cox2 activity. When Gem was added to MIA Paca‐2 and then infected with AdCox2Luc, there was a statistically significant increase in Cox2 activity (Fig. 3B). On the contrary, when Gem was added to PANC‐1 and then infected with AdCox2Luc there was a decrease in Cox2 activity (data not shown). When 5‐FU was added to MIA Paca‐2 (Fig. 2B) or PANC‐1 (data not shown) and then infected with AdCox2Luc, there was a statistically significant decrease in Cox2 activity.
Figure 3.

Cyclooxygenase (COX) 2 promoter activity and adenovirus infectivity in MIA Paca‐2 cells in the presence of chemotherapy. (A,B) In order to assess the effect of chemotherapeutics on COX2 promoter activity, the target cells were treated with COX2 promoter‐controlled luciferase‐expressing vector (AdCOX2Luc) at 100 vp/cell combined with gemcitabine (Gem) (250 or 500 nM) or 5‐fluorouracil (FU) (10 or 20 μM). (A) AdCOX2Luc→Chemo. AdCOX2Luc→ Gem significantly increased Cox2 activity. (B) Chemo→AdCOX2Luc. Gem→AdCOX2Luc significantly increased Cox2 activity, and 5‐FU→AdCOX2Luc decreased Cox2 activity. (C,D) To assess the effect of chemotherapeutics on viral infectivity, the target cells were treated with CMV promoter‐controlled luciferase‐expressing vector (AdCMVLuc) at 100 vp/cell combined with Gem (250 or 500 nM) or 5‐FU (10 or 20 μM). (C) AdCMVLuc→Chemo. With either Gem or 5‐FU there was a statistically significant increase in infectivity. (D) Chemo→AdCMVLuc. Pretreatment with either 5‐FU or Gem increased infectivity (*P < 0.05; **P < 0.001). RLU, relative light unit.
When the same analyses were carried out in Hs766T cells, there was no significant effect on Cox2 promoter activity regardless of the combination administration order (Fig. 4A,B).
Figure 4.

Cyclooxygenase (COX) 2 promoter activity and adenovirus infectivity in Hs766T cells in the presence of chemotherapy. (A,B) In order to assess the effect of chemotherapeutics on COX2 promoter activity, the target cells were treated with AdCOX2Luc combined with gemcitabine (Gem) or 5‐fluorouracil (FU). (A) AdCOX2Luc→Chemo. There was no significant change in Cox2 activity. (B) Chemo→ AdCOX2Luc. No significant change in Cox2 promoter activity was observed. (C,D) In order to assess the effect of chemotherapeutics on viral infectivity, the target cells were treated with AdCMVLuc combined with Gem or 5‐FU. (C) AdCMVLuc→Chemo. Either Gem or 5‐FU post‐treatment increased infectivity. (D) Chemo→AdCMVLuc. Pretreatment with Gem or 5‐FU had no effect in infectivity (*P < 0.05; **P < 0.001). RLU, relative light unit.
Effect of chemotherapeutics on cell infectivity. When four pancreatic cancer cell lines were infected with AdCMVLuc and subsequently treated with either Gem or 5‐FU, there was a statistically significant increase in luc expression demonstrating increased infectivity (Figs 3C,D,4C,D). When Gem was added to cells prior to AdCMVLuc, there was a significant increase in infectivity of MIA Paca‐2 cells (Fig. 3D), a decrease in PANC‐1 (data not shown), and no significant change in Hs766T (Fig. 4C). Similarly, when cells were treated with 5‐FU and then AdCMVLuc, there was an increase of infectivity in MIA Paca‐2 (Fig. 3D) and Capan‐1 (data not shown), a decrease in PANC‐1 (data not shown), and no significant change in Hs766T (Fig. 4D).
Effect of chemotherapeutics on CAR and integrin expression. Table 1 shows the percentage of positive cells by FACS analysis. In the presence of Gem and 5‐FU, there was an increase in αvβ5 and α5β1 integrins except in Hs766T, which showed minimal change in integrin expression. Interestingly, the decrease in CAR in Capan‐1 and Hs766T with Gem treatment was notable. Also, there was a decrease in CAR with the treatment of Hs766T with 5‐FU. CAR increased when Capan‐1 was treated with 5‐FU. PANC‐1 and MIA Paca‐2 had a significant increase in CAR in the presence of Gem or 5‐FU.
Table 1.
Expression of αvβ5 and α5β1 integrins and CAR after treatment with chemotherapeutics†
| Positive cell (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| αvβ5 | α5β1 | CAR | |||||||
| Control | Gem | 5‐FU | Control | Gem | 5‐FU | Control | Gem | 5‐FU | |
| Capan1 | 0.85 | 1.08 | 3.41 | 11.41 | 23.63 | 34.58 | 3.12 | 1.15 | 4.76 |
| PANC‐1 | 12.75 | 22.80 | 17.23 | 57.96 | 58.89 | 61.46 | 51.90 | 92.00 | 85.90 |
| HS766t | 1.11 | 1.42 | 2.42 | 2.13 | 3.91 | 7.24 | 0.73 | 0.31 | 0.44 |
| MIA Paca‐2 | 1.98 | 19.38 | 8.38 | 10.97 | 90.93 | 45.43 | 2.95 | 14.84 | 12.96 |
†Data are presented as the percentage of surface marker‐positive cells after each treatment. FU, fluorouracil; Gem, gemcitabine.
Combination effect in vivo. In Hs766T, when 5‐FU was combined with CRAd there was no significant difference in tumor volume when compared with single therapies during this experiment (Fig. 5). Possible therapeutic potential was shown in the tumors treated with 5/3COX2CRAdF and then treated 5‐FU, and with 5‐FU first and then 5/3COX2CRAdF, but the effect was not statistically significant compared with 5‐FU alone (P = 0.113 and P = 0.067, respectively).
Figure 5.

In vivo combination effect in Hs766T cells. HS766t subcutaneous tumors were established on the flanks of 40 NCR‐nu nude mice. Upon tumor establishment, mice were divided into eight groups: untreated, gemcitabine (Gem) only, 5‐fluorouracil (FU) only, conditionally replicative adenovirus (CRAd) + Gem, CRAd + 5‐FU, CRAd only, Gem + CRAd, and 5‐FU + CRAd. CRAd was injected intratumorally at a dose of 1 × 1010 vp. Gem and 5‐FU were injected intraperitoneally at a dose of 40 and 25 mg/kg respectively. When tumors were treated with 5/3COX2CRAdF and then Gem there was a significant decrease in tumor volume compared with Gem alone (P = 0.001) or 5/3COX2CRAdF alone (P = 0.012) 12 days after treatment. Tumor‐bearing mice treated with Gem had significantly larger tumor volumes on day 12 than mice treated with 5/3COX2CRAdF (P = 0.05).
The tumor‐bearing mice first treated with Gem followed by 5/3COX2CRAdF showed a significant reduction in tumor size compared to Gem alone after 12 days (P = 0.014). The most significant decrease in tumor volume occurred when tumors were treated with 5/3COX2CRAdF and then Gem compared with Gem alone (P = 0.001) or 5/3COX2CRAdF alone (P = 0.012) 12 days after treatment (Fig. 5). The same difference was also observed on days 16 and 18. Amongst all combinations, only the treatment first with 5/3COX2CRAdF and then Gem showed a significantly stronger antitumor effect than 5/3COX2CRAdF single therapy.
In MIA Paca‐2, both 5‐FU and Gem showed combination benefit compared to single chemotherapy when administered prior to CRAd injection (P = 0.03 and P = 0.01, respectively). By contrast, only 5‐FU showed benefit if chemotherapeutics were administered after CRAd injection (5‐FU, P = 0.01; Gem, P = 0.56). No combination benefit was observed compared with CRAd monotherapy (Fig. 6).
Figure 6.
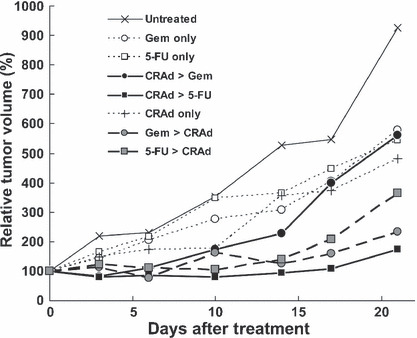
In vivo combination effect in MIA Paca‐2 cells. MIA Paca‐2 subcutaneous tumors were treated in the same way as Hs766T tumors. Conditionally replicative adenovirus (CRAd) was injected intratumorally at a dose of 1 × 1010 vp. Gemcitabine (Gem) and 5‐fluorouracil (FU) were injected intraperitoneally at a dose of 40 and 25 mg/kg respectively. Both 5‐FU and Gem showed combination benefits compared with single chemotherapy when administered prior to CRAd injection, whereas only 5‐FU showed benefit if the chemotherapeutics were administered after CRAd injection (P = 0.01). There was no combination benefit observed compared to CRAd monotherapy.
None of the tested combinations of CRAd with the chemotherapeutics (Gem and 5‐FU) showed inhibition of the therapeutic effect.
Discussion
We have developed a capsid‐modified CRAd controlled with a COX2 promoter vector (RGDCOX2CRAdF and 5/3COX2CRAdF) for pancreatic cancer.( 9 ) This vector is moving toward clinical trials with support of the National Cancer Institute/Rapid Access to Intervention Development (NCI/RAID) program at the University of Minnesota. The planning of the clinical protocol raised an important clinical question about possible combination therapies with chemotherapeutics. Most patients with unresectable pancreatic cancer have been receiving chemotherapy with 5‐FU, and more recently with Gem.( 22 , 23 ) It is likely that combination therapy of CRAd and those chemotherapies shows enhanced therapeutic effect in pancreatic cancer, as seen with herpes simplex virus.( 12 ) On the other hand, viral replication may be inhibited because Gem and 5‐FU are nucleotide analogs. Thus, we need to know whether the combination of CRAd therapy and these standard chemotherapies demonstrates synergistic or antagonistic effects. Also, combination therapy may provide an enhanced antitumor effect with the CRAd protocol. In the case of ONYX‐015, the most impressive therapeutic effect was observed in a head and neck squamous cell carcinoma patient when combined treatment included 5‐FU and radiation.( 24 ) To date, there has been no investigation to determine the effect of Gem or 5‐FU on CRAd for pancreatic cancer.
When the chemotherapeutics were introduced before viral infection of the cells, three out of four cell lines (MIA Paca‐2, Capan‐1, and PANC‐1) showed synergistic effects whereas the combination was antagonistic in Hs766T (1, 2). When the chemotherapeutics were added after viral infection, Gem showed synergy only in HS766T (Fig. 2A). By comparison, 5‐FU showed mixed results (1, 2).
The increased combined effectiveness may arise from several possibilities: (1) chemotherapeutics affect the promoter that controls replication; or (2) chemotherapeutics affect viral infectivity by controlling the expression of viral receptor on cell surfaces. To assess the effect of chemotherapeutics on the COX2 promoter, the cells were infected with non‐replicative COX2 promoter‐controlled luciferase‐expressing adenoviral vector. As shown in 3, 4, some cell lines showed increasing or decreasing promoter activity. However, the COX2 promoter activity profile did not match the combination effect. The viral infectivity profile tended to correlate with primary (CAR) and secondary (integrins) receptor expression (Table 1) but did not provide a general rule applicable in all tested cell lines. Thus, at this time, neither promoter induction nor infectivity increase can fully determine the in vitro combination effects.
The in vitro studies showed that Gem and 5‐FU affected 5/3COX2CRAdF function in a sequence‐dependent manner. Thus, depending on the sequence of administration, pancreatic cancer standard chemotherapeutics may hamper the in vivo function of 5/3COX2CRAdF.
In order to know the in vivo effects of the combination regiment, experiments were carried out with two pancreatic cancer cell lines, Hs766T and MIA Paca‐2, in nude mice. The results in both Hs766T and MIA Paca‐2 (5, 6) showed that combination therapy always exhibited stronger antitumor effects compared with single therapy. No inhibitory effect has been observed in any combination or administration order. The data suggests that removal of standard chemotherapy upon CRAd administration will not be necessary.
In Hs766T, although single chemotherapy did not change the tumor growth, combination therapies showed significant effects compared to all single therapies. The strongest antitumor effect was observed in the tumor treated with CRAd before Gem administration. The in vitro data with this cell line indicated that the combination therapy showed synergy when CRAd was administered before Gem, whereas pretreatment with Gem inhibited the cytocidal effect (Fig. 2), and this may explain the stronger in vivo effect in the CRAd→Gem group compared with the Gem→CRAd group. Although 5‐FU (pre‐ and post‐treatment) and pretreatment with Gem caused an antagonistic effect in vitro in Hs766T, such combination administration orders showed enhanced therapeutic effect in vivo.
Contrary to Hs766T, MIA Paca‐2 showed some response to single therapies. In this cell line, the effect of the CRAd→Gem sequence remained comparable to those of single therapies. When chemotherapeutics were administered prior to CRAd injection, both Gem and 5‐FU showed benefit. Both chemotherapeutics enhance adenovirus receptor expression in MIA Paca‐2 in vitro (Table 1), which may explain the improved effect when cells were treated with chemotherapeutics first.
When in vivo data in these two cell lines were compared, there was a clear contrast in the preferential sequence for combination therapy. Gem showed the strongest combination effect when it was administered after CRAd injection in Hs766T (Fig. 5), whereas Gem→CRAd was strongest in MIA Paca‐2 (Fig. 6). This same contrast was already shown in vitro (1, 2). The difference may be explained by the increased CAR and integrin expression after pretreatment with Gem in MIA Paca‐2, but not in Hs766T (Table 1). As a consequence, enhancement of infectivity was observed with Gem in MIA Paca‐2 but not in Hs766T (3, 4).
In the present study, we have tested the combination therapies of CRAd (5/3COX2CRAdF) and standard chemotherapeutics for pancreatic cancer. Although further analyses are required for the mechanism of the in vivo combination effect, all combinations showed enhanced therapeutic effect compared to single therapy although some of them were not statistically significant. No combination showed in vivo interference in the pancreatic cancer model. In this sense, patients receiving standard chemotherapy are able to receive CRAd therapy without terminating chemotherapies; the combination may show improved clinical output. Based on this data, the combination of 5/3COX2CRAd and standard chemotherapeutics (Gem or 5‐FU) is planned for clinical protocol.
Acknowledgment
This work was supported in part by National Institutes of Health grants R01CA94084, R01 DK63615, and P20CA101955 (Developmental Project), and a University of Minnesota AHC‐Translational Research Grant to M.Y.
References
- 1. Jemal A, Siegel R, Ward E et al. Cancer statistics, 2008. CA Cancer J Clin 2008; 58: 71–96. [DOI] [PubMed] [Google Scholar]
- 2. Sener SF, Fremgen A, Menck HR, Winchester DP. Pancreatic cancer: a report of treatment and survival trends for 100 313 patients diagnosed from 1985–1995, using the National Cancer Database. J Am Coll Surg 1999; 189: 1–7. [DOI] [PubMed] [Google Scholar]
- 3. Yamamoto M. Conditionally replicative adenovirus for gastrointestinal cancers. Expert Opin Biol Ther 2004; 4: 1241–50. [DOI] [PubMed] [Google Scholar]
- 4. Yamamoto M, Curiel DT. Gene therapy and virotherapy for pancreatic cancer. In: Von Hoff DD, Evans DB, Hurban RH, eds. Panceatic Cancer. Sulbury: Jones and Bartlett Publishers, 2005; 525–39. [Google Scholar]
- 5. Hemminki A, Alvarez RD. Adenoviruses in oncology: a viable option? BioDrugs 2002; 16: 77–87. [DOI] [PubMed] [Google Scholar]
- 6. Immonen A, Vapalahti M, Tyynela K et al. AdvHSV‐tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Mol Ther 2004; 10: 967–72. [DOI] [PubMed] [Google Scholar]
- 7. Xia ZJ, Chang JH, Zhang L et al. [Phase III randomized clinical trial of intratumoral injection of E1B gene‐deleted adenovirus (H101) combined with cisplatin‐based chemotherapy in treating squamous cell cancer of head and neck or esophagus.]. Ai Zheng 2004; 23: 1666–70. [PubMed] [Google Scholar]
- 8. Davydova J, Le L, Gavrokova T, Wang M, Krasnykh V, Yamamoto M. Infectivity‐enhanced cyclooxygenase‐2‐based conditionally replicative adenoviruses for esophageal adenocarcinoma. Cancer Res 2004; 64: 4319–27. [DOI] [PubMed] [Google Scholar]
- 9. Yamamoto M, Davydova J, Wang M et al. Infectivity enhanced, cyclooxygenase‐2 promoter‐based conditionally replicative adenovirus for pancreatic cancer. Gastroenterology 2003; 125: 1203–18. [DOI] [PubMed] [Google Scholar]
- 10. Mini E, Nobili S, Caciagli B, Landini I, Mazzei T Cellular pharmacology of gemcitabine. Ann Oncol 2006; 17 (Suppl 5): v7–12. [DOI] [PubMed] [Google Scholar]
- 11. Giovannetti E, Danesi R, Mey V, Nannizzi S, Pasqualetti G, Del Tacca M. In vitro studies on gemcitabine combinations with other antiblastics. Ann Oncol 2006; 17 (Suppl 5): v17–19. [DOI] [PubMed] [Google Scholar]
- 12. Eisenberg DP, Adusumilli PS, Hendershott KJ et al. 5‐Fluorouracil and gemcitabine potentiate the efficacy of oncolytic herpes viral gene therapy in the treatment of pancreatic cancer. J Gastrointest Surg 2005; 9: 1068–77; discussion 1077–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guichard S, Cussac D, Hennebelle I, Bugat R, Canal P. Sequence‐dependent activity of the irinotecan‐5FU combination in human colon‐cancer model HT‐29 in vitro and in vivo. Int J Cancer 1997; 73: 729–34. [DOI] [PubMed] [Google Scholar]
- 14. Wadler S, Mao X, Bajaj R, Hallam S, Schwartz EL. N‐(Phosphonacetyl)‐l‐aspartate synergistically enhances the cytotoxicity of 5‐fluorouracil/interferon‐alpha‐2a against human colon cancer cell lines. Mol Pharmacol 1993; 44: 1070–6. [PubMed] [Google Scholar]
- 15. Ramírez PJ, Vickers SM, Ono HA et al. Optimization of conditionally replicative adenovirus for pancreatic cancer and its evaluation in an orthotopic murine xenograft model. Am J Surg 2008; 195: 481–90. [DOI] [PubMed] [Google Scholar]
- 16. Yamamoto M, Alemany R, Adachi Y, Grizzle WE, Curiel DT. Characterization of the cyclooxygenase‐2 promoter in an adenoviral vector and its application for the mitigation of toxicity in suicide gene therapy of gastrointestinal cancers. Mol Ther 2001; 3: 385–94. [DOI] [PubMed] [Google Scholar]
- 17. Prichard MN, Shipman C Jr. Analysis of combinations of antiviral drugs and design of effective multidrug therapies. Antivir Ther 1996; 1: 9–20. [PubMed] [Google Scholar]
- 18. Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev 1995; 47: 331–85. [PubMed] [Google Scholar]
- 19. Prichard MN, Shipman C Jr. A three‐dimensional model to analyze drug–drug interactions. Antiviral Res 1990; 14: 181–205. [DOI] [PubMed] [Google Scholar]
- 20. Cardillo TM, Blumenthal R, Ying Z, Gold DV. Combined gemcitabine and radioimmunotherapy for the treatment of pancreatic cancer. Int J Cancer 2002; 97: 386–92. [DOI] [PubMed] [Google Scholar]
- 21. Van Moorsel CJ, Pinedo HM, Veerman G, Vermorken JB, Postmus PE, Peters GJ. Scheduling of gemcitabine and cisplatin in Lewis lung tumour bearing mice. Eur J Cancer 1999; 35: 808–14. [DOI] [PubMed] [Google Scholar]
- 22. Nieto J, Grossbard ML, Kozuch P. Metastatic pancreatic cancer 2008: is the glass less empty? Oncologist 2008; 13: 562–76. [DOI] [PubMed] [Google Scholar]
- 23. Stathopoulos GP, Androulakis N, Souglakos J, Stathopoulos J, Georgoulias V. Present treatment and future expectations in advanced pancreatic cancer. Anticancer Res 2008; 28: 1303–8. [PubMed] [Google Scholar]
- 24. Khuri FR, Nemunaitis J, Ganly I et al. A controlled trial of intratumoral ONYX‐015, a selectively‐replicating adenovirus, in combination with cisplatin and 5‐fluorouracil in patients with recurrent head and neck cancer [see comments]. Nat Med 2000; 6: 879–85. [DOI] [PubMed] [Google Scholar]