

Abstract
Coxiella burnetii is a Gram-negative, obligate intracellular bacterium and the causative agent of Q fever. Infections are usually acquired after inhalation of contaminated particles, where C. burnetii infects its cellular target cells, alveolar macrophages. Respiratory pathogens encounter the C-type lectin surfactant protein D (SP-D) during the course of natural infection. SP-D is a component of the innate immune response in the lungs and other mucosal surfaces. Many Gram-negative pulmonary pathogens interact with SP-D, which can cause aggregation, bactericidal effects and aid in bacterial clearance. Here we show that SP-D binds to C. burnetii in a calcium-dependent manner with no detectable bacterial aggregation or bactericidal effects. Since SP-D interactions with bacteria often alter macrophage interactions, it was determined that SP-D treatment resulted in a significant decrease in C. burnetii interactions to a mouse alveolar macrophage model cell line MH-S indicating SP-D causes a significant decrease in phagocytosis. The ability of SP-D to modulate macrophage activation by C. burnetii was tested and it was determined that SP-D does not alter the correlates measured for macrophage activation. Taken together these studies support those demonstrating limited activation of alveolar macrophages with C. burnetii and demonstrate interactions with SP-D participate in reduction of phagocyte attachment and phagocytosis.
Introduction
Coxiella burnetii is a Gram-negative zoonotic bacterial pathogen that is the etiologic agent of Q Fever in humans and coxiellosis in animals [1]. Q fever is usually acquired after contact with infected animals. While infection with C. burnetii often results in asymptomatic seroconversion, Q Fever can also present as an acute febrile illness, which can either resolve or result in chronic infection most often manifesting as endocarditis [2]. C. burnetii is subject to an phenomenon that occurs in many Gram-negative bacteria, the transition from “smooth” to “rough” LPS, which occurs after serial passage in a non-immunocompetent host [3]. This transition can occur via the deletion of O-antigen or core carbohydrate biosynthesis genes. C. burnetii RSA439 a clonal isolate that serves as a model for this phenomenon contains a deletion of a 26 kDa fragment of the genome, which renders it unable to produce O-antigen [4]. Smooth variants, designated phase I are virulent during animal infection, whereas, rough variants designated phase II are attenuated in immunocompetent animal infection. However, both phase I and phase II infect and multiply equally in a variety of cell types [5].
Infection by C. burnetii occurs via inhalation of aerosolized bacteria into the alveolar space within the lungs where C. burnetii proceeds to infect and survive within a variety of target cells, predominantly alveolar macrophages. Alveolar macrophages represent a unique lineage due to environmental conditions within the alveolar space [6]. Recent studies conducted with isolated alveolar macrophages from bronchial alveolar lavage have shown that virulent and avirulent strains of C. burnetii are able to infect alveolar macrophages and stimulate the production of TNF-α, IL-6, and IL-10 [7]. In addition to this alveolar macrophage study, studies using other monocytes or lineage macrophages have demonstrated that infection with either virulent or avirulent C. burnetii results in the production of TNF-α and IL-6 [5]. Therefore, C. burnetii infection triggers an early robust pro-inflammatory response.
Pulmonary pathogens are exposed to many components of the innate immune system that are usually involved in clearance of these invading organisms. For example, respiratory pathogens that reach the alveolar space encounter C-type lectins including surfactant proteins, often modeled by surfactant protein D (SP-D) [8]. SP-D is a hydrophilic protein that has been shown to be involved in both pulmonary surfactant homeostasis and the innate immune response [9]. Structurally SP-D is a tetramer composed of homotrimeric subunits that interact via their N-terminal domain, have a collagen-like domain and a C-terminal carbohydrate recognition domain (CRD) [10]. SP-D binds to various self and non-self ligands through its CRD in a calcium-dependent manner and then interacts with immune cells through its collagen-like domain to activate immune cells for clearance of the pathogen [10]. SP-D has been shown to directly interact with a number of bacteria including Pseudomonas aeruginosa, Streptococcus pneumoniae, Escherichia coli, and Mycobacterium tuberculosis leading to a number of physiologically relevant processes associated with bacterial clearance including agglutination, phagocytosis, and growth inhibition [11–15]. It is probable that C. burnetii interacts with SP-D in the lungs before invasion of alveolar macrophages, which may regulate these interactions. In this study, we show that SP-D binds directly to C. burnetii but does not lead to aggregation or cause cytotoxic affects. In addition, we show that the interaction between SP-D and C. burnetii modulates bacterial attachment and phagocytosis by alveolar macrophages, but that the level of macrophage activation is not altered.
Materials and Methods
Bacteria and cell culture
C. burnetii RSA439 and RSA493 were grown in either axenic ACCM-2 media or L-929 mouse fibroblasts (ATCC CCL-1) and genome equivalent were determined by real-time PCR, as previously described [16,17]. Briefly, DNA was purified from C. burnetii at indicated time points using Roche High Pure PCR Template Prep Kit and then 2 uL of purified DNA was used in 20 uL real time PCR reactions using ABI Sybergreen master mix (Applied Biosystems by Life Technologies, Foster City, CA, USA) and primers specific for com1 (CBU_1910). Prior to use, the C. burnetii were centrifuged and washed once in PBS to remove residual ACCM-2 or SP Buffer. Escherichia coli DH5α were grown in LB broth and quantified using OD600 or viable counts. Prior to use, E. coli were washed once with PBS to remove residual LB. RSA493 experiments were conducted under BSL3 containment according to Texas A&M University Office of Biosafety standard operating procedures. The murine alveolar macrophage cell line (MH-S) [18] were propagated in RPMI supplemented with 1X GlutaMAX (Gibco by Life Technologies, Foster City, CA, USA), 10% heat-inactivated fetal bovine serum, 18 mM sodium bicarbonate, 10 mM HEPES, 25 mM glucose, and 0.05 mM 2-mercaptoethanol at 37 degrees in 5% CO2. For C. burnetii infections MH-S cells were seeded at 1 x 105 per mL and allowed to adhere overnight.
SP-D binding, agglutination and antimicrobial assays
Binding to SP-D was determined using a previously described solution phase assay with some modifications [19]. Briefly, ~1 x 108 C. burnetii or E. coli were incubated with 1mg/mL recombinant human SP-D (R&D Systems Inc, Minneapolis, MN, USA) in PBS + 5 mM CaCl2, PBS + 5 mM CaCl2 + 0.1 M galactose or PBS + 5 mM CaCl2 +10 mM EDTA overnight at 37°C. Bacteria were washed three times with PBS via centrifugation and resuspended in PBS + 10X sample buffer. Samples were briefly boiled and then subjected to 12.5% SDS-PAGE and transferred onto nitrocelluloase membrane at 100 V for 1 hour. The membrane was blocked with 10% powdered milk in PBS and then probed with polyclonal anti-SP-D (My BioSource, San Diego, CA, USA), polyclonal anti-C. burnetii, or E. coli antibodies for 1 hour at 37°C. The membranes were washed 3 times with TBS + 0.05% Tween 20 and probed with HRP- goat anti-rabbit secondary antibody (BioRad, Hercules, CA, USA). The protein bands were visualized using chemiluminescence reagent (GE Healthcare Life Sciences, Pittsburgh, PA, USA).
Agglutination assays were performed by incubated ~1 x 108 C. burnetii or E. coli with 1mg/mL SP-D in PBS + 5mM CaCl2 in a cuvette that remained stationary for the duration of the assay. At selected time points the OD600 of the bacterial suspension was measured.
Growth inhibition assays were performed using 1 x 103 E. coli or 1 x 105 C. burnetii. E. coli were treated with 10mg/mL SP-D for 1 hr at 37°C and then subjected to viability counts. C. burnetii were treated with 10mg/mL SP-D for 12 hrs at 37°C. C. burnetii suspensions were added ACCM-2 and incubated at 37°C in a microaerophilic incubator at 2.5% O2 and 5% CO2. Every two days, 1 ml bacterial culture was removed and DNA was extracted and quantified via real-time PCR as previously described [16].
Attachment and Phagocytosis assay
MH-S cells were seeded at 1 x 105 cells per ml onto glass cover slips in 24 well tissue culture plates. MH-S cells were infected with CFSE-stained (Molecular Probes by Life Technologies, Foster City, CA, USA) C. burnetii at MOI 100 and incubated at 37°C. At the indicated time post infection, media was removed and MH-S cells were washed 3 times with RPMI. Coverslips were then fixed with 2% para-formaldehyde and the nuclei were stained with Hoescht (Molecular Probes by Life Technologies, Foster City, CA, USA). Infected MH-S cells were visualized using a Nikon-A1 confocal microscope with 450 and 488 nm filters using the 60X oil immersion objective. A minimum of 100 nuclei from MH-S cells were counted and CFSE C. burnetii were enumerated within those cells. The infectivity index was calculated as the number of infected cells (bound and internalized C. burnetii) divided by the total number of counted cells, multiplied by 100.
Cytokine production
MH-S cells were infected with C. burnetii at an MOI of 100 and at the indicated times post infection, RNA was extracted from MH-S cells using QiaShredder columns (Qiagen, Valencia, CA, USA) followed by RNEasy kit (Qiagen, Valencia, CA, USA) treatment according to manufacturer’s protocol. Residual DNA was removed using DNA-free kit (Ambion, by Life Technologies, Foster City, CA, USA). RNA was then quantified using a nanophotometer (Implen Inc, Westlake Village, CA, USA) and 500 ng was applied to each reverse transcription reaction using TaqMan Reverse Transcription kit (Applied Biosystems by Life Technologies, Foster City, CA, USA). After reverse transcription, 2.5 uL was used for real-time PCR using primers listed in Table 1. The fold change in expression for each gene was determined using the 2-ΔCt method and murine gapdh as a reference [20]. Average gadph Ct values among all samples did not deviate more than ± 1 Ct over the 3 independent experiments run with each sample in triplicated to verify reproducibility of results and to comply with MIQE standards [21]. Student t-tests were conducted to determine significance. Nitrite production in MH-S culture supernatant was determined using Griess reagents (Invitrogen by Life Technologies, Foster City, CA, USA) according to the manufacturer’s protocol.
Table 1. Real-time polymerase chain reaction primers used in this study.
| Primer | Sequence |
|---|---|
| Il1b F | GCA ACT GTT CCT GAA CTC AAC T |
| Il1b R | ATC TTT TGG GGT CCG TCA ACT |
| Il4 F | TTT GAA CGA GGT CAC AGG AG |
| Il4 R | TTC TTC GTT GCT GTG AGG AC |
| Il6 F | TAG TCC TTC CTA CCC CAA TTT CC |
| Il6 R | TTG GTC CTT AGC CAC TCC TTC |
| Il10 F | GCT CTT ACT GAC TGG CAT GAG |
| Il10 R | CGC AGC TCT AGG AGC ATG TG |
| Il12b F | CCA GAG ACA TGG AGT CAT AG |
| Il12b R | AGA TGT GAG TGG CTC AGA GT |
| Csf2 F | CAG CTT CTC AGA CTG CTG CT |
| Csf2 R | CTT GGT CCC TTT AAG GCA GA |
| Tnfa F | CCC TCA CAC TCA GAT CAT CTT CT |
| Tnfa R | GCT ACG ACG TGG GCT ACA G |
| Nos2 F | GTT CTC AGC CCA ACA ATA CAA GA |
| Nos2 R | GTG GAC GGG TCG ATG TCA C |
| Gapdh F | TGT GTC CGT CGT GGA TCT GA |
| Gapdh R | CCT GCT TCA CCA CCT TCT TGA |
Results
SP-D binds C. burnetii in a calcium-dependent manner, but does not cause aggregation or inhibition of growth
To determine if SP-D binds to C. burnetii, we used a solution phase assay. As shown in Fig 1, SP-D bound to C. burnetii. The addition of EDTA, a calcium chelator, abolished the interaction between C. burnetii and SP-D, which confirms that the binding was calcium dependent, a hallmark of most SP-D interactions (Fig 1). In addition, the interaction between SP-D and C. burnetii was inhibited in the presence of galactose, a monosaccharide with a high binding affinity for SP-D (Fig 1) [22]. The ability of SP-D to aggregate C. burnetii was determined using a spectrophotometry assay. SP-D was able to aggregate E. coli in agreement with previous results, however, SP-D was unable to aggregate C. burnetii (Fig 2a and 2b) [14]. To determine if SP-D was bactericidal towards C. burnetii we incubated the bacteria with SP-D and the ability to replicate was monitored in ACCM-2. SP-D did not significantly effect the viability of C. burnetii, however, incubation with SP-D did significantly decrease the viability of E. coli in agreement with previous results (Fig 3a and 3b) [14].
Fig 1. SP-D binds to C. burnetii in a calcium dependent fashion that can be inhibited by monosaccride.

SP-D was incubated with C. burnetii or E. coli with or without EDTA or galactose, in PBS + CaCl2 overnight at 37°C. Bacterial suspensions were then washed 3x with PBS by centrifugation. All samples were subjected to SDS-PAGE followed by blotting to detect SP-D that co-sedimented with the bacteria. SP-D was loaded as a positive control (left lane each blot labeled SP-D). Representative results are shown from 3 independent experiments.
Fig 2. SP-D does not cause aggregation of C. burnetii.

(A) E. coli or (B) C. burnetii were incubated with or without SP-D in PBS + CaCl2 in an undisturbed suspension for 3 or 4 hrs, respectively. The ability of SP-D to aggregate the bacteria was determined using spectrometry readings at OD600. The data are representative of 3 independent experiments ± standard deviation. Student t-tests compare data sets +/- SP-D ** p<0.005, *** p<0.0005.
Fig 3. SP-D is not bactericidal towards C. burnetii.

(A) E. coli were treated with or without SP-D for one hour at 37°C followed by viable counts on LB. Data displayed are mean ± standard error of three independent experiments. Student t-test compares SP-D to PBS treated E. coli. * p<0.05. (B) C. burnetii were treated with 0 (triangles), 5 (squares) or 10 mg/ml (squares) SP-D overnight and transferred to ACCM-2. Every 2 days genome equivalents (GE) were calculated by real-time PCR. Data displayed are mean ± standard deviation of GE/mL, representative of three independent experiments.
SP-D causes a decrease in C. burnetii interactions with macrophages
SP-D interactions are known to modulate attachment and uptake of many Gram- negative bacteria by a variety of cell types [11,23,24]. To determine whether SP-D was able to modulate the adherence and uptake of C. burnetii by MH-S cells, a murine alveolar macrophage cell line, the amount of adherent and internalize C. burnetii was determined using fluorescently labeled bacteria [18]. We used both C. burnetii grown in axenic ACCM-2 media and C. burnetii grown in L-929 cells to determine if growth in axenic media alters the physiological properties of the bacterium. As shown in Fig 4, SP-D treatment significantly reduced the C. burnetii infectivity index (bound and internalized C. burnetii) at 8 hrs of infection for both ACCM-2 and L-929 grown bacteria. Interestingly, we observed that SP-D was able to modulate interactions of L-929 grown C. burnetii at 4 hrs, but had no effect at this time for ACCM-2 grown C. burnetii (Fig 4). This suggests that the passage history (media vs cell line infection) of the C. burnetii may affect binding interactions with cells via surface receptor that interact with the ligand SP-D.
Fig 4. SP-D treatment results in a decrease in infectivity.
ACCM-2 or L-929 passaged C. burnetii were stained with CSFE and treated overnight with SP-D or the equivalent volume of buffer alone (PBS). MH-S cells were infected at MOI 100 for 4 or 8 hours and fixed. MH-S nuclei were stained with Hoescht, the number of infected cells and bacteria was quantified via confocal microscopy, and the infectivity index was calculated (bound and internalized C. burnetii). Data are displayed as mean ± standard deviation of infectivity from one representative of 5 experiments, n = 3. Student t-tests compare SP-D to PBS treated groups. *, p<0.05, ***, p<0.0005.
SP-D treatment does not result in modulation of macrophage activation
Our goals were two-fold in the comparison of C. burnetii infection of this alveolar macrophage line. First we wanted to determine if MH-S cells mimic the activation observed by human alveolar macrophages due to C. burnetii infection to assess their usefulness as model cell line and then to determine if SP-D alters this activation [7]. MH-S cells have been used as an in vitro model for Legionella pneumophila infection the most closely related bacterial species to C. burnetii as an alternative to hard to obtain primary alveolar macrophages [25,26]. Therefore, we determined the transcriptional responses of MH-S cells to both virulent phase I and avirulent phase II C. burnetii. The following genes were selected to evaluate macrophage activation: Il1b, Il4, Il6, Il10, Il12b, Cfs2, Nos2 and Tnfa. E. coli lipopolysaccharide (LPS) was used as a positive control for macrophage activation. As shown in Fig 5, MH-S infected with either phase I or phase II C. burnetii resulted in a significant induction of Il1b, Il6, Il12b, Csf2, Nos2 and Tnfa when compared to uninfected control. Infection with C. burnetii did not induce significant production of Il4 or Il10 (Fig 5). Although infection with C. burnetii increase the expression of Nos2 there was no production of nitrites by these cells, which agrees with previous results using human (Fig 6) [27]. Pre-incubation of phase II C. burnetii with SP-D prior to infection did not affect the transcriptional response of MH-S cells (Fig 6).
Fig 5. SP-D treatment does not alter C. burnetii stimulated transcriptional patterns in MH-S cells.

MH-S cells were infected with PBS or SP-D treated C. burnetii RSA439 (phase II), RSA493 (phase I) or treated with E. coli LPS for 0, 4, and 8 hours. RNA was extracted as described and applied to real-time PCR. Data represent the mean ± standard deviation of fold change in RNA expression of select genes compared to expression of host cell Gapdh, n = 3. RSA439 and LPS data are representative of three independent experiments, RSA493 data are representative of two independent experiments. Student t-tests compare each time and condition to its corresponding 0 time point, all RSA439 and RSA439 + SP-D were significantly up-regulated * p<0.05. There was no significant difference in expression between untreated and SP-D treated C. burnetii RSA439 as determined by student t-tests.
Fig 6. SP-D treatment does not alter C. burnetii stimulated nitric oxide secretion.
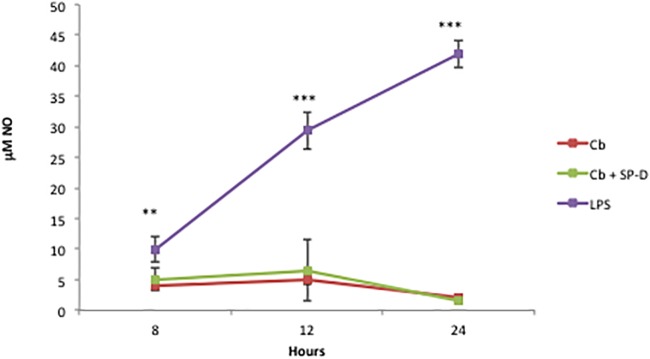
MH-S cells were infected with PBS or SP-D treated C. burnetii (Cb or Cb + SP-D respectively) or E. coli LPS (LPS) for 8, 12, and 24 hours at which point cell supernatants were removed and evaluated for nitrate production via the Greiss assay. The data represent the mean ± standard deviation of NO mM in cell culture supernatant n = 3 and are compared to uninfected and untreated controls using student t-tests ** p<0.005, *** p<0.0005.
Discussion
Our data demonstrate that C. burnetii interacts with SP-D in a calcium-dependent manner that can be inhibited by carbohydrate. SP-D has been previously shown to bind to the LPS of many diverse pathogenic Gram-negative bacteria such as Pseudomonas aeruginosa, uropathogenic E. coli, Klebsiella pneumoniae and Helicobacter pylori in a calcium-dependent manner that can be inhibited by carbohydrate [11,19,28,29]. Furthermore, it was determined C. burnetii interactions with SP-D did not cause bacterial agglutination or bactericidal effects. Most bacterial pathogens that bind SP-D are also agglutinated by SP-D including Streptococcus pneumoniae, Mycobacterium tuberculosis, H. pylori and approximately 50% of clinical Pseudomonas aeruginosa isolates [12,13,30,31]. However, other examples of SP-D binding without aggregation exist, such as with Chlamydia trachomatis [32]. Thus the interactions of C. burentii with SP-D do not result in aggregation similar to C. trachomatis. Bactericidal effects of SP-D have only been demonstrated for E. coli or rough LPS variants of other bacteria such as Bordetella pertussis [14,33]. Therefore, it is somewhat surprising that phase II C. burnetii, which is a rough variant is resistant to the bactericidal effects of SP-D.
Interactions with SP-D can also modulate adherence and phagocytosis; therefore, we investigated the effects of SP-D on C. burnetii interactions with MH-S cells. We observed that SP-D decreases the infectivity of C. burnetii to MH-S cells (bound and internalized bacteria). It is most likely that a decrease in adherence is responsible for this phenotype, but since this study did not distinguish between bound and internalized C. burnetii that remains to be determined. These interactions most closely resemble those observed for M. tuberculosis where SP-D decreases adherence and therefore phagocytosis [30]. Other outcomes of SP-D interaction include increased adherence to macrophages but decreases phagocytosis as for Pneumocystis carinii or increased phagocytosis as seen with C. trachomatis [24,32]. Alternatively, SP-D can cause increased uptake and killing of P. aerugionsa and K. pneumoniae [11,34].
Our data also indicate that C. burnetii infection drives MH-S alveolar macrophages towards classical activation and the resulting transcriptional patterns are not altered with SP-D treatment. Several other studies have evaluated the effects of SP-D on bacterial activation in macrophages. SP-D has no effect on the secretion of TNFα induced by P. aeruginosa [35]. On the other hand, SP-D coated K. pneumophila cause an increase in expression of IL-6, IL-10, and IL-12 [36]. LPS induces classical macrophage activation that results in resistance to intracellular pathogens and is characterized by secretion of IL-1β, TNF, IL-12, and IL-6 [27]. The activation patterns between C. burnetii and E. coli LPS were similar, although the magnitude of induction was different hence the conclusion of classic activation. Interestingly, we observed a greater induction of cytokine expression for phase I C. burnetii versus phase II C. burnetii at very early time points (4 and 8 hrs) after infection. This is in contrast to the cytokine release profiles observed after 24 hours of infection in human derived alveolar macrophages, where phase II causes the largest release of cytokines [7]. These differences may be due to the time points observed or due to analyzing expression profiles over cytokine release. The expression profiles after infection of L. pneumophila in MH-S cells and human derived alveolar macrophages were found to be similar [26]. We therefore conclude that MH-S cells represent a viable model for studying C. burnetii alveolar macrophage interactions.
In conclusion, SP-D binding to C. burnetii is calcium-dependent and can be inhibited by carbohydrate. SP-D decreases the interactions of C. burnetii to MH-S cells, these interactions are most similar to M. tuberculosis. Future experiments will help define the role of SP-D in the innate response to C. burnetii. For example, it will be important to determine if SP-D coated C. burnetii are viable once phagocytosed or if coated of SP-D causes intracellular killing as it does for the closely related L. pneumophila [37].
Acknowledgments
We would like to thanks Laura Hendrix and Robert Faris for helpful discussions. We would also like to thank Liz Case for critical review of this manuscript.
Data Availability
All relevant data are within the paper.
Funding Statement
Support was provided by the National Institutes of Health AI088430 awarded to JS; and by National Institutes of Health AI090142 awarded to JS [http://grants.nih.gov/grants/oer.htm]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. van Schaik EJ, Chen C, Mertens K, Weber MM, Samuel JE (2013) Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii . Nat Rev Micro advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maurin M, Raoult D (1999) Q Fever. Clinical Microbiology Reviews 12: 518–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moos A, Hackstadt T (1987) Comparative virulence of intra- and interstrain lipopolysaccharide variants of Coxiella burnetii in the guinea pig model. Infection and Immunity 55: 1144–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Narasaki C, Toman R (2012) Lipopolysaccharide of Coxiella burnetii In: Toman R, Heinzen RA, Samuel JE, Mege J-L, editors. Coxiella burnetii: Recent Advances and New Perspectives in Research of the Q Fever Bacterium: Springer Netherlands; pp. 65–90. [Google Scholar]
- 5. Howe D, Shannon JG, Winfree S, Dorward DW, Heinzen RA (2010) Coxiella burnetii Phase I and II Variants Replicate with Similar Kinetics in Degradative Phagolysosome-Like Compartments of Human Macrophages. Infection and Immunity 78: 3465–3474. 10.1128/IAI.00406-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu HM, Jin M, Marsh CB (2005) Toward functional proteomics of alveolar macrophages. American Journal of Physiology—Lung Cellular and Molecular Physiology 288: L585–L595. [DOI] [PubMed] [Google Scholar]
- 7. Graham JG, MacDonald LJ, Hussain SK, Sharma UM, Kurten RC, Voth DE (2013) Virulent Coxiella burnetii pathotypes productively infect primary human alveolar macrophages. Cellular Microbiology 15: 1012–1025. 10.1111/cmi.12096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sano H, Kuroki Y (2005) The lung collectins, SP-A and SP-D, modulate pulmonary innate immunity. Molecular Immunology 42: 279–287. [DOI] [PubMed] [Google Scholar]
- 9. Sano H, Kuronuma K, Kudo K, Mitsuzawa H, Sato M, Murakami S, et al. (2006) Regulation of inflammation and bacterial clearance by lung collectins. Respirology 11: S46–S50. [DOI] [PubMed] [Google Scholar]
- 10. Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, et al. (2006) Surfactant proteins SP-A and SP-D: Structure, function and receptors. Molecular Immunology 43: 1293–1315. [DOI] [PubMed] [Google Scholar]
- 11. Restrepo CI, Dong Q, Savov J, Mariencheck WI, Wright JR (1999) Surfactant Protein D Stimulates Phagocytosis of Pseudomonas aeruginosa by Alveolar Macrophages. American Journal of Respiratory Cell and Molecular Biology 21: 576–585. [DOI] [PubMed] [Google Scholar]
- 12. Griese M, Starosta V (2005) Agglutination of Pseudomonas aeruginosa by Surfactant Protein D. Pediatric Pulmonology 40: 378–384. [DOI] [PubMed] [Google Scholar]
- 13. Jounblat R, Kadioglu A, Iannelli F, Pozzi G, Eggleton P, Andrew PW (2004) Binding and Agglutination of Streptococcus pneumoniae by Human Surfactant Protein D (SP-D) Vary between Strains, but SP-D Fails To Enhance Killing by Neutrophils. Infection and Immunity 72: 709–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu H, Kuzmenko A, Wan S, Schaffer L, Weiss A, Fisher JH, et al. (2003) Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. The Journal of Clinical Investigation 111: 1589–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferguson JS, Voelker DR, Ufnar JA, Dawson AJ, Schlesinger LS (2002) Surfactant Protein D Inhibition of Human Macrophage Uptake of Mycobacterium tuberculosis Is Independent of Bacterial Agglutination. The Journal of Immunology 168: 1309–1314. [DOI] [PubMed] [Google Scholar]
- 16. Samuel JE, Hendrix LR (2009) Laboratory Maintenance of Coxiella burnetii Current Protocols in Microbiology: John Wiley & Sons, Inc. [DOI] [PubMed] [Google Scholar]
- 17. Omsland A, Beare PA, Hill J, Cockrell DC, Howe D, Hansen B, et al. (2011) Isolation from Animal Tissue and Genetic Transformation of Coxiella burnetii Are Facilitated by an Improved Axenic Growth Medium. Applied and Environmental Microbiology 77: 3720–3725. 10.1128/AEM.02826-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mbawuike IN, Herscowitz HB (1989) MH-S, a murine alveolar macrophage cell line: morphological, cytochemical, and functional characteristics. Journal of Leukocyte Biology 46: 119–127. [DOI] [PubMed] [Google Scholar]
- 19. Kurimura Y, Nishitani C, Ariki S, Saito A, Hasegawa Y, Takahashi M, et al. (2012) Surfactant Protein D Inhibits Adherence of Uropathogenic Escherichia coli to the Bladder Epithelial Cells and the Bacterium-induced Cytotoxicity: A POSSIBLE FUNCTION IN URINARY TRACT. Journal of Biological Chemistry 287: 39578–39588. 10.1074/jbc.M112.380287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Livak KJ, Schmittgen TD (2001) Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 21. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. (2009) The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clinical Chemistry 55: 611–622. 10.1373/clinchem.2008.112797 [DOI] [PubMed] [Google Scholar]
- 22. Persson A, Chang D, Crouch E (1990) Surfactant protein D is a divalent cation-dependent carbohydrate-binding protein. Journal of Biological Chemistry 265: 5755–5760. [PubMed] [Google Scholar]
- 23. Hogenkamp A, Herías MV, Tooten PCJ, Veldhuizen EJA, Haagsman HP (2007) Effects of surfactant protein D on growth, adhesion and epithelial invasion of intestinal Gram-negative bacteria. Molecular Immunology 44: 3517–3527. [DOI] [PubMed] [Google Scholar]
- 24. O'Riordan DM, Standing JE, Kwon KY, Chang D, Crouch EC, Limper AH (1995) Surfactant protein D interacts with Pneumocystis carinii and mediates organism adherence to alveolar macrophages. The Journal of Clinical Investigation 95: 2699–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yan L, Cirillo JD (2004) Infection of murine macrophage cell lines by Legionella pneumophila . FEMS Microbiology Letters 230: 147–152. [DOI] [PubMed] [Google Scholar]
- 26. Matsunaga K, Klein TW, Friedman H, Yamamoto Y (2001) Alveolar Macrophage Cell Line MH-S Is Valuable as an In Vitro Model for Legionella pneumophila Infection. American Journal of Respiratory Cell and Molecular Biology 24: 326–331. [DOI] [PubMed] [Google Scholar]
- 27. Benoit M, Barbarat B, Bernard A, Olive D, Mege JL (2008) Coxiella burnetii, the agent of Q fever, stimulates an atypical M2 activation program in human macrophages. European Journal of Immunology 38: 1065–1070. 10.1002/eji.200738067 [DOI] [PubMed] [Google Scholar]
- 28. Yokota S, Amano K, Nishitani C, Ariki S, Kuroki Y, Fujii N (2012) Implication of Antigenic Conversion of Helicobacter pylori Lipopolysaccharides That Involve Interaction with Surfactant Protein D. Infection and Immunity 80: 2956–2962. 10.1128/IAI.00345-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lim BL, Wang JY, Holmskov U, Hoppe HJ, Reid KBM (1994) Expression of the Carbohydrate Recognition Domain of Lung Surfactant Protein D and Demonstration of Its Binding to Lipopolysaccharides of Gram-Negative Bacteria. Biochemical and Biophysical Research Communications 202: 1674–1680. [DOI] [PubMed] [Google Scholar]
- 30. Ferguson JS, Voelker DR, McCormack FX, Schlesinger LS (1999) Surfactant Protein D Binds to Mycobacterium tuberculosis Bacilli and Lipoarabinomannan via Carbohydrate-Lectin Interactions Resulting in Reduced Phagocytosis of the Bacteria by Macrophages1. The Journal of Immunology 163: 312–321. [PubMed] [Google Scholar]
- 31. Khamri W, Moran AP, Worku ML, Karim QN, Walker MM, Annuk H, et al. (2005) Variations in Helicobacter pylori Lipopolysaccharide To Evade the Innate Immune Component Surfactant Protein D. Infection and Immunity 73: 7677–7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oberley RE, Ault KA, Neff TL, Khubchandani KR, Crouch EC, Snyder JM (2004) Surfactant proteins A and D enhance the phagocytosis of Chlamydia into THP-1 cells. Lung Cellular and Molecular Physiology 287: 296–306. [DOI] [PubMed] [Google Scholar]
- 33. Schaeffer LM, McCormack FX, Wu H, Weiss AA (2004) Interactions of Pulmonary Collectins with Bordetella bronchiseptica and Bordetella pertussis Lipopolysaccharide Elucidate the Structural Basis of Their Antimicrobial Activities. Infection and Immunity 72: 7124–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ofek I, Mesika A, Kalina M, Keisari Y, Podschun R, Sahly H, et al. (2001) Surfactant Protein D Enhances Phagocytosis and Killing of Unencapsulated Phase Variants of Klebsiella pneumoniae . Infection and Immunity 69: 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bufler P, Schmidt B, Schikor D, Bauernfeind A, Crouch EC, Griese H (2003) Surfactant Protein A and D Differently Regulate the Immune Response to Nonmucoid Pseudomonas aeruginosa and Its Lipopolysaccharide. American Journal of Respiratory Cell and Molecular Biology 28: 249–256. [DOI] [PubMed] [Google Scholar]
- 36. Keisari Y, Wang H, Mesika A, Matatov R, Nissimov L, Crouch E, et al. (2001) Surfactant protein D-coated Klebsiella pneumoniae stimulates cytokine production in mononuclear phagocytes. Journal of Leukocyte Biology 70: 135–141. [PubMed] [Google Scholar]
- 37. Sawada K, Ariki S, Kojima T, Saito A, Yamazoe M, Nishitani C, et al. (2010) Pulmonary Collectins Protect Macrophages against Pore-forming Activity of Legionella pneumophila and Suppress Its Intracellular Growth. Journal of Biological Chemistry 285: 8434–8443. 10.1074/jbc.M109.074765 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.