

Abstract
Background
Tumor necrosis factor (TNF)-α and matrix metalloproteinases (MMPs) are elevated in pleural fluids of tuberculous pleuritis (TBP) where pleural mesothelial cells (PMCs) conduct the first-line defense against Mycobacterium tuberculosis (MTB). However, the clinical implication of TNF-α and MMPs in TBP and the response of PMCs to MTB infection remain unclear.
Methods
We measured pleural fluid levels of TNF-α and MMPs in patients with TBP (n = 18) or heart failure (n = 18) as controls. Radiological scores for initial effusion amount and residual pleural fibrosis at 6-month follow-up were assessed. In vitro human PMC experiments were performed to assess the effect of heat-killed M. tuberculosis H37Ra (MTBRa) on the expression of TNF-α and MMPs.
Results
As compared with controls, the effusion levels of TNF-α, MMP-1 and MMP-9 were significantly higher and correlated positively with initial effusion amount in patients with TBP, while TNF-α and MMP-1, but not MMP-9, were positively associated with residual pleural fibrosis of TBP. Moreover, effusion levels of TNF-α had positive correlation with those of MMP-1 and MMP-9 in TBP. In cultured PMCs, MTBRa enhanced TLR2 and TLR4 expression, activated ERK signaling, and upregulated TNF-α mRNA and protein expression. Furthermore, knockdown of TLR2, but not TLR4, significantly inhibited ERK phosphorylation and TNF-α expression. Additionally, both MTBRa and TNF-α markedly induced MMP-1 and MMP-9 synthesis in human PMCs, and TNF-α neutralization substantially reduced the production of MMP-1, but not MMP-9, in response to MTBRa stimulation.
Conclusion
MTBRa activates TLR2/ERK signalings to induce TNF-α and elicit MMP-1 and MMP-9 in human PMCs, which are associated with effusion volume and pleural fibrosis and may contribute to pathogenesis of TBP. Further investigation of manipulation of TNF-α and MMP expression in pleural mesothelium may provide new insights into the mechanisms and rational treatment strategies for TBP.
Introduction
Tuberculosis (TB) remains a major global public health issue and continues to cause significant morbidity and mortality worldwide [1]. It has been well documented that tumor necrosis factor (TNF)-α and matrix metalloproteinases (MMPs) are essential in the pathogenesis of tuberculosis [2, 3]. Tuberculous infection commences when activated macrophages engulf Mycobacterium tuberculosis (MTB) and produce TNF-α and MMP-9 to recruit more macrophages, other immune cells and stromal cells such as fibroblasts, astrocytes or epithelial cells, to aggregate around the infected cells and form granulomas [4]. Within the granuloma, the cell-cell interactions develop to amplify the immune responses and enhance TNF-α, MMP-1 and MMP-9 expression in immune cells and stromal cells [3]. During reactivation, granulomas break down and extracellular matrix (ECM) were degraded by highly upregulated MMP-1, leading to MTB transmission, tissue remodeling and subsequent fibrosis [5].
Tuberculous pleuritis (TBP) is the most common form of extrapulmonary tuberculosis and a usual cause of pleural fibrosis [6]. The pleura space is lined by a metabolically active monolayer of pleural mesothelial cells (PMCs), which may serve as the first-line defense against invading microorganisms [7]. In response to pleural infection, PMCs not only provide physical barriers but also produce cytokines, chemokines, ECM and proteases that participate in the induction and resolution of inflammation and tissue repair [8]. The TNF-α level in the pleural fluid from patients with TBP are significantly higher than in those with pleural malignancy [9]. Moreover, patients with TBP who go on to develop pleural fibrosis have substantially higher TNF-α level in the pleural fluid than those who do not [9]. On the other hand, MMP-1 and MMP-9 are elevated in TBP as compared with transudative effusions [10], and MMP-9 is prominently expressed in the granulomas of tuberculous pleural tissues [11]. All these findings suggest that PMCs and its relationship with TNF-α and MMPs may contribute to the pathogenesis of TBP.
There have been studies on the expression of TNF-α and MMPs in monocytes and fibroblasts composing the pulmonary tuberculous granuloma [12, 13]. However, the role of PMCs in TBP and the regulation of TNF-α expression and MMP elaboration in PMCs upon MTB infection have never been investigated. We hypothesized that TNF-α can be induced by MTB in PMCs and may drive PMCs to elicit MMPs expression and secretion. Therefore, we explored the effect of heat-killed M. tuberculosis H37Ra on TNF-α expression and subsequent production of MMPs in human PMCs, the underlying mechanisms, and the clinical implications in TBP.
Materials and Methods
Patient Recruitment
Consecutive patients with pleural effusions of unknown causes admitted to Taipei Medical University Hospital who underwent thoracentesis and/or closed pleural biopsy were eligible for this study. TBP was diagnosed as previously described [14]. Patients with congestive heart failure (CHF) and a transudative pleural effusion were enrolled as controls [10]. Ethics approval (CRC-05-11-01) was obtained from the Institutional Review Board of Taipei Medical University (Taipei, Taiwan), and all patients gave written informed consent before entering the study.
Chest Radiograph Scoring
The posteroanterior chest radiographs (CXR) were performed at admission, on discharge and 6 months later, respectively, and were scored by two radiologists blind to any clinical information to determine (a) the largest linear width of pleural opacity and (b) pleural CXR score: the estimated overall percentage of pleural shadowing in the hemithorax. Residual pleural thickening (RPT) was defined as a lateral pleural thickening of ≥ 10 mm shown on CXR at the end of 6-month follow-up [15].
Reagents
Heat-killed M. tubuculosis H37Ra (MTBRa) obtained from Difco Lab (Detroit, Mich) was dissolved in phosphate-buffered saline (PBS) and used as a stimulant [16]. Recombinant human TNF-α were purchased from Pepro Tech EC (London, UK), respectively. Except for ERK (Cell Signaling Technology, Beverly, MA) and HRP conjugated anti-mouse and anti-rabbit IgG (Jackson Immunoresearch, USA), other antibodies were purchased from GeneTex (GeneTex Inc). SB203580, SP600125, PD98059, LY294002 and parthenolide were obtained from Calbiochem (San Diego, CA).
Human Pleural Mesothelial Cells (PMCs)
The cell culture of Met-5A human PMC line (#CRL-9444TM) was obtained from American Type Culture Collection (ATCC, Manassas, VA) and primary human PMCs were collected from the pleural fluids of patients with congestive heart failure. The ethics approval (IRB No.: CRC-05-11-01) for the collection and generation of the primary-cultured human PMCs was obtained from the Institutional Review Board of Taipei Medical University (Taipei, Taiwan), and the written informed consent was acquired from all patients recruited. The human pleural fluids were centrifuged and cells were grown in medium 199 containing 10% FBS at 37°C in the humidified incubator of 5% CO2. Mesothelial cells were used at passages three to six and were characterized by the cobblestone morphology, the presence of cytokeratin, and the absence of factor VIII, as described in our previous study [17].
Enzyme-linked Immunosorbent Assay (ELISA)
Pleural fluid samples from patients with TBP and CHF were collected as described in our previous report [18]. The commercially available ELISA kits were used to measure effusion levels of TNF-α, MMP-1 MMP-7, and MMP-9 (R & D System; Minneapolis, MN, USA) [18]. The levels of TNF-α in the supernatant of human PMCs after MTBRa stimulation were analyzed with ELISA methods as well.
Western Blot
The proteins were separated in denaturing sodium dodecyl sulfate (SDS) polyacrylamide gels and electrophoretically transferred onto nitrocellulose membranes. Blotting membranes were incubated with specific primary and HRP-conjugated secondary antibodies. Bound antibody was visualized by chemiluminescence and exposure of the membrane to light-sensitive film. The quantitative densitometric analysis was performed as previously described [19].
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from Met-5A cells using the TRIsure®reagent (Bioline, London, UK) and RNA (1 μg) was used for cDNA synthesis (Super Script On-Step RT-PCR system, GIBCOTM). The following primers were used: for TNF-α, sense 5′-AGCCCATGTT GTAGCAAACC-3′ and antisense 5′-CCAAAGTAGACCTGCCCAGA-3′; and for GAPDH, sense 5′-GCCGCCTGGTCACCAGGGCTG-3′ and antisense 5′-ATGGACTGTGGTCATGAGCCC-3′. The PCR was performed with the following conditions: 30 cycles of a 15-s denaturation step at 95°C, a 60-s annealing step at 48°C, and a 60-s extension step at 72°C to amplify TNF-α cDNA, followed by 25 cycles of a 15-s denaturation step at 95°C, a 30-s annealing step at 60°C, and a 60-s extension step at 72°C to amplify GAPDH cDNA. The quantitative analyses were performed as previously described [20].
Flow Cytometry
Met-5A cells were collected and washed with 2% FBS flow cytometry wash buffer. The resulting cells were sequentially stained with primary antibodies against control IgG, TLR2 and TLR4 for 30 min at room temperature, respectively. After additional washing steps, cells were incubated with the secondary antibody, FITC-conjugated goat anti-rabbits IgG, for another 30 min. The cells were washed and 10,000 events were analyzed by flow cytometry using FACSCalibur flow cytometer (Becton Dickinson).
RNA Interference
MeT-5A cells were transfected with a final concentration of 25 nmol of small interfering RNA (siRNA) against TLR2 or TLR4, or scrambled siRNA using the DharmaFECT® siRNA transfection reagent (Thermo Scientific, USA) according to the manufacturer’s instructions. After 48 h incubation with the transfection reagent, cells were stimulated with MTBRa for different times. Specific gene silencing was confirmed by Western blotting.
Statistical Analyses
Data analyses were performed with SigmaStat 3.5 (SYSTAT Software, San Jose, CA). Data were expressed as mean ± SD, median or frequency (%), as indicated. Comparisons of continuous data were made using an unpaired t test or Mann–Whitney U test between two groups, where appropriate, and one-way analysis of variance among three groups. Categorical variables between two groups were examined using χ2 method. Correlations were analyzed with the Spearman rank correlation coefficients. p < 0.05 was considered statistically significant.
Results
Patient Characteristics
Clinical data and pleural fluid characteristics of 18 patients with TBP and 18 control patients with CHF were shown in Table 1. No statistical differences among the two groups were found in terms of age, gender, duration of illness before management. The initial amount of effusion, measured as area of effusion shadowing on CXR, the effusion pH value, and the effusion glucose level were significantly lower in TBP than in CHF-related effusions. In contrast, as compared with the effusion of CHF patients, the leukocyte count, the levels of lactate dehydrogenase (LDH) and ADA were significantly higher in TBP. After 6-months anti-TB medications, 6 of the 18 patients with TBP developed significant RPT (≥10 mm).
Table 1. Clinical Data and Pleural Fluid characteristics*.
Definition of abbreviations: TBP, tuberculous pleuritis; CHF, congestive heart failure; LDH, lactate dehydrogenase; ADA, adenosine deaminase; RPT, residual pleural thickening.
| Characteristics | TBP | CHF | p Value |
|---|---|---|---|
| Subjects, n | 18 | 18 | |
| Mean age, years | 62 ± 24 | 72 ± 20 | 0.197 |
| Male, n (%) | 15 (83) | 10 (56) | 0.073 |
| Symptom onset to management, days | 12 ± 8 | 10 ± 4 | 0.130 |
| Area of effusion shadowing, † % | 47.8 ± 17.6 | 62.4 ± 20.4 | 0.038 |
| Pleural effusion | |||
| pH value | 7.33 ± 0.12 | 7.38 ± 0.04 | 0.019 |
| Glucose, mg/dL | 109 ± 44 | 169 ± 82 | 0.023 |
| LDH, IU/dL | 467 ± 160 | 141 ± 84 | 0.003 |
| Leukocyte count, cells/μL | 2304 ± 870 | 300 ± 53 | 0.028 |
| ADA, IU/L | 140 ± 60 | 25 ± 8 | < 0.001 |
| Area of pleural thickening at 6 months, † % | 4.5 ± 2.2 | - | |
| RPT ≥ 10 mm, n (%) | 6 (33) | - |
* Values are presented as mean ± SD unless otherwise specified.
† on posteroanterior chest radiography.
Pleural Fluid Levels of TNF-α and MMPs in TBP and Relationship with Radiological Scores
As shown in Fig 1A, 1B and 1C, the median levels of TNF-α, MMP-1, MMP-9, in pleural fluids from patients with TBP were significantly higher than in those with CHF (TNF-α: 77.4 vs. 4.2 pg/ml, p<0.001; MMP-1: 4.4 vs. 0.1 ng/ml, p<0.001; MMP-9: 1.1 vs. 0.2 ng/ml, p<0.001), respectively. By contrast, the effusion levels of MMP-7 were comparable between the two groups.
Fig 1. Effusion levels of TNF-α, MMPs and their relationship with effusion radiological scores in TBP patients.

Levels of (A), TNF-α, (B), MMP-1 and (C), MMP-9 in pleural effusion of patients with TBP (n = 18) or CHF (n = 18), and the correlation between effusion levels of (D), TNF-α, (E), MMP-1 or (F), MMP-9 and effusion shadowing on chest radiograph in patients with TBP. TNF, tumor necrosis factor; MMP, matrix metalloproteinase; TBP, tuberculous pleuritis; CHF, congestive heart failure.
In TBP, the pleural fluid levels of TNF-α, MMP-1 and MMP-9 all correlated positively with the effusion shadowing on CXR, though MMP-1 had a weak correlation (Fig 1D, 1E and 1F). Additionally, the levels of TNF-α and MMP-1, but not MMP-9, correlated positively with the pleural thickening on CXR after 6-months anti-TB medications (Fig 2A, 2B and 2C). Moreover, the effusion levels of TNF-α and MMP-1 were significantly higher in TBP patients with RPT ≥ 10 mm than those with RPT <10 mm, whereas the MMP-9 values were comparable between two groups (Fig 2D, 2E and 2F). These findings may indicate that all these effusion mediators, especially TNF-α, are essential in the pathogenesis of TBP, in which their cross relationship with PMCs may play an important role.
Fig 2. Relationship between effusion levels of TNF-α and MMPs and residual pleural thickening in TBP patients.
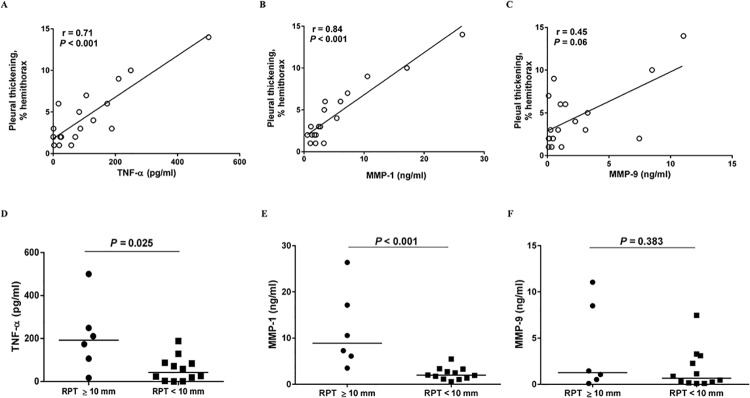
Correlation between pleural fluid levels of (A), TNF-α, (B), MMP-1 or (C), MMP-9 and pleural thickening on chest radiograph in patients with TBP (n = 18), and pleural fluid levels of (D), TNF-α, (E), MMP-1 and (F), MMP-9 between TBP patients with RPT ≥10 mm (n = 6) and those with RPT <10 mm (n = 12). TNF, tumor necrosis factor; MMP, matrix metalloproteinase; TBP, tuberculous pleuritis; RPT, residual pleural thickening.
MTBRa Upregulates TNF-α Production in Human PMCs
Furthermore, we explored the production of TNF-α in human PMCs upon exposure to MTBRa. As shown in Fig 3A, the addition of MTBRa (1 ng/ml) for 2 to 6 hours markedly stimulated TNF-α production by Met-5A cells. Besides, the TNF-α level in the supernatants increased substantially when Met-5A cells were stimulated with different concentrations of MTBRa for 6 h as compared with the resting condition (Fig 3B). In Fig 3C, MTBRa (1 ng/ml) significantly induced TNF-α protein expression in MeT-5A cells in a time-dependent manner. Moreover, TNF-α synthesis increased substantially after stimulation with different concentrations of MTBRa (0.1, 1, and 10 ng/ml) for 6 h (Fig 3D). Furthermore, MTBRa significantly increased the expression of TNF-α mRNA in MeT-5A cells, as compared with the resting condition (Fig 3E). Consistently, MTBRa concentration (1, 10, and 100 pg/ml)-dependently enhanced the TNF-α production in the primary-cultured human PMCs (Fig 3F). These findings suggest that, upon M. tuberculosis infection, human PMCs may substantially contribute to the increased production of TNF-α in TBP. Since the number of times primary human PMCs can be passage is limited and the cellular responses of MeT-5A cells were in parallel with those of primary PMCs, that has been validated in several previous studies [18, 19, 21], the following experiments on cellular signaling mediating MTBRa action were performed by using MeT-5A cells as a model of human pleural biology.
Fig 3. Expression and release of TNF-α in MTBRa-Stimulated Human PMCs.

Levels of TNF-α in the culture supernatants of MeT-5A cells stimulated by (A), MTBRa (1 ng/ml) for the indicated times or (B), MTBRa with various concentrations (0.1–10 ng/ml) for 6 h. TNF-α protein expression in MeT-5A cells treated with (C), MTBRa (1 ng/ml) for the indicated times or (D), MTBRa (0.1–10 ng/ml) for 6 h. (E), TNF-α mRNA expression in MeT-5A cells treated with MTBRa (0.1–10 ng/ml) for 4 h. (F), TNF-α protein expression in primary-cultured human PMCs stimulated with MTBRa (0.1–10 ng/ml) for 6 h. Supernatant TNF-α level was analyzed by ELISA and cell lysate TNF-α protein level was assessed by Western blot. TNF-α mRNA concentrations were analyzed by semiquantitative reverse transcriptase PCR and normalized with GAPDH mRNA. Data are shown as mean ± SD of three to five independent experiments. #p<0.05 and ###p<0.001 compared with the control or resting group. PMC, pleural mesothelial cell; MTBRa, heat-killed M. tuberculosis H37Ra; TNF, tumor necrosis factor; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Signal pathways mediating TNF-α expression in MTBRa-stimulated human PMCs
To clarify the signalings mediating TNF-α expression in MTBRa-stimulated human PMCs, we examined the several signaling pathways, including NF-κB, PI3K/AKT or MAPKs, by using their specific pharmacologic inhibitors. As shown in Fig 4A and 4B, both the protein and mRNA expression of TNF-α induced by MTBRa were markedly attenuated by pretreatment with MEK inhibitor (PD98059, 10 μM). Similar inhibitory effect by MEK inhibitor on MTBRa–induced TNF-α secretion was shown in Fig 4C. However, other inhibitors did not affect MTBRa–induced TNF-α protein synthesis. Consistently, MTBRa significantly induced phosphorylation of ERK 1/2 within 30 min, compared with the resting condition (Fig 4D). These results demonstrated MTBRa induced TNF-α expression via ERK-dependent pathway in human PMCs.
Fig 4. Signal pathways mediating TNF-α expression in MTBRa-stimulated human PMCs.

(A), MeT-5A cells were pretreated with vehicle, SP600125 (SP), SB20358 (SB), LY294002 (LY), Parthenolide (Par), PD98059 (PD), respectively, then stimulated with MTBRa (1 ng/ml) for 24 h. TNF-α protein expression was assessed by Western blot. (B), MeT-5A cells were pretreated with PD98059 (PD) (10 μM), followed by stimulation with MTBRa (1 ng/ml) for 4h. TNF-α mRNA concentrations were analyzed by RT-PCR. (C), MeT-5A cells were pretreated with PD98059 (PD) (5 and 10 μM) following stimulation with MTBRa (1 ng/ml) for 6 h, supernatant TNF-α level was analyzed by ELISA. (D), MeT-5A cells were treated with MTBRa (1 ng/ml) for the indicated times. ERK expression was analyzed by Western blotting with antibodies specific for phosphorylated or total proteins. ##p <0.01, ###p<0.001 compared with the resting group; **p<0.001, ***p<0.001 compared with the vehicle (DMSO) group. PMC, pleural mesothelial cell; TLR, toll-like receptor; MTBRa, heat-killed M. tuberculosis H37Ra; TNF, tumor necrosis factor; ERK, extracellular-signal-regulated kinase.
MTBRa Activates TLR2/ERK Signaling to Induce TNF-α Expression
M. tuberculosis is known to induce proinflammatory cytokines expression through cell surface receptors, especially Toll-like receptors (TLRs) [22–23]. Firstly, we examined the basal and MTBRa-induced expression of TLRs in MeT-5A. The flow cytometric analyses revealed that, compared with controls, MTBRa significantly upregulated the expression of both TLR2 and TLR4 on MeT-5A cell surface (Fig 5A).
Fig 5. TLR/ERK Signaling Activating TNF-α Expression in MTBRa-Stimulated Human PMCs.

(A), Flow cytometric analyses of expression of TLR2 or TLR4 (green) on cell surface of MeT-5A cells treated with MTBRa (1 ng/ml) for 3 h or left untreated (control). Isotype control IgG (red). Quantitative analysis of TLR2 or TLR4 expression was expressed as percentage of the total cells. A representative of three experiments is depicted. ###p<0.001 compared with the control group. MeT-5A cells were transfected with scrambled siRNA, TLR2 siRNA (25 nM) or TLR4 siRNA (25 nM). After 30 min, 3 and 6 h of stimulation with MTBRa, the cellular extracts were prepared and the protein amounts of (B), TLR2 and TLR4, (C) ERK, and (D), TNF-α were determined by Western blotting. The data represent four independent experiments. ##p <0.01, ###p<0.001 compared with the control (scrambled RNA) group; **p<0.01, ***p<0.001 compared with scrambled RNA-transfected MTBRa-treated group. PMC, pleural mesothelial cell; TLR, toll-like receptor; MTBRa, heat-killed M. tuberculosis H37Ra; ERK, extracellular-signal-regulated kinase; TNF, tumor necrosis factor.
Furthermore, we used specific siRNA for TLR2 or TLR4 to confirm whether and which TLRs are involved in MTBRa-induced TNF-α expression in human PMCs. The knockdown efficiencies of siRNAs were demonstrated as the marked reduction of TLR2/4 protein levels in human PMCs transfected with TLR2/4 siRNA, as compared with those transfected with a non-targeting scrambled siRNA (Fig 5B). Pretreatment with TLR2 siRNA significantly decreased ERK phosphorylation and TNF-α expression at 30 min and 6 hours after MTBRa administration, respectively (Fig 5C and 5D, upper panel). In contrast, although TLR4 expression increased significantly upon MTBRa stimulation (Fig 5A), knockdown of TLR4 did not abrogate MTBRa-induced ERK phosphorylation and TNF-α production in MeT-5A cells (Fig 5C and 5D, lower panel). These findings suggest that TLR2, but not TLR4, is the main receptor for MTBRa to mediate ERK activation and TNF-α expression in human PMCs.
Relationship between TNF-α and MMP-1/-9 in TBP
Since MTBRa could stimulate human PMCs to elaborate TNF-α, we further explored the relationship between TNF-α and MMPs in TBP. The levels of TNF-α were positively correlated with those of MMP-1 and MMP-9 (r = 0.78, p<0.001 and r = 0.70, p<0.01), respectively (Fig 6A and 6B). To determine whether this significant correlation was a causal or associational finding in TBP, further tests of the effect of MTBRa or TNF-α on the expression of MMP-1 and -9 in PMCs were mandatory.
Fig 6. Relationship between pleural fluid levels of TNF-α and MMP-1/-9 in TBP.

Correlation between TNF-α and (A), MMP-1 and (B), MMP-9 in TBP (n = 18). Effects of Heat-killed M. tuberculosis H37Ra or TNF-α on the expression of MMP-1 and MMP-9 in human pleural mesothelial cells. (C) and (D), MeT-5A cells were pretreated with vehicle (control), control IgG (12 μg/ml) or anti-TNF-α Ab (12 μg/ml) for 30 min, then stimulated with MTBRa for 24 hours. (E) and (F), MeT-5A cells were stimulated with TNF-α (5–20 ng/ml) for 24 h. The level of MMP-1 and MMP-9 was assessed by Western blot. Data are shown as mean ± SD of three to four independent experiments. ###p<0.001 compared with the resting group; ***p<0.001 compared with the MTBRa-treated group (control). TNF, tumor necrosis factor; MMP, matrix metalloproteinase; TBP, tuberculous pleuritis; MTBRa, heat-killed M. tuberculosis H37Ra.
Accordingly, we investigated whether MTBRa or TNF-α can induce MMP-1 and MMP-9 expression in PMCs. As shown in Fig 6C and 6D, MTBRa (10 ng/ml) significantly increased MMP-1 and MMP-9 expression in PMCs as compared with the resting condition. By contrast, pretreatment with anti-TNF-α Ab (12.5 μg/ml) significantly reduced MTBRa-induced MMP-1 expression. However, MMP-9 production is not suppressed by neutralization of TNF-α. Furthermore, administration of recombinant human TNF-α alone substantially increased MMP-1 and MMP-9 production in PMCs in a concentration-dependent manner (Fig 6E and 6F). Collectively, these data suggest that TNF-α is a potent stimulator of MMP-1 and MMP-9 production by PMCs, and that the mycobacterial induction of MMPs in TBP, especially MMP-1, may be mediated by PMC elaboration of TNF-α.
Discussion
The present clinical data demonstrated that effusion levels of TNF-α, MMP-1 and MMP-9 were significantly elevated in TBP. TNF-α, MMP-1 and MMP-9 were associated with pleural fluid volume, while TNF-α and MMP-1, but not MMP-9, correlated with area of pleural fibrosis. Moreover, substantial positive correlation existed between TNF-α and MMP-1/-9 in TBP. The in vitro data showed that MTBRa upregulated TNF-α expression in human PMCs through activation of TLR2/ERK signaling. Both MTBRa and TNF-α significantly induced the synthesis of MMP-1 and MMP-9, and neutralization of TNF-α substantially reduced MMP-1 production elicited by MTBRa. To the best of our knowledge, this is the first study to show that MTB could stimulate human PMCs to elaborate TNF-α and drives the expression of MMP-1 and MMP-9, which indicates the potential role of PMCs in the pathogenesis of TBP.
The clinical data in the present study demonstrate the elevated effusion levels of TNF-α, MMP-1 and MMP-9 in TBP and the association of these mediators with amount of pleural effusion and area of pleural fibrosis. These results highlighted the role of TNF-α, MMP-1 and MMP-9 in the pathogenesis of TBP. TNF-α is the key cytokine for early inflammatory responses during MTB infection [2]. However, in TBP, the production of TNF-α and the activated signalings in PMCs upon exposure to MTB has never been investigated. Our in vitro data revealed that MTBRa time- and concentration-dependently induced TNF-α production in human PMCs, implying that PMCs may initiate the immune response against MTB infection in the pleural space. Consequently, the mechanism by which PMCs identify MTB as a pathogen needs to be explored.
Previous studies have been demonstrated that TLR2 is the principal receptor for activation of macrophage or alveolar epithelial cells by MTB infection [24–25]. Moreover, MAPKs such as p38 and ERK are signaling mediators that play key roles in the elaboration of proinflammatory cytokines such as TNF-α and IL-10 [26]. In line with the earlier report [27], our data showed that the expression of both TLR2 and TLR4 on PMCs were substantially enhanced upon MTBRa stimulation, and that MTBRa further activated ERK signaling to elicit TNF-α production. However, only knockdown of TLR2, but not TLR4, could significantly reduce MTBRa-induced ERK phosphorylation and TNF-α expression, suggesting that MTBRa upregulates TNF-α expression from human PMCs via TLR2-ERK-dependent signaling pathway. To our knowledge, this is the first study to demonstrate the participation of TLR signalings in the inflammatory responses of human PMCs to MTB infection.
MMP-9 is abundantly increased and associated with granuloma formation in TBP [28], signifying its role in early inflammatory responses to MTB infection of the pleura. In keeping with the earlier report [29], our data showed that MMP-9 was elevated, and moreover, correlated substantially with the pleural fluid volume of TBP. This may be ascribed to the implication of MMP-9 in the proteolytic processes during granuloma formation, which may degrade basement membranes of endothelial cells or PMCs and facilitate fluid entry into the pleural space. On the other hand, MMP-1 is the main protease for tissue destruction and remodeling in tuberculosis [5]. Moreover, it was reported that patients with MMP-1 (-1607G) gene polymorphism are prone to develop lung fibrosis after pulmonary MTB infection [30]. In accordance with the previous studies [29–31], our results showed that the MMP-1 level in TBP was significantly associated with the degree of residual pleural fibrosis and was substantially higher in patients with RPT ≥ 10 mm than those without. In contrast, MMP-9 level did not relate to pleural fibrosis, suggesting the differential roles of MMP-1 and MMP-9 in the pathological processes of TBP. Collectively, these findings indicate that MMP-1 and MMP-9 are closely associated with clinical and radiological markers, are implicated in MTB-driven pleura matrix destruction, and may be key factors in the pathogenesis of TBP.
Accordingly, the mechanism regulating MMP expression in TBP needs to be investigated. Previous studies showed that MTB infection can induce MMP-1 and MMP-9 expressions in vivo and in vitro [12, 13]. In addition, TNF-α is responsible for driving the gene expression of MMP-1 and MMP-9 in MTB-infected airway epithelial cells and monocytes, respectively [12, 13]. Our data showed that either MTBRa or TNF-α alone could stimulate protein expression of MMP-1 and MMP-9 in human PMCs, and neutralization of TNF-α substantially reduced MMP-1, but not MMP-9, elicited by MTBRa, suggesting that TNF-α is critical for MMP-1 expression in human PMCs. The previous study ever demonstrated that TNF-α is a key autocrine and paracrine regulator of MMP-9 secretion by human monocytes [12]. As our results showed that MTBRa remarkably stimulated TNF-α expression and which significantly induced MMP-1 and MMP-9 production, TNF-α may serve as an autocrine trigger for MMP secretion by human PMCs in response to MTB infection and may play a critical role in the whole pathological process of TBP (Fig 7).
Fig 7. Schematic diagram of the activated signalings in human pleural mesothelial cells upon Mycobacterium tuberculosis infection.

MTBRa induces TNF-α expression in human pleural mesothelial cells through activation of TLR2/ERK signaling pathway, and increases MMP-1 and MMP-9 production (see text for further explanation). MTBRa, heat-killed M. tuberculosis H37Ra; TLR, toll-like receptor; ERK, extracellular-signal-regulated kinase; TNF, tumor necrosis factor; MMP, matrix metalloproteinase; Ⓟ, phosphorylate
However, as MTB-infected monocytes or macrophages are proposed to be the major source of TNF-α and MMP-9 in tuberculosis [12, 28], and there is emerging evidence that cell-cell interaction between immune cells and stromal cells are implicated the intercellular network to drive MMP production in tuberculosis [3]. Therefore, the current study is limited by lack of information on the relative contribution of PMCs and other immune cells to the production of TNF-α and MMPs in TBP. Further studies on the interaction between PMCs and other immune cells during the development of TBP are needed to verify this issue.
In conclusion, the present study demonstrated that MTBRa upregulates TNF-α expression through activation of TLR2/ERK signalings, and increases MMP-1 and MMP-9 production in human PMCs, which are associated with effusion volume and pleural fibrosis and may contribute to the pathogenesis of TBP. Further investigation of the manipulation of TNF-α and MMP expression in pleural mesothelium may provide new insights into the mechanisms and rational treatment strategies for TBP.
Data Availability
All relevant data are within the paper.
Funding Statement
This study was supported by grants from the National Science Council of Taiwan (NSC 101-2314-B-038–044-MY3 and NSC 102-2628-B-038– 009-MY3). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Dye C, Williams BG. (2010) The population dynamics and control of tuberculosis. Science 328: 856–861. 10.1126/science.1185449 [DOI] [PubMed] [Google Scholar]
- 2. Mootoo A, Stylianou E, Arias MA, Reljic R. (2009) TNF-alpha in tuberculosis: a cytokine with a split personality. Inflamm Allergy Drug Targets 8: 53–62. [DOI] [PubMed] [Google Scholar]
- 3. Elkington PT, Ugarte-Gil CA, Friedland JS. (2011) Matrix metalloproteinases in tuberculosis. Eur Respir J 38: 456–464. 10.1183/09031936.00015411 [DOI] [PubMed] [Google Scholar]
- 4. Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, Ramakrishnan L. (2010) Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science 327: 466–469. 10.1126/science.1179663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elkington P, Shiomi T, Breen R, Nuttall RK, Ugarte-Gil CA, Walker NF, et al. (2011) MMP-1 drives immunopathology in human tuberculosis and transgenic mice. J Clin Inves 121: 1827–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Porcel JM. (2009) Tuberculous pleural effusion. Lung 187: 263–270. 10.1007/s00408-009-9165-3 [DOI] [PubMed] [Google Scholar]
- 7. Mutsaers SE. (2004) The mesothelial cell. Int J Biochem Cell Biol 36: 9–16. [DOI] [PubMed] [Google Scholar]
- 8. Mutsaers SE. (2002) Mesothelial cells: their structure, function and role in serosal repair. Respirology 7: 171–191. [DOI] [PubMed] [Google Scholar]
- 9. Hua CC, Chang LC, Chen YC, Chang SC. (1999) Proinflammatory cytokines and fibrinolytic enzymes in tuberculous and malignant pleural effusions. Chest 116: 1292–1296. [DOI] [PubMed] [Google Scholar]
- 10. Hoheisel G, Sack U, Hui DS, Huse K, Chan KS, Chan KK, et al. (2001) Occurrence of matrix metalloproteinases and tissue inhibitors of metalloproteinases in tuberculous pleuritis. Tuberculosis 81: 203–209. [DOI] [PubMed] [Google Scholar]
- 11. Park KJ, Hwang SC, Sheen SS, Oh YJ, Han JH, Lee KB. (2005) Expression of matrix metalloproteinase-9 in pleural effusions of tuberculosis and lung cancer. Respiration 72: 166–175. [DOI] [PubMed] [Google Scholar]
- 12. Price NM, Gilman RH, Uddin J, Recavarren S, Friedland JS. (2003) Unopposed matrix metalloproteinase-9 expression in human tuberculous granuloma and the role of TNF-alpha-dependent monocyte networks. J Immunol 171: 5579–5586. [DOI] [PubMed] [Google Scholar]
- 13. O'Kane CM, Elkington PT, Jones MD, Caviedes L, Tovar M, Gilman RH, et al. (2010) STAT3, p38 MAPK, and NF-kappaB drive unopposed monocyte-dependent fibroblast MMP-1 secretion in tuberculosis. Am J Respir Cell Mol Biol 43: 465–474. 10.1165/rcmb.2009-0211OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Diacon AH, Van de Wal BW, Wyser C, Smedema JP, Bezuidenhout J, Bolliger CT, et al. (2003) Diagnostic tools in tuberculous pleurisy: a direct comparative study. Eur Respir J 22: 589–591. [DOI] [PubMed] [Google Scholar]
- 15. de Pablo A, Villena V, Echave-Sustaeta J, Encuentra AL. (1997) Are pleural fluid parameters related to the development of residual pleural thickening in tuberculosis? Chest 112: 1293–1297. [DOI] [PubMed] [Google Scholar]
- 16. Tchou-Wong KM, Tanabe O, Chi C, Yie TA, Rom WN. (1999) Activation of NF-kB in Mycobacterium tuberculosis-induced interleukin-2 receptor expression in mononuclear phagocytes. Am J Respir Crit Care Med 159: 1323–1329. [DOI] [PubMed] [Google Scholar]
- 17. Chen WL, Sheu JR, Hsiao CJ, Hsiao SH, Chung CL, Hsiao G. (2014) Histone deacetylase inhibitor impairs plasminogen activator inhibitor-1 expression via inhibiting TNF-α-activated MAPK/AP-1 signaling cascade. Biomed Res Int 2014: 231012 10.1155/2014/231012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chung CL, Chen CH, Sheu JR, Chen YC, Chang SC. (2005) Proinflammatory cytokines, transforming growth factor-beta1, and fibrinolytic enzymes in loculated and free-flowing pleural exudates. Chest 128: 690–697. [DOI] [PubMed] [Google Scholar]
- 19. Chung CL, Sheu JR, Liu HE, Chang SC, Chou YC, Chen WL, et al. (2009) Dynasore, a dynamin inhibitor, induces PAI-1 expression in MeT-5A human pleural mesothelial cells. Am J Respir Cell Mol Biol 40: 692–700. 10.1165/rcmb.2008-0087OC [DOI] [PubMed] [Google Scholar]
- 20. Chou YC, Sheu JR, Chung CL, Hsiao CJ, Hsueh PJ, Hsiao G. (2011) Hypertonicity-enhanced TNF-α release from activated human monocytic THP-1 cells requires ERK activation. Biochim Biophys Acta 1810: 475–484. 10.1016/j.bbagen.2011.01.004 [DOI] [PubMed] [Google Scholar]
- 21. Chung CL, Sheu JR, Chen WL, Chou YC, Hsiao CJ, Hsiao SH, et al. (2012) Histone Deacetylase Inhibitor CBHA Attenuates PAI-1 Expression in Human Pleural Mesothelial Cells. Am J Respir Cell Mol Biol 46: 437–445. 10.1165/rcmb.2011-0118OC [DOI] [PubMed] [Google Scholar]
- 22. Ryffel B, Fremond C, Jacobs M, Parida S, Botha T, Schnyder B, et al. (2005) Innate immunity to mycobacterial infection in mice: critical role for toll-like receptors. Tuberculosis 85: 395–405. [DOI] [PubMed] [Google Scholar]
- 23. Quesniaux VJ, Nicolle DM, Torres D, Kremer L, Guérardel Y, Nigou J, et al. (2004) Toll-like receptor 2 (TLR2)-dependent-positive and TLR2-independent-negative regulation of proinflammatory cytokines by mycobacterial lipomannans. J Immunol 172: 4425–4434. [DOI] [PubMed] [Google Scholar]
- 24. Underhill DM, Ozinsky A, Smith KD, Aderem A. (1999) Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A 96: 14459–14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Petrikin JE, Gaedigk R, Leeder JS, Truog WE. (2010) Selective Toll—like receptor expression in human fetal lung. Pediatr Res 68: 335–338. 10.1203/00006450-201011001-00656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Song CH, Lee JS, Lee SH, Lim K, Kim HJ, Park JK, et al. (2003) Role of mitogen-activated protein kinase pathways in the production of tumor necrosis factor-alpha, interleukin-10, and monocyte chemotactic protein-1 by Mycobacterium tuberculosis H37Ra-infected human monocytes. J Clin Immunol 23: 194–201. [DOI] [PubMed] [Google Scholar]
- 27. Means TK, Wang S, Lien E, Yoshimura A, Golenbock DT, Fenton MJ. (1999) Human toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J Immunol 163: 3920–3927. [PubMed] [Google Scholar]
- 28. Sheen P, O'Kane CM, Chaudhary K, Tovar M, Santillan C, Sosa J, et al. (2009) High MMP-9 activity characterizes pleural tuberculosis correlating with granuloma formation. Eur Respir J 33: 134–141. 10.1183/09031936.00127807 [DOI] [PubMed] [Google Scholar]
- 29. Iglesias D, Alegre J, Alemán C, Ruíz E, Soriano T, Armadans LI, et al. (2005) Metalloproteinases and tissue inhibitors of metalloproteinases in exudative pleural effusions. Eur Respir J 25: 104–109. [DOI] [PubMed] [Google Scholar]
- 30. Wang CH, Lin HC, Lin SM, Huang CD, Liu CY, Huang KH, et al. (2010) MMP-1 (-1607G) polymorphism as a risk factor for fibrosis after pulmonary tuberculosis in Taiwan. Int J Tuberc Lung Dis 14: 627–634. [PubMed] [Google Scholar]
- 31. Ong CW, Elkington PT, Friedland JS. (2014) Tuberculosis, pulmonary cavitation, and matrix metalloproteinases. Am J Respir Crit Care Med 190: 9–18. 10.1164/rccm.201311-2106PP [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.