

Abstract
Glioblastoma multiforme (GBM) is the most aggressive type of brain tumor, and the prognosis remains poor. Rearrangement of ROS1 gene, which was shown to have an oncogenic potential, was previously discovered in GBM cell lines. In this pilot study, we aimed to identify the incidence of ROS1 rearrangement in GBM patient tissues to explore novel biomarkers for therapeutic strategy. Formalin-fixed and paraffin-embedded (FFPE) tissue sections from 109 patients with GBM were screened for ROS1 rearrangement by anti-ROS immunohistochemistry (IHC) and ROS1 break-apart fluorescent in situ hybridization (FISH) assays. O6-methylguanine-DNA methyltransferase (MGMT) gene promoter methylation and Isocitrate dehydrogenase 1 (IDH1) mutation status were also assessed. All samples were interpreted by two experienced pathologists who were blinded to the clinical data. A total of 109 samples were collected and all samples were examined for ROS1 rearrangement by IHC and FISH assays, and none was found to harbor ROS1 rearrangement. MGMT gene methylation was found in 42 (39.2%) cases, and IDH1 mutation was found in 6 (5.5%) cases. In this study, ROS1 rearrangement was not identified in GBM patients, and thus it is difficult to classify ROS1 rearrangement as a novel molecular subset in GBM patients for now.
Introduction
Glioblastoma multiforme (GBM) is the most common type of primary brain tumors and the most aggressive subtype of high-grade gliomas. It is classified as grade IV in the World Health Organization classification of tumors of the central nervous system [1, 2]. The current standard treatment strategy for GBM patients consists of surgery followed by concurrent adjuvant radiotherapy in combination with temozolomide. However, still less than 5% of patients survive longer than 5 years after diagnosis. The median overall survival is only 14.6 months with radiotherapy plus temozolomide and 12.1 months with radiotherapy alone [3].
During recent years, comprehensive molecular profiling studies have broadened our knowledge of the underlying genetic and epigenetic aberrations that are associated with initiation and progression of GBM. The incidence of chromosomal rearrangements such as interchromosoal, intrachromosoal and intragenic rearrangements is significantly higher in GBM than in other tumor types [4]. For instance, epidermal growth factor receptor (EGFR) was shown to be amplified in 43% of adult GBM, and intragenic deletions in EGFR associated with amplification were commonly found in GBM [5, 6]. Besides chromosomal aberrations, frequent mutations of PTEN (29%), TP53 (29%), EGFR (20%), NF1 (9%), RB1 (8%), phosphatidylinositol-4, 5-bisphospate 3-kinase, catalytic subunit-α (PIK3CA; 7%), and IDH1 (5%) have been reported [7]. However, majority of drugs that target key signaling pathways of GBM have not proved a significant survival benefit in previous GBM patient cohorts [8].
Regarding epigenomic aberrations, the promoter methylation status of the O6-methylguanine-DNA methyltransferase (MGMT) gene has been suggested as a distinct subset of GBMs. Epigenetic MGMT gene silencing by promoter methylation is associated with loss of MGMT expression and diminished DNA repair activity, which leads to increased sensitivity to temozolomide, and thus longer survival [9–11].
ROS1 is a receptor tyrosine kinase of the insulin receptor family with constitutive kinase activity. ROS1 was found to be expressed in most glioblastoma cell lines, and characterization of ROS1 cDNA revealed a structural class of transmembrane protein kinase [12–14]. Rearrangement of ROS1 gene, involving ROS1 carboxy-terminal kinase fused to the amino-terminal portion of a protein called FIG (Fused in Glioblastoma) was also found in glioblastoma cell line [15]. When 10 different cell lines from all grades of astrocytomas were screened for this fusion transcript, FIG-ROS1 was found in two GBM cell lines (U118MG and U138MG).Interestingly, this FIG-ROS1 fusion transcript retained the active kinase domain with oncogenic potential. Recently, Stransky et al. reported the identification of CEP85L-ROS1 in a glioblastoma patient sample [16].
ROS1 rearrangement was also found in other solid tumors such as non-small-cell lung cancer (NSCLC) and cholangiocarcinoma [17–20]. To date, nine different ROS1 fusion partners have been identified in NSCLC, all of which are potentially targetable due to the same cytoplasmic portion of the ROS1 tyrosine kinase domain [21]. Due to the biologic similarity of ROS1 and ALK, several ALK inhibitors have been shown to inhibit ROS1 [22]. Preliminary data from a phase 1 trial of crizotinib in the ROS1-positive NSCLC expansion cohort demonstrated an overall response rate of 61% [23]. Therefore, identification of ROS1 rearrangement in GBM could offer a new therapeutic option to tackle this fatal disease.
In this study, we aimed to identify the incidence of ROS1 rearrangement and evaluate clinicopathological features associated with ROS1 rearrangement in GBM patients.
Methods
Patient characteristics
Patients with histologically proven GBM (World Health Organization grade 4) with newly diagnosed GBM were identified consecutively between January 2001 and December 2013. Of these, 109 patients who had available tissue samples for biomarker analyses were selected for this study. All patients provided written informed consent. Study protocol and informed consent forms were approved by the ethics committee and the institutional review board of Severance Hospital.
ROS1 Fluorescence in situ Hybridization
To identify ROS1 rearrangement, fluorescent in situ hybridization (FISH) assays were carried out on formalin-fixed and paraffin-embedded (FFPE) tumors by using a break-apart probe to ROS1 (Break-Apart Rearrangement Probe; Abbott Molecular) according to manufacturer’s instructions. At least 100 nuclei per case were evaluated. FISH positivity for ROS1 rearrangement was defined as > 15% of tumor cells with a split signal. FISH studies were interpreted by two experienced evaluators (SHK & JC) who were blinded to the clinical data.
ROS1 immunohistochemistry
For ROS1 immunohistochemistry (IHC) analysis, FFPE tissues sectioned at a thickness of 4uM and stained using Ventana automated immunostainer BenchMark XT. The slides were dried at 60°C for 1h and deparaffinized using EZ Prep at 75°C for 4 min. Cell conditioning was carried out using CC1 solution at 100°C for 64 min. ROS1 antibody (rabbit monoclonal, clone D4D6, Cell Signaling Technology) was diluted to 1:50, treated, and incubated at 37°C for 32 mins. Signals were detected using OptiView DAB IHC Detection Kit (Ventana Medical Systems). Counterstaining was carried out with Hematoxylin for 4 min at room temperature. Immunostained slides were scored with intensities of 0, 1+, 2+, and 3+ as follows: intensity 0 was defined as no detectable staining. Intensity 1+was defined as reactivity only detectableat high magnification (x 20–40 objective). More intense reactivity was divided into moderate(2+) and strong (3+) based on the ease of detectionat low magnification (x 4 objective).For interpretation of ROS1 expression, 3+ perinuclear staining was considered positive. IHC studies were interpreted by two experienced evaluators (SHK & JC) who were blinded to the clinical data.
MGMT gene promoter methylation assay
MGMT gene promoter methylation was assessed in patients with available tissue. Genomic DNA was extracted from 107 paraffin-embedded samples and the DNA methylation status of CpG islands at the MGMT promoter was assessed by methylation-specific polymerase chain reaction as previously described [11]. Unmethylated control DNA and methylated control DNA with bisulfite treatment (Qiagen, Germany) were used as negative and positive controls, respectively. Polymerase chain reaction products were separated on 8% polyacrylamide gels, stained with ethidium bromide, and examined under ultraviolet illumination by investigators blinded to clinical information.
Isocitrate dehydrogenase 1 (IDH1) sequencing analysis
IDH1 assay was performed according to the method described by previously [24]. DNA was isolated from each FFPE tumor tissue using a QIAamp DNA FFPE tissue kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. The quantity of isolated genomic DNA was evaluated using a NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). The detection of IDH1 mutation was performed by polymerase chain reaction (PCR) using forward and reverse primers that were designed to amplify exon 4 (codon R132) of the IDH1 gene. IDH1 forward primer (5’-ACC AAA TGG CAC CAT ACG A-3’) and reverse primer (5’-GCA AAA TCA CAT TAT TGC CAA C-3’) generated a 130-bp PCR product. PCR amplification was performed using an AmpliTaq Gold PCR Master Mix (Applied Biosystems, Foster City, CA, USA). The reaction mixture was subjected to an initial denaturation at 95 C for 10 min, followed by 35 cycles of amplification consisting of denaturation at 95 for 30s, annealing at 55 C for 30 s, and extension at 72 C for 60 s. After purification and sequencing amplification, the sequencing products were analyzed by a 3730XL DNA sequencer (Applied Biosystems).
Cell line
For positive control of ROS1 FISH assay, U118MG cell line (ATCC, Manassas, VA) maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS and the 1% antibiotics streptomycin at 37°C in a 5% CO2 environment, was used.
Statistical analysis
Progression-free survival (PFS) and overall survival (OS) were measured from the time of surgery to disease progression or death, or date of last follow-up visit, and were analyzed using the Kaplan-Meier method. Log-rank test was used to compare MGMT promoter methylation status with survival. Cox proportional hazards model was used to perform univariate and multivariate analyses. P values of ≤ 0.05 were considered statistically significant.
Results
Patient characteristics
A total of 109 patient samples with histologically proven GBM were available for analysis (Table 1, Fig 1). The median age of all patients was 56 years (range, 20–84 years), and there were 61 males (56%) and 48 females (48%). Total surgical resection was performed in 82 patients (75%), partial resection in 20 patients (18%) and biopsy was performed in 7 patients (7%). With the median follow-up period of 24 months, the median OS was 21.0 months (95% CI, 17.6–24.4) and the median PFS was 11.0 months (95% CI, 9.0–12.9) (Fig 2A and 2B).
Table 1. Clinicopathological characteristics of patients (n = 109).
| Characteristics | No (%) |
|---|---|
| Age, years | |
| Median | 56 |
| Range | 20–84 |
| Sex | |
| Male | 61 (56) |
| Female | 48 (44) |
| Surgery | |
| Biopsy | 7 (6.5) |
| Partial resection | 20 (18.3) |
| Total resection | 82 (75.2) |
| ROS1 rearrangement | |
| Yes | 0 (0) |
| No | 109 (100) |
| MGMT promoter status | |
| Methylated | 42 (38.5) |
| Unmethylated | 65 (59.6) |
| Unknown (invalid, indeterminate) | 2 (1.9) |
| IDH1 mutation | |
| Mutated (R132H) | 6 (5.5) |
| Wild type | 103 (94.5) |
| Overall survival (months) | 21 |
| 95% CI | 17.6–24.4 |
| Progression-free survival (months) | 11 |
| 95% CI | 9.0–12.9 |
Abbreviations: MGMT, O6-methylguanine-DNA methyltransferase; IDH1, Isocitrate dehydrogenase 1
Fig 1. Classical histologic findings of glioblastoma showing prominent pseudopalisading necrosis are seen (hematoxylin and eosin x 200).

Fig 2. Kaplan-Meier curve of (A) overall survival and (B) progression-free survival of all patients.

Analysis of ROS1 rearrangement
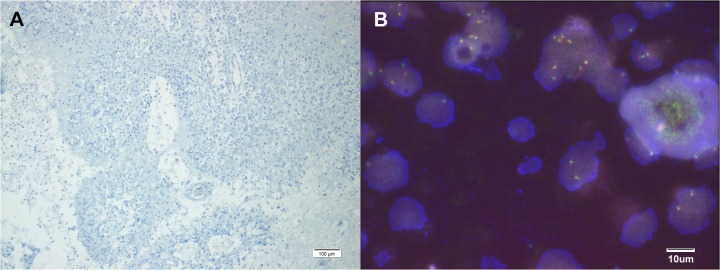
FISH analysis of U118MG showed ROS1 rearrangement (Fig 3). We performed IHC in 109 GBM patient samples and there was no positive staining for ROS1 (Fig 4A). Next, we tested with ROS1 FISH break-apart probes, but none met the criteria to be considered FISH-positive. No sample had separation of 5' (green) and 3' (red) signals, and no sample had isolated 3' (red) signals detected (Fig 4B).
Fig 3. ROS1 break-apart fluorescent in situ hybridization shown in U118MG cells.

Fig 4. (A) Immunohistochemical staining of ROS1 showing negative immunoreactivity (B) ROS1 break-apart fluorescent in situ hybridization showing negativity for ROS1 rearrangement.
Analysis of MGMT methylation and IDH1 mutation
Among 107 patients whose MGMT methylation status was available, 42 (38.5%) patients had methylated MGMT promoter and 65 (59.6%) patients had unmethylated MGMT promoter. The methylation status could not be determined in the remaining 2 (1.9%) patients (S1 Fig). IDH1 mutation was found in 6 (5.5%) patients, all of whom had point mutations affecting codon 132 of the IDH1, located on chromosome locus 2q33. This mutation resulted in arginine to histidine substitution (R132H mutation) in all four samples (S2 Fig).
Prognostic factors of survival
We analyzed univariate and multivariate analysis to identify prognostic factors of OS. Age, sex, extent of resection and MGMT gene promoter methylation status were included in analysis. Univariate analysis revealed that age, extent of resection, and MGMT gene promoter methylation status were significant prognostic factors. Multivariate analysis revealed that age and MGMT gene promoter methylation status were independent prognostic factors for OS (Table 2).
Table 2. Univariate and multivariate analyses of prognostic factor of overall survival.
| Univariate analysis | Multivariate analysis | |||||
|---|---|---|---|---|---|---|
| Overall survival | Overall survival | |||||
| Variable | No. of patients | Median, month (95% CI) | HR (95% CI) | P | HR (95% CI) | P |
| Age (y) | ||||||
| ≤50 | 42 | 23 (17–28) | 0.033 | 0.005 | ||
| >50 | 67 | 17 (14–21) | 1.54 (1.04–2.31) | 1.71 (1.14–2.58) | ||
| Sex | ||||||
| M | 61 | 19 (15–22) | 0.988 | - | ||
| F | 48 | 17 (11–22) | 0.99 (0.68–1.47) | - | ||
| Extent of resection | ||||||
| Total resection | 82 | 21 (16–25) | 0.04 | 0.022 | ||
| Partial + biopsy | 27 | 15 (12–17) | 1.45 (1.04–2.32) | 1.76 (1.09–2.86) | ||
| MGMT gene | ||||||
| Methylated | 42 | 24 (19–28) | 0.031 | 0.009 | ||
| Unmethylated | 65 | 17 (15–18) | 1.55 (1.04–2.32) | 1.72 (1.14–2.59) | ||
| Unknown | 2 | 5 (4–7) | - | - | ||
Discussion
In this study, we screened ROS1 rearrangement in GBM patients by both FISH and IHC for the first time, and reported that ROS1 rearrangement was not discovered in this GBM cohort. To date, ROS1 rearrangement has been only identified in GBM cell lines and it is unclear if other fusion variants exist in clinical samples.
Treatment of patients with GBM evolved slowly in the last decades. Although genetic and epigenetic alterations have been found in GBM, drugs that specifically target signaling pathways such as receptor tyrosine kinase have not proved a significant benefit in survival in unselected GBM patient cohorts [25]. Bevacizumab, an angiogenesis inhibitor, showed in two recent randomized phase III trials to bring about 3 to 4month prolongation of progression-free survival, without significant effect on overall survival [26, 27]. Therefore, efforts to find genetic alterations that drive gliomagenesis and identify molecularly defined patient subgroups for targeted therapies are imperative.
The discovery and characterization of ROS1 rearrangement in solid tumors have raised significant clinical interest because small molecule inhibitors may be effective to these tumors. Currently, 9 fusion partners to ROS1 have been identified (FIG, CCDC6, CD74, EZR, KDELR2, LRIG3, SLC34A2, SDC4, TPM3) all of which retain the ROS1 cytoplasmic kinase domain. The oncogenic ROS1 gene fusion in lung adenocarcinomas, which is identified in up to 3.4% of patients, has expanded the list of themolecular subsets of lung cancers [21–23, 28–30]. Since ALK and ROS1 share an approximately 49% amino acid sequence in the kinase domain, ALK inhibitors have been proved to be effective in inhibiting ROS1 activity [22]. Currently, there are ongoing clinical trials of drugs targeting ROS1 for non-small cell lung cancer patients with ROS1 rearrangement.
For cholangiocarcinoma (CCA), only FIG-ROS1 fusion transcript has been identified so far. Gu et al. found out the presence of ROS1 rearrangement in 8.7% of CCA patients and demonstrated inhibition of growth by ALK inhibitor in ROS1 rearranged CCA cells. These results suggest that ROS1 rearrangement in CCA is a promising druggable target with considerable incidence [20]. ROS1 rearrangement was also found in gastric cancer patients where IHC analysis revealed 23 (4.6%) positive cases among which 3 (0.6%) were FISH positive [31].
In our study, we used both FISH and IHC analyses for screening because RT-PCR method can only detect known fusion variants. For screening methods, FISH and reverse transcriptase-polymerase chain reaction (RT-PCR) have been used more commonly, although they are time consuming, costly, and not suitable for rapid screening. Immunohistochemical analyses using an anti-ROS1 rabbit monoclonal antibody (D4D6) have recently shown to accurately identify ROS1-rearranged cancers showing 100% (8/8) sensitivity and 100% (138/138) specificity when compared with break-apart FISH [28]. To date, only FIG-ROS1 has been identified in GBM cell lines and it is unclear whether ROS1 fusion variants with oncogenic activity exist in clinical samples.
The limitation of this study arises from a relatively small sample size. Reflecting upon the incidences of ROS1 rearrangements found in other tumor types, it is a rare phenomenon that requires large-scaled screening efforts. Moreover, there may be a possibility of false-negative IHC test results due to a low level of the expressed ROS1 fusion transcripts.
Although ROS1 rearrangement was not identified in our study cohort, it is notable that the initial discovery of ROS1 rearrangement in NSCLC was based on the identification in 1 cell line [17]. It is uncertain that ROS1 rearrangement may represent a potentialnew therapeutic opportunity for now, but biomarker discovery efforts should be continued to develop molecular tumor classification and to improve outcome and management of patients with GBM.
Supporting Information
(TIF)
(TIF)
Acknowledgments
We deeply appreciate Cell Signaling Technology for providing ROS1 immunohistochemistry antibody and Abbott Molecular for providing ROS1 FISH probe and positive control image.
Data Availability
Data cannot be made publicly available because the ethics committee of Severance Hospital Institutional Review Board has restrictions on releasing personal information. However this information can be released upon request via contacting Sun Min Lim (sunmin83@gmail.com).
Funding Statement
This work was funded by the Novartis Pharmaceuticals and a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI12C1186 to BCC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Jinyoung Sohn is employed by JEUK Co., Ltd. John Schulz is employed by Abbott Molecular Diagnostics. JEUK Co., Ltd and Abbott Molecular Diagnostics provided support in the form of salaries for authors Jinyoung Sohn and John Schulz respectively, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, et al. (2007) The 2007 WHO Classification of Tumors of the Central Nervous System. Acta Neuropathol 114(2): 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, et al. (2014) Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Review Cancer 14: 92–104. 10.1038/nrc3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, et al. (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352: 987–996. [DOI] [PubMed] [Google Scholar]
- 4. Malhotra A, Lindberg M, Faust GG, Leibowitz ML, Clark RA, et al. (2013) Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements sprawned by homology-independent mechanisms. Genome Res 23: 762–776. 10.1101/gr.143677.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 17: 98–110. 10.1016/j.ccr.2009.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brennan CW, Verhakk RG, McKenna A, Campos B, Noushmehr H, et al. (2013) The somatic genomic landscape of glioblastoma. Cell 2013; 155: 462–477. 10.1016/j.cell.2013.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061–1068. 10.1038/nature07385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J (2013) Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nature Rev Clin Oncol 10: 14–26. [DOI] [PubMed] [Google Scholar]
- 9. Kim YS, Kim SH, Cho J, Kim JW, Chang JH, et al. (2012) MGMT gene promoter methylation as a potent prognostic factor in glioblastoma treated with temozolomide chemoradiotherapy: a single-institution study. Int J Radiat Oncol Biol Phys 84(3): 661–667. 10.1016/j.ijrobp.2011.12.086 [DOI] [PubMed] [Google Scholar]
- 10. Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG (1999) Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 59: 793–79. [PubMed] [Google Scholar]
- 11. Esteller M, Garcia Foncillas J, Andion E, Goodman SN, Hidalgo OF, et al. (2000) Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 343: 1350–1354. [DOI] [PubMed] [Google Scholar]
- 12. Birchmeier C, Sharma S, Wigler M (1987) Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc Natl Acad Sci USA 84:9270–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Birchmeier C, O’Neill K, Riggs M, Wigler M (1990) Characterization of ROS1 cDNA from a human glioblastoma cell line. Proc Natl Acad Sci USA 87: 4799–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharma S, Birchmeier C, Nikawa J, O’Neill K, Rodgers L, et al. (1989) Characterization of the ros1-gene products expressed in human glioblastoma cell lines. Oncogene Res 5(2): 91–100. [PubMed] [Google Scholar]
- 15. Charest A, Lane K, McMahon K, Park J, Preisinger E, Conroy H, et al. (2003) Fusion of FIG to the receptor tyrosine kinase ROS in a Glioblastoma with an interstitial del (6)(q21q21). Genes Chromosomes Cancer 37: 58–71 [DOI] [PubMed] [Google Scholar]
- 16. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun 2014; 5: 4846 10.1038/ncomms5846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rikova K, Gluo A, Zeng Q, Possemato A, Yu J, Haack H, et al. (2007) Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131: 1190–1203 [DOI] [PubMed] [Google Scholar]
- 18. Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, et al. (2012) ROS1 rearrangements define a unique molecular class of lung cancers. J ClinOncol 30:863–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saborowski A, Saborowski M, Davare MA, Druker BJ, Klimstra DS, et al. (2013) Mouse model of intrahepatic cholangiocarcinoma validates FIG-ROS as a potent fusion oncogene and therapeutic target. Proc Natl Acad Sci USA 110(48): 19513–8. 10.1073/pnas.1311707110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gu TL, Deng X, Huang F, Tucker M, Crosby K, et al. (2011) Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One 6; e15640 10.1371/journal.pone.0015640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chin LP, Soo RA, Soong R, Ou SH (2012) Targeting ROS1 with anaplastic lymphoma kinase inhibitors: a promising therapeutic strategy for a newly defined molecular subset of non-small-cell lung cancer. J Thorac Oncol 7(11): 1625–1630. 10.1097/JTO.0b013e31826baf83 [DOI] [PubMed] [Google Scholar]
- 22. Ou SH, Tan J, Yen Y, Soo RA (2012) ROS1 as a ‘druggable’ receptor tyrosine kinase: lessons learned from inhibiting the ALK pathway. Expert Rev Anticancer Ther 12(4): 447–456. 10.1586/era.12.17 [DOI] [PubMed] [Google Scholar]
- 23.Ou SH, Kim DW, Camidge DR. (2013) Crizotinib therapy for patients with advanced ROS1-rearranged non-small cell lung cancer. Presented at the 15th World Conference on Lung Cancer, Sydney, Australia; October 28.
- 24. Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN (2009) Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol 68: 1319–1325. 10.1097/NEN.0b013e3181c391be [DOI] [PubMed] [Google Scholar]
- 25. Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J (2013) Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nat Rev Clin Oncol 10: 14–26. 10.1038/nrclinonc.2012.204 [DOI] [PubMed] [Google Scholar]
- 26. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, et al. (2014) Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 370(8): 709–722. 10.1056/NEJMoa1308345 [DOI] [PubMed] [Google Scholar]
- 27. Gilbert MR, Dignamm JJ, Armstrong TS, Wefel JS, Blumenthal DT, et al. (2014) A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370(8): 699–708. 10.1056/NEJMoa1308573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Davies KD, Le AT, Theodoro MF, Skokan MC, Aisner DL, et al. (2012) Identifying andtargeting ROS1 gene fusions in non-small cell lungcancer. Clin Cancer Res 18:4570–4579. 10.1158/1078-0432.CCR-12-0550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rimkunas VM, Crosby KE, Li D, Hu Y, Kelly ME, et al. (2012) Analysis ofreceptor tyrosine kinase ROS1 positive tumors in nonsmall cell lung cancer: identification of a FIG-ROS1fusion. Clin Cancer Res 18:4449–4457. 10.1158/1078-0432.CCR-11-3351 [DOI] [PubMed] [Google Scholar]
- 30. Kim HR, Lim SM, Kim HJ, Hwang SK, Park JK, et al. (2013) The frequency and impact ofROS1rearrangement on clinical outcomes in never smokers with lung adenocarcinoma. Ann Oncol 24(9): 2364–2370. 10.1093/annonc/mdt220 [DOI] [PubMed] [Google Scholar]
- 31. Lee J, Lee SE, Kang SY, Do IG, Lee S, et al. (2013) Identification of ROS1 rearrangement in gastric adenocarcinoma. Cancer 119: 1627–1635. 10.1002/cncr.27967 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(TIF)
Data Availability Statement
Data cannot be made publicly available because the ethics committee of Severance Hospital Institutional Review Board has restrictions on releasing personal information. However this information can be released upon request via contacting Sun Min Lim (sunmin83@gmail.com).