

N-glycosylation is identified as a novel regulator of JAM-A function. Human JAM-A carries a single N-glycan at N185, which regulates the protein’s role in barrier function, migration, and leukocyte binding.
Abstract
Junctional adhesion molecule-A (JAM-A) is an adherens and tight junction protein expressed by endothelial and epithelial cells. JAM-A serves many roles and contributes to barrier function and cell migration and motility, and it also acts as a ligand for the leukocyte receptor LFA-1. JAM-A is reported to contain N-glycans, but the extent of this modification and its contribution to the protein’s functions are unknown. We show that human JAM-A contains a single N-glycan at N185 and that this residue is conserved across multiple mammalian species. A glycomutant lacking all N-glycans, N185Q, is able to reach the cell surface but exhibits decreased protein half-life compared with the wild- type protein. N-glycosylation of JAM-A is required for the protein’s ability to reinforce barrier function and contributes to Rap1 activity. We further show that glycosylation of N185 is required for JAM-A–mediated reduction of cell migration. Finally, we show that N-glycosylation of JAM-A regulates leukocyte adhesion and LFA-1 binding. These findings identify N-glycosylation as critical for JAM-A’s many functions.
INTRODUCTION
Junctional adhesion molecule-A (JAM-A) was originally described as a platelet receptor (Naik et al., 1995) and later as an adherens and tight junction protein (Martin-Padura et al., 1998). JAM-A belongs to the immunoglobulin (Ig) superfamily of cell adhesion receptors and is characterized by two extracellular N-terminal Ig-like domains, a transmembrane domain, and a short C-terminal cytoplasmic tail that terminates with a PDZ-binding domain (Severson and Parkos, 2009). The first extracellular Ig-like domain regulates cis- and trans-homophilic interactions (Severson et al., 2008; Monteiro et al., 2014), and the second Ig-like domain serves as a ligand for the leukocyte adhesion receptor LFA-1 (Shaw et al., 2004) and stabilizes the aforementioned homophilic interactions (Wojcikiewicz et al., 2009). The C-terminus of JAM-A interacts with numerous proteins including zona occludens-1, AF-6 (Ebnet et al., 2000), Par3 (Ebnet et al., 2001), and CD9 (Peddibhotla et al., 2013). JAM-A has been implicated in a number of cellular functions, including leukocyte diapedesis (Martin-Padura et al., 1998; Ostermann et al., 2002; Shaw et al., 2004), platelet activation (Naik et al., 1995), angiogenesis (Naik and Naik, 2006), and barrier function (Aurrand-Lions et al., 2001; Laukoetter et al., 2007), and is a receptor for reovirus (Barton et al., 2001). The protein has been implicated in a number of pathologies, including vascular diseases (Babinska et al., 2007; Schmitt et al., 2014), inflammatory bowel disease (Vetrano and Danese, 2009), and cancers of the breast (Naik et al., 2008; McSherry et al., 2011; Murakami et al., 2011), lung (Zhang et al., 2013), and pancreas (Fong et al., 2012).
JAM-A, like most immunoglobulin superfamily proteins, is predicted to carry N-glycans on its extracellular domain. Protein N-glycosylation is an enzyme-driven co/posttranslational modification by which a core saccharide is covalently attached to the amide residue of asparagine in an N-X-S/T/C motif (where X cannot be P; Stanley et al., 2009). Subsequent processing in the endoplasmic reticulum and Golgi complex generates an array of N-glycan structures, and thus numerous “glycoforms” of any given glycoprotein exist on an individual cell (Moremen et al., 2012; Breitling and Aebi, 2013; Scott et al., 2013). The importance of N-glycosylation in regulating the function of adhesion molecules such as E-cadherin (Pinho et al., 2011; Langer et al., 2012), PECAM-1 (Kitazume et al., 2010), VE-cadherin (Geyer et al., 1999), and ICAM-1 (Diamond et al., 1991; Scott et al., 2013; He et al., 2014) has been reported. No studies examined the extent to which human JAM-A is N-glycosylated or whether this modification is important for the protein’s function. In a proteome-wide screen of N-glycosylated proteins, it was suggested that human JAM-A is N-glycosylated at two positions, N185 and N191 (Chen et al., 2009). However, the corresponding sequon at N191 is –NPT- and is in conflict with the consensus motif, suggesting that this site is unlikely to carry an N-linked oligosaccharide. Intriguingly, N185 is the site of a somatic mutation in breast cancer reported in the COSMIC database (Forbes et al., 2008).
Our studies were undertaken to determine whether human JAM-A is N-glycosylated and whether this modification is required for JAM-A function. We show that human JAM-A carries a single N-glycan at N185. This modification is not required for protein expression or transport to the membrane. However, removal of the N-glycan abrogates the effect of JAM-A on barrier function, Rap1 activity, and collective and single-cell migration and prevents leukocyte binding and interaction with LFA-1. Collectively these data demonstrate that N-glycosylation is required for JAM-A function.
RESULTS
Human JAM-A contains one N-glycan
It was suggested that JAM-A carries N-glycans (Naik et al., 1995; Martin-Padura et al., 1998), but these studies failed to determine precisely how many N-glycans are on human JAM-A. It is well established that N-glycan site occupancy varies across species (Scott and Patel, 2013), so we first wanted to determine how many N-glycans are on human JAM-A, as well as JAM-A expressed by other vertebrates. As can be seen in Figure 1A, multispecies alignment demonstrates the potential for an N-glycan in the first Ig-like domain of rodent JAM-A that is absent in other vertebrates. A consensus N-glycan sequon –NSS- is found in the second Ig-like domain of all species analyzed and corresponds to N185 of human JAM-A. There are reports of a potential N-glycan on N191 of human JAM-A (Chen et al., 2009), but the corresponding sequon is –NPT-, which is in conflict with the consensus motif. Indeed, none of the species analyzed expresses a consensus N-glycan motif at the residues corresponding to N191 in humans, suggesting that this site is not likely to be N-glycosylated. To determine the extent to which human JAM-A is N-glycosylated, we cultured human umbilical vein endothelial cells (HUVECs), which are widely reported to express JAM-A (Martin-Padura et al., 1998; Naik and Naik, 2006), with or without tunicamycin, an inhibitor of N-glycosylation, for 36 h and assessed JAM-A expression by Western blot analysis. As seen in Figure 1B, tunicamycin treatment resulted in a lower–molecular weight (∼4 kDa) form of JAM-A, indicating that the protein contains N-linked sugars. We next wanted to determine how many N-glycans human JAM-A supported and performed site-directed mutagenesis of N185 and N191. CHO cells were transfected with empty vector pCDNA3.1 (EV), wild-type human JAM-A (wt), JAM-A point mutant with asparagine 185 mutated to glutamine (JAM-A N185Q), or JAM-A N191Q. As can be seen in Figure 1C, JAM-A N185Q ran at a reduced molecular weight as compared with wt JAM-A. This reduced molecular weight was identical to wt JAM-A protein produced in the presence of tunicamycin. On the other hand, the N191Q mutant migrated at the same molecular weight as wt JAM-A, indicating that there is no modification at this residue. These data demonstrate that human JAM-A carries a single N-glycan at N185.
FIGURE 1:
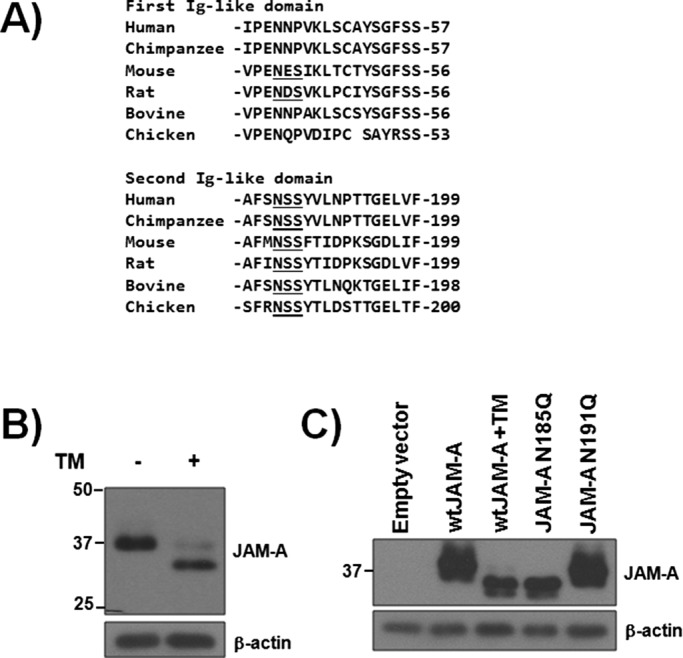
Human JAM-A contains a single N-glycan at N185. (A) Multispecies alignment of potential N-glycan sites. N-X-S/T consensus motifs are underlined. (B) Western blot analysis of JAM-A expression in HUVECs with or without tunicamycin (TM) treatment for 36 h. (C) Western blot analysis of CHO cells stably transfected with empty vector control or vectors expressing wild-type, N185Q, or N191Q human JAM-A. Some cells were pretreated with TM for 48 h. Data are representative of at least three separate experiments.
N-glycosylation regulates JAM-A dimerization and protein half-life
In some instances, N-glycosylation is required for protein folding and is critical for transport of proteins to the plasma membrane (Stanley et al., 2009). We next determined whether the N185Q mutant was able to reach the cell surface. CHO cells expressing wt and N185Q were incubated with the cell-impermeable sulfo-NHS-biotin tag, which allows for the detection of surface-exposed proteins when precipitated against streptavidin. As can be seen in Figure 2A, both wt and N185Q JAM-A were labeled with sulfo-NHS-biotin, indicating that N-glycosylation is not required for surface transport. As a separate readout, surface JAM-A expression was assessed by flow cytometric analysis. Both wt and N185Q were able to reach the cell surface in CHO cells, HUVECs, and MDA-MB-231 cells (Supplemental Figure S1).
FIGURE 2:

N-glycosylation is not required for JAM-A transport but regulates homodimerization and stability. (A) CHO cells expressing wt or N185Q human JAM-A were incubated with sulfo-NHS-biotin to label surface-exposed proteins. Biotinylated proteins were precipitated using streptavidin–agarose and detected by Western blot analysis. The data are representative of three separate experiments. (B) CHO cells expressing wt, N185Q, or 6163 JAM-A and some cells were incubated with BS3 to cross-link proteins. Cells were processed for Western blot analysis. (C) CHO cells expressing wt or N185Q human JAM-A were incubated with 100 μM cycloheximide (CHX) for up to 24 h. Lysates were subjected to Western blot analysis for JAM-A and β-actin and densitometric analysis conducted using ImageJ. Statistical comparisons were assessed between the samples at each time point using a two-tailed Student’s t test. *p < 0.05 between the samples from four separate experiments.
JAM-A forms homodimers, which are critical to the protein’s function (Severson et al., 2008; Monteiro et al., 2014). Moreover, the second Ig-like domain, where N185 resides, has been implicated in stabilizing these dimers (Wojcikiewicz et al., 2009). Thus we next determined whether N-glycosylation was important for JAM-A dimerization. As seen in Figure 2B, wt JAM-A dimerized in the presence of the cell-impermeable cross-linker BS3, whereas the previously described dimerization-deficient mutant, 6163 (Mandell et al., 2004), showed no evidence of dimerization. In the presence of BS3, N185Q was able to form homodimers but at a reduced level compared with wt protein. To confirm this finding, we tested the ability of CHO cells expressing EV or wt or N185Q JAM-A to adhere and spread on JAM-A/fc, as well as the ability of wt to adhere and spread on JAM-A/fc treated with PNGaseF to remove N-glycans. The wt protein, but not N185Q, was able to adhere and spread to JAM-A/fc (Supplemental Figure S2A) only when N-glycans were present on the chimeric protein (Supplemental Figure S2B). Finally, junctional accumulation between wt and N185Q was assessed by confocal microscopy. Junctional accumulation was seen only in cells expressing both wt and N185Q JAM-A (Supplemental Figure S1D). Collectively these data demonstrate that N-glycans regulate JAM-A homophilic interactions.
Protein dimerization and N-glycans can both increase a protein’s half-life by prolonging surface residency through interactions with other saccharides on the cell’s surface. To determine whether N-glycosylation was a regulator of JAM-A half-life, we incubated cells with the translation inhibitor cycloheximide and monitored JAM-A depletion within whole-cell lysates. As can be seen in Figure 2C, the decay of N185Q was significantly increased compared with wt protein. Collectively these data show that N-glycosylation is not required for JAM-A transport but is a critical regulator of dimerization and protein stability.
N-glycosylation is required for JAM-A’s effects on barrier function
Previously it was reported that expression of JAM-A in CHO cells reduces paracellular passage of fluorescein isothiocyanate–dextran (Martin-Padura et al., 1998; Aurrand-Lions et al., 2001). To determine whether glycosylation of JAM-A is required to support this function, we measured FITC-dextran flux across CHO cell monolayers expressing EV or wt or N185Q JAM-A. As seen in Figure 3A, expression of wt JAM-A reduced paracellular leak of FITC-dextran compared with EV control. However, N185Q JAM-A demonstrated a similar level of flux as EV cells, indicating that the N-glycan residue was critical for this increase in barrier function. As a separate readout of barrier function, cell impedance was determined. CHO cells were grown on gold-plated microtiter wells, and monolayer integrity was assessed using a real-time cell analyzer (RTCA; Wittchen et al., 2011). As can be seen in Figure 3B, expression of wt JAM-A caused a small but significant increase in cell index, an arbitrary index of impedance, above both EV and N185Q JAM-A after 18 h, indicating an increase in barrier function.
FIGURE 3:

N-glycosylation regulates JAM-A–mediated increases in barrier integrity. (A) CHO cells expressing empty vector or wt or N185Q JAM-A were grown on 0.4-μm Transwell inserts for 48 h. FITC-dextran (10 kDa) was added to the upper chamber and measured from the lower chamber after 2 h of incubation. Data shown are representative of three separate experiments run in triplicate. Statistical differences were determined by one-way analysis of variance (ANOVA) with Tukey’s posttest. *p < 0.05 vs. empty vector and N185Q. (B) The same cells as in A were grown on RTCA plates, and impedance was assessed for 30 h. Data shown are representative of four separate experiments run in quadruplicate. Statistical differences were determined by two-way ANOVA with Bonferroni posttest against empty vector. (C) CHO cells transfected with empty vector or wt or N185Q human JAM-A were assayed for Rap1 activity by pull down using GST-RalGDS-RBD. (D) Quantification. *p < 0.05 vs. EV; ***p < 0.01 vs. EV; #p < 0.05 vs. wt by one-way ANOVA with Tukey’s posttest from four separate experiments.
It has been reported that JAM-A mediates barrier function by controlling Rap1 activity. We next determined Rap1 activity in CHO cells expressing EV or wt or N185Q human JAM-A that had been confluent for 24 h. As seen in Figure 3, C and D, expression of wt JAM-A significantly increased Rap1 activity above EV levels. N185Q JAM-A increased Rap1 activity compared with EV levels but to a lesser extent than wt JAM-A. Collectively these data show that N-glycosylation of JAM-A is required for the protein’s ability to increase barrier function.
N-glycosylation controls JAM-A’s effects on cell migration
There are numerous reports that JAM-A expression controls cell spreading, single-cell motility, and collective cell migration, with the effects being cell-type specific (Bazzoni et al., 2005; Naik and Naik, 2006; McSherry et al., 2009; Severson et al., 2009). We first determined whether expression of wt or N185Q altered cell spreading. As seen in Figure 4, A and B, expression of wt or N185Q JAM-A had no effect on CHO cell spreading on fibronectin as assessed by total cell area or cell index by RTCA. Overexpression of wt caused an increase in HUVEC spreading but a decrease in MDA-MB-231 cell spreading (Supplemental Figure S3), highlighting the cell type–specific effects of the protein.
FIGURE 4:

N-glycosylation controls JAM-A–mediated cell motility. Cell spreading was assessed by determining total cell area (A) or measuring cell impedance using RTCA (B). The total number of cells used in A was as follows: EV, 47; wt, 49; N185Q, 46. RTCA data are representative of three separate experiments run in quadruplicate. CHO cells expressing empty vector or wt or N185 JAM-A were plated on FN-coated glass-bottom dishes, and single-cell motility was assessed as velocity (C) and persistence (directionality/time) (D) using time-lapse images. The data are averages ± SEM from three different experiments. The total number of cells was as follows: EV, 52; wt, 53; N185Q, 46. Statistical differences were determined by one-way ANOVA with Tukey’s posttest. *p < 0.05 vs. EV and N185Q.
We next determined whether wt or N185 altered cell motility. Expression of wt JAM-A caused a significant decrease in single-cell velocity of CHO cells (Figure 4C; Supplemental Videos 1–3), as well as of HUVECs and MDA-MB-231 cells (Supplemental Figure S4), as compared with EV and N185Q. However, there was no effect on persistence of migration (Figure 4D).
Because expression of wt JAM-A reduced single-cell motility and this effect was glycosylation dependent, we examined whether a similar phenomenon occurred in collective migration of cells. As seen in Figure 5, expression of wt JAM-A significantly decreased wound closure compared with EV and N185Q. There are reports that overexpression of JAM-A increases rates of directed migration in HUVEC but only on vitronectin (Naik and Naik, 2006). We next determined whether this effect was controlled by N-glycosylation of JAM-A. As previously reported, overexpression of wt JAM-A increased the rate of haptotaxis of HUVECs to vitronectin but not fibronectin (Supplemental Figure S5). In contrast, N185Q migrated at the same rate as EV control toward both matrix proteins. Taken together, these data demonstrate that N-glycosylation controls JAM-A–mediated cell motility and migration. There are reports that JAM-A regulates β1 integrin (CD29) expression in some lines (McSherry et al., 2009; Severson et al., 2009) but not others (Huang et al., 2006; Cera et al., 2009), which could explain some of the foregoing observations. Knockdown or overexpression of JAM-A with siRNA did not alter CD29 expression in CHO cells, HUVECs, or MDA-MB-231 cells (Supplemental Figure S6).
FIGURE 5:
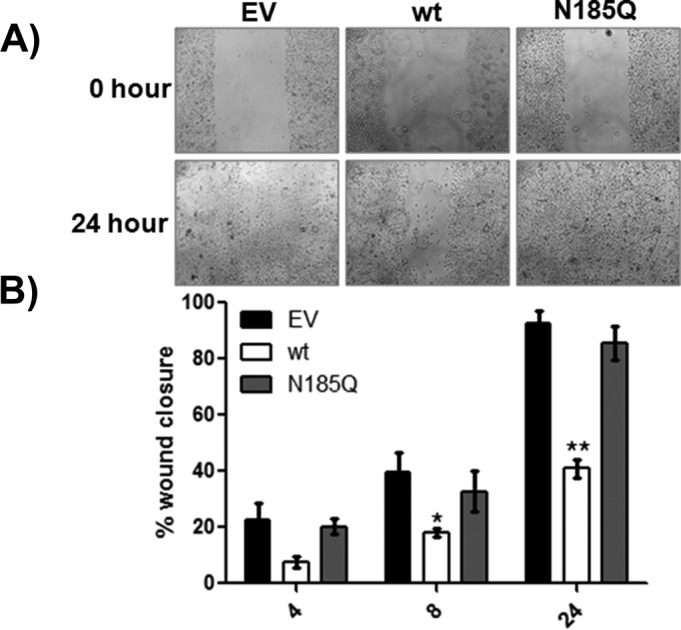
N-glycosylation controls JAM-A–mediated decrease in wound closure. CHO cells expressing EV or wt or N185Q JAM-A were grown to confluence on FN-coated glass-bottom dishes. A wound was made across the entire length of the glass insert using the tip of a 200-μl pipette tip, and closure was assessed over a 24-h period. (A) Representative micrographs at the beginning (0 h) and end (24 h) of an experiment. (B) Quantitation of wound closure. Five separate regions from each dish were monitored for closure. Data presented are the average ± SEM of the five regions from one dish and are representative of four separate experiments. Statistical differences were determined by one-way ANOVA with Tukey’s posttest. *p < 0.05 vs. EV; **p < 0.05 vs. EV and N185Q.
JAM-A N-glycosylation controls leukocyte binding
JAM-A supports leukocyte adhesion (Ostermann et al., 2002). Specifically, the second Ig-like domain, where N185 is located, interacts with LFA-1 (Fraemohs et al., 2004). As can be seen in Figure 6A, CHO cells expressing wt JAM-A supported significantly increased leukocyte adhesion compared with EV control cells. Strikingly, N185Q-expressing cells did not support leukocyte binding above EV control, suggesting that glycosylation of this residue is involved in LFA-1 binding.
FIGURE 6:
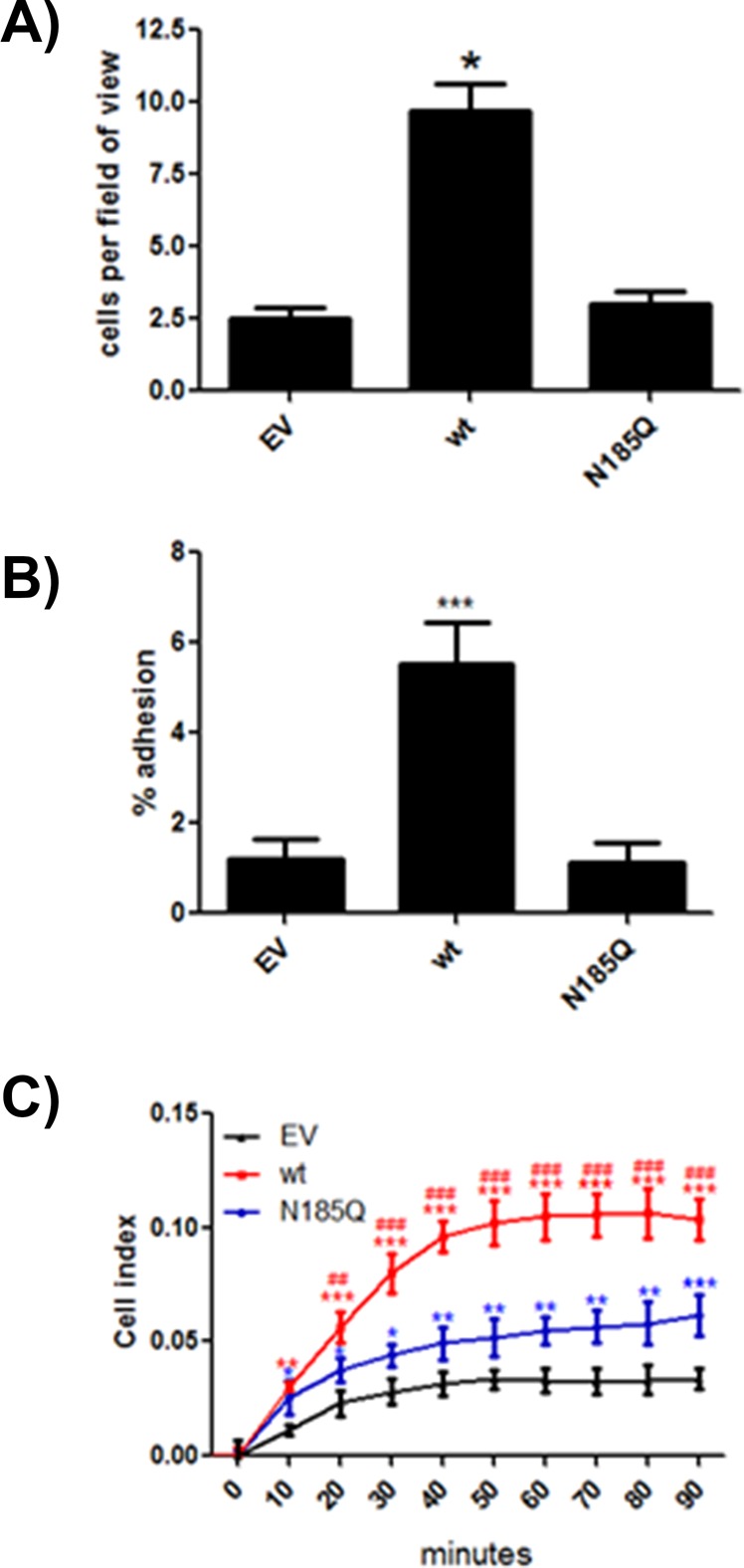
N-glycosylation of JAM-A is required for interactions with LFA-1. (A) Equal numbers of HL-60 cells were added to glass coverslips covered with confluent CHO cells expressing empty vector or wt or N185Q JAM-A in the presence of 100 μg/ml PMA for 30 min. Adherent HL-60 cells were counted after washing. Data are representative of three separate experiments run in triplicate. Statistical differences were determined by one-way ANOVA with Tukey’s posttest. *p < 0.05 vs. EV and N185Q. (B) CHO cells labeled with CellTracker Green and expressing empty vector or wt or N185Q human JAM-A were allowed to adhere to microtiter plates coated with LFA-1/fc chimera (20 μg/ml). After washing, adherent cells were assessed on a fluorometer. Data are representative of three separate experiments. *p < 0.05 vs. EV and N185Q. (C) CHO cells expressing empty vector or wt or N185Q JAM-A were allowed to adhere and spread on RTCA plates coated with LFA-1/fc chimera (20 μg/ml) for 90 min. Data are representative of two independent experiments run in quadruplicate. Statistical differences were assessed by two-way ANOVA with Bonferroni posttest against EV and N185Q. *p < 0.05, **p < 0.01, and ***p < 0.001 vs. EV. ##p < 0.05 and ###p < 0.01 vs. N185Q.
To confirm this interaction, we tested the ability of CHO cells with or without JAM-A proteins to bind to LFA-1. CHO cells will not bind to LFA-1/fc chimeras unless they express JAM-A (Fraemohs et al., 2004). As can be seen in Figure 6, B and C, cells expressing wt JAM-A demonstrated significantly more adhesion to LFA-1/fc than did EV cells. In contrast, cells expressing N185Q failed to adhere at levels above EV, indicating that N-glycosylation of JAM-A is required for LFA-1 binding. To confirm the specificity of LFA-1 binding, we assessed CHO cells expressing EV, JAM-A, or ICAM-1 for their ability to bind LFA-1/fc–coated beads. Cells expressing JAM-A and ICAM-1 bound significantly more beads than EV control (Supplemental Figure S7), confirming specificity of the ligand.
JAM-A glycosylation patterns are cell-type specific
Having confirmed that JAM-A contains N-glycans that regulate the protein’s function, and in light of both our findings and previously published data demonstrating that JAM-A’s effects are cell-type specific, we investigated whether the glycoprotein’s saccharide content varied across a panel of endothelial and epithelial cells, as well as when the protein was exogenously expressed, using lectin affinity pull down (lectin specificity outline in Table 1). To test for sialic acid, a common capping sugar on N-glycan structures, we incubated human cells lines with Sambucus nigra lectin (SNA), a lectin that recognizes α-2,6-linked sialic acid, which is added to cells via the enzyme ST6GAL1. CHO cells express low levels of ST6GAL1 and were thus also tested with Maackia amurensis lectin (MAA), a lectin that recognizes α-2,3 sialic acid, the predominant sialic acid structure in these cells (Lee et al., 1989; Xu et al., 2011). As seen in Figure 7, N-glycans from all cell lines tested contained bianternary (LCA), trianternary (PHA-E), and sialyated structures (SNA or MAA). Of interest, UEA-1, a lectin specific for fucose, interacted with JAM-A produced in epithelial but not endothelial cells or cell expressing exogenous JAM-A. These data demonstrate a conserved N-glycan profile of JAM-A, including the global presence of sialic acid and epithelial cell–specific presence of fucose.
TABLE 1:
Lectin specificity.
| Lectin | Specificity |
|---|---|
| Lens culinaris agglutinin (LCA) | Biantennary N-glycan |
| Phaseolus vulgaris erythroagglutinin (PHA-E) | Triantennary N-glycan |
| P. vulgaris leucoagglutinin (PHA-L) | Tetraantennary N-glycan |
| Ulex europaeus agglutinin I (UEA-1) | α-Linked fucose |
| S. nigra lectin (SNA) | α-2,6-Linked sialic acid |
| M. amurensis lectin II (MAA) | α-2,3-Linked sialic acid |
FIGURE 7:
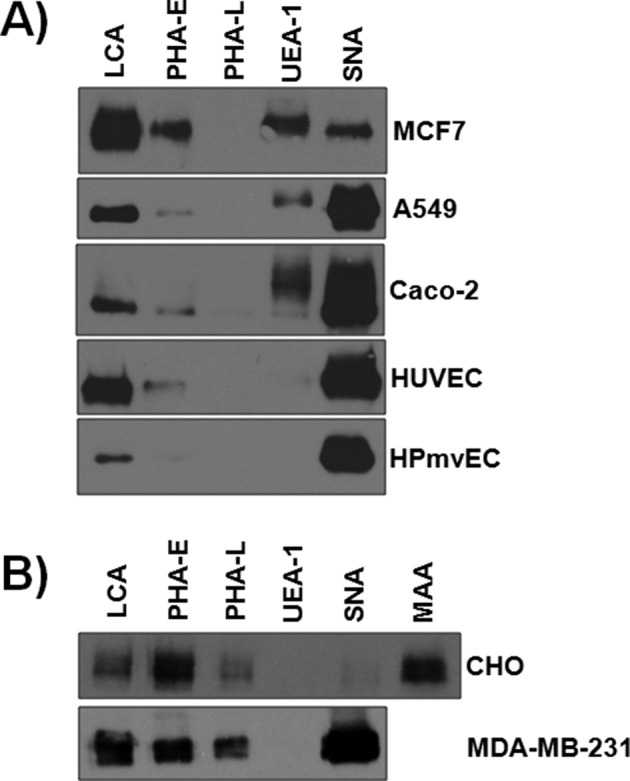
JAM-A N-glycan content differs between epithelial and endothelial cells. Endogenous expression of JAM-A from MCF7, A549, Caco-2, HUVEC, and pulmonary microvascular endothelial cells (HPmvEC) (A) and exogenous expression of the protein in CHO and MDA-MB-231 (B) was analyzed for N-glycan content after lysis and incubation with agarose-bound lectins as described in the Materials and Methods. Bound proteins were subjected to Western blot analysis for detection of JAM-A. Data are representative of two to four experiments per cell type.
DISCUSSION
Before this study, the extent of N-glycosylation on JAM-A and its involvement in the protein’s functions were unknown. We have shown that human JAM-A carries a single N-glycan at residue N185 but not on the previously implicated residue N191 (Figure 1). Glycosylation of N185 is not required for surface transport but regulates dimerization and protein half-life (Figure 2). N-glycosylation of JAM-A at N185 is also a critical regulator of barrier function (Figure 3) and cell migration (Figures 4 and 5). Our data also indicate that N-glycans at N185 of JAM-A are required for leukocyte binding via LFA-1 (Figure 6). Finally, we report cell-specific JAM-A N-glycan signatures (Figure 7). Taken together, these data indicate that N-glycosylation of JAM-A is critical for the protein’s function.
JAM-A is known to control barrier function in CHO cells (Martin-Padura et al., 1998; Aurrand-Lions et al., 2001), as well as in some epithelial cells (Monteiro and Parkos, 2012; Monteiro et al., 2014). The reported mechanism is believed to be by controlling Rap1/2 activity, which is also dependent on JAM-A homodimerization (Severson et al., 2008; Monteiro et al., 2014). In the present study, we also found that JAM-A is able to increase barrier function and increase Rap1 activity in CHO cells. The N-glycan deficient JAM-A N185Q mutant does not increase barrier function and only slightly increases Rap1 activity. Dimerization of JAM-A is reported to occur through key residues in the first Ig-like domain (Ostermann et al., 2002; Mandell et al., 2004). Atomic force microscopy experiments elegantly demonstrated, however, that these dimers are stabilized by the second Ig-like domain, the region where N185 is located (Wojcikiewicz et al., 2009), although it was unclear what portion of the second Ig-like domain was involved. We now identify N-glycans bound at N185 as regulators of dimerization. There are numerous reports of N-glycans regulating homodimers to control protein function, such as E-cadherin, platelet endothelial cell adhesion molecule 1 (PECAM-1), and N-cadherin (Guo et al., 2009; Pinho et al., 2011; Langer et al., 2012). For example, expression of PECAM-1 is reduced in cells that are missing ST6GAL1, a gene responsible for the addition of α-2,6 sialic acid on N-glycans. Loss of this enzyme results in decreased PECAM-1 surface residency and a loss of antiapoptotic signaling associated with the protein (Kitazume et al., 2014). In addition, it has been reported that these sialic acid residues, which are similar to the ones on JAM-A, regulate dimerization of PECAM-1 (Kitazume et al., 2010).
Several reports suggested that JAM-A controls cell motility and migration (Bazzoni et al., 2005; Naik et al., 2008; McSherry et al., 2009; Gotte et al., 2010; Murakami et al., 2011; Cao et al., 2014). However, the results from these studies have been conflicting. For example, it was reported that JAM-A expression decreases migration of murine endothelial and MDA-MB-231 cells but increases the migration of murine neutrophils, HUVECs, and MCF7 cells (Bazzoni et al., 2005; Naik and Naik, 2006; Naik et al., 2008; Cera et al., 2009; McSherry et al., 2009). In Figure 4 and Supplemental Figure S3, we show that JAM-A regulates cell spreading of HUVECs and MDA-MB-231 but not CHO cells. In addition, Figure 4 and Supplemental Figure S4 demonstrate that expression of exogenous wt JAM-A reduces random motility of all cells tested. One possible explanation for these cell type–specific effects could be unique N-glycan profiles associated with the different cells. Figure 7 demonstrates that cell-specific N-glycan profiles of JAM-A do exist; if and how these specific modifications regulate protein function are worthy of additional investigation.
Changes in N-glycosylation are often associated with cellular transformation and inflammation and could influence JAM-A function (Couldrey and Green, 2000; Chacko et al., 2011; Guo et al., 2012; Scott et al., 2012; Liu et al., 2013). It is intriguing to speculate that changes in JAM-A N-glycosylation as a result of disease progression could result in a loss-of-function state, such as is reported her earlier with the N185Q mutant. In fact, N185S is reported as a somatic mutation in breast cancer in the COSMIC database (Bamford et al., 2004). This mutation would likely result in a loss of JAM-A function and lead to a more aggressive cell phenotype, which has been described in breast cancer cells lacking JAM-A (Naik et al., 2008). Additional studies are needed to determine whether N185S behaves as a dominant negative when compared with the results obtained with wt JAM-A, which restored a normal breast epithelial phenotype (Naik et al., 2008).
One of the first reported functions of JAM-A was as a leukocyte receptor and regulator of diapedesis (Martin-Padura et al., 1998). In support of this, JAM-A−/− mice have been shown to have defects in leukocyte trafficking in disparate models of disease (Azari et al., 2010; Lakshmi et al., 2012; Schmitt et al., 2014). JAM-A controls diapedesis by interacting with LFA-1 expressed on leukocytes (Fraemohs et al., 2004). It has been known for some time that this interaction involves the second Ig-like domain of JAM-A, but the precise region of binding was unknown. The present data suggest that the N-glycan at N185 is required for this function. At least two possible scenarios exist by which this N-glycan could control LFA-1 binding. First, it is possible that this N-glycan residue is required for positioning the amino acids within JAM-A that serve as a ligand for LFA-1. The second possibility is that the N-glycans themselves directly interact with LFA-1 or with an associated molecule on the surface of the leukocyte that controls LFA-1 to mediate the interaction. As examples, N-glycans of intercellular adhesion molecule 1 (ICAM-1) regulate the binding of both LFA-1 and MAC-1. In the case of MAC-1, complex N-glycans at positions N240 and N269 block the binding motif on the third Ig-like domain of ICAM-1, which can be abolished by site-directed mutagenesis or production of the protein under conditions in which N-glycan complexity is limited. (Diamond et al., 1991; Scott et al., 2013). Further, the CD18 integrin, which is shared by LFA-1 and Mac1, binds ICAM-1 in a glycosylation-dependent manner (Sriramarao et al., 1993). N-glycosylation at a single residue of ICAM-1, N103, is required for the proper positioning of the binding domain that interacts with LFA-1 (Diamond et al., 1991).
This report has demonstrated that N-glycosylation of human JAM-A at N185 is required for the protein’s known functions. These findings identify a novel regulator of JAM-A function and one that is altered during disease. It will be interesting to determine whether changes in glycosylation of JAM-A could affect JAM-A function during disease, leading to compromised barrier function and altered junctional stability. Future studies are needed to determine the N-glycan composition of JAM-A and understand how this changes during inflammation and disease.
MATERIALS AND METHODS
Cell culture and transfections
CHO-K1, HL-60, MDA-MB-231, A549, Caco-2, and MCF7 cells were purchased from the American Type Culture Collection (Manassas, VA). CHO-K1 and MDA-MB-231 were propagated in high-glucose DMEM (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO) and antibiotic/antimycotic solution (Invitrogen). MCF7 and HL-60 cells were propagated in RPMI-1640 containing 10% FBS and antibiotic/antimycotic solution. Caco-2 cells were propagated in MEM containing 20% FBS and antibiotic/antimycotic solution. HUVECs and human pulmonary microvascular endothelial cells were purchased from Lonza and cultured in EGM-2 + bullet kit (Lonza, Walkersville, MD). Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s recommendations. To generate stable lines, transfected cells were grown in the foregoing media supplemented with 1000 μg/ml G418 (Invitrogen) for 3 wk, at which time, individual colonies were selected and expanded. For knockdown of JAM-A, cells were transfected with SMARTpool ON-TARGET PLUS siRNA against human f11r (Dharmacon, Lafayette, CO) at a final concentration of 10 nM.
Plasmid and site-directed mutagenesis
The pCDNA3.1 HA-JAM-A plasmid was previously described (Naik et al., 2001) and generously provided by Ulhas Naik (University of Delaware, Newark, DE). The pCMV ICAM-1 plasmid was previously described (Scott et al., 2013). Site-directed mutagenesis to generate N185Q, N191Q, and 6163 was achieved by overlap PCR and confirmed by sequencing using the following primers. N185Q: forward, 5′-cacccgtgccttcagccagtcttcctatgtcctga-3′, and reverse, 5′-tcaggacataggaagactggctgaaggcacgggtg-3′. N191Q: forward, 5′-aactcttcctatgtcctgcagcccacaacaggagagctg-3′, and reverse, 5′-cagctctcctgttgtgggctgcaggacataggaagagtt-3′. 6163: forward, 5′-gtctccttggtcaaacgcccacgccacacggggagaaga-3′, and reverse, 5′-tcttctccccgtgtggcgtgggcgtttgaccaaggagac-3′.
Rap1 activity assay
Rap1 activity was determined as previously described (Wittchen et al., 2005). Briefly, cells were lysed (50 mm Tris, pH 7.4, 75 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS containing protease inhibitors and 1 μM sodium vanadate) and clarified by centrifugation. Active Rap1 was captured by incubation with 50 μg of glutathione S-transferase (GST)–RalGDS-RBD immobilized on glutathione–Sepharose beads. Active Rap1 bound to beads was compared with total levels (from reserved input) by Western blot.
Western blot analysis
Lysates were resolved on SDS–PAGE gels and proteins transferred to nitrocellulose membranes. Blots were blocked with 3% bovine serum albumin (BSA) in Tris-buffered saline containing Tween-20 (TBST; 25 mM Tris, pH 7.6, 150 mM NaCl, 0.2% Tween-20). Blots were probed with primary antibodies against JAM-A (612120; BD Biosciences, San Jose, CA), Rap1 (2399; Cell Signaling, Danvers, MA), CD29 (9699, for human cell lines; Cell Signaling), CD29 (MAB2405, CHO cells; R&D Biosystems, Minneapolis, MN), ICAM-1 (4915; Cell Signaling), and β-actin (14-1116; Millipore, Billerica, MA). After washing with TBST, blots were incubated with species-appropriate horseradish peroxidase–conjugated secondary antibodies (Jackson Laboratories, West Grove, PA), and signals were detected using enhanced chemiluminescence substrate with x-ray film or a Bio-Rad Chemidoc system. Densitometric analysis was conducted on scanned films using ImageJ (National Institutes of Health, Bethesda, MD).
RTCA and cell spreading
Before plating, cells were incubated in DMEM containing 0.5% delipidated BSA and antibiotic-antimycotic solution for 1 h. Cells were then resuspended in the same medium for 1 h with gentle rotation. For the RTCA xCELLigence System (Roche Applied Sciences), 2000 cells/well were added to an E-plate 16 coated with fibronectin (FN; 25 μg/ml). Attachment and spreading were monitored as cell index, an arbitrary unit of cell impedance. For analysis of total spread area, cells were prepared as described and added to glass coverslips coated with 25 μg/ml FN. Cells were fixed with ice-cold ethanol and stained with Alexa 488–phalloidin (Invitrogen). Micrographs were captured using a Zeiss Axiovert 200M microscope equipped with a 63× objective lens and a Hamamatsu ORCA-ERAG digital camera. Area was calculated using ImageJ.
Wound-healing assay
Cells were plated onto FN-coated (25 μg/ml) glass-bottom dishes at a confluent density. After a 24-h incubation, a wound was created using a 200-μl pipette tip. Images were acquired at 0, 4, 8, 12, and 24 h after wounding. Each cell type was tested three times, with five regions per wound assessed. Percentage wound closure was calculated using ImageJ.
Lectin affinity pull down
Cells were lysed (50 mM Tris, 150 mM NaCl, 1% SDS, 5 μM 2-mercaptoethanol) and boiled for 10 min. Lysates were diluted 1:20 in TBS and incubated with 50 μl of lectin-conjugated agarose (Vector Laboratories, Burlingame, CA) for 4 h at room temperature. For MAA pull downs, 50 μg of biotin-tagged MAA (Vector Laboratories) was incubated with 50 μl of streptavidin agarose for 1 h at room temperature before addition of the lysates. Beads were washed five times with TBS, and bound proteins were released by boiling in sample buffer and processed for Western blot analysis as described.
Flow cyotometric analysis
Cells were trypsinized and incubated with Alexa Fluor 488–conjugated anti–JAM-A (FAB1103G; R&D Biosystems). Cells were washed three times in PBS, resuspended in sorting medium (2% FBS in PBS) containing 10 μg/ml DNase I and 5 mM EDTA, and passed through a 30-μm filter (04-004-2326; Partec). Cell populations were profiled for Alexa Fluor 488 signal with 488/525 ± 20 nm (excitation/emission) settings on a Bio-Rad S3 cell sorter. Cytometry data were analyzed with FlowJo software.
Haptotaxis assay
The bottoms of 8-μm-pore Transwell inserts (Corning, Corning, NY) were coated with FN (20 μg/ml) or vitronectin (20 μg/ml; Advanced Biomatrix, San Diego, CA). Cells were serum starved for 2 h before being added to the wells and given 4 h to migrate. Cells were fixed in ice-cold ethanol, cells from the upper chamber were gently removed with a cotton swab, and the migrated cells on the lower chamber were stained with Alexa 488–conjugated phalloidin. Three fields of view from three Transwells per condition were captured and averaged per experiment.
Immunostaining
Cells plated onto glass coverslips were fixed with ice-cold methanol before being blocked with 5% BSA in PBS. Cells were stained with antibody against JAM-A (sc-53624; Santa Cruz Biotechnology) and counterstained with Alexa Fluor 488–conjugated secondary and Hoechst 33342 (Invitrogen). Samples were mounted with Fluoro-Gel (17985-11; Electron Microscopy Sciences, Hatfield, PA) and imaged on an Olympus FV1000 confocal microscope using a 40× (numerical aperture 1.3) objective with 1.7× digital magnification. Image channels were pseudocolored and merged using ImageJ.
Single-cell tracking of motility
Cells were plated onto FN-coated (25 μg/ml) glass-bottom culture dishes 4–6 h before imaging. Time-lapse microscopy was performed on an incubator-housed microscope (20× objective; VivaView FL) with a camera (Orca ER/AG type c4742-80-12AG) with image acquisition every 15 min for 12 h. Cell velocity and persistence were measured with ImageJ using the Manual Tracking plug-in.
Monocyte adhesion assay
CHO cells were grown on FN-coated (10 μg/ml) 12-mm glass coverslips in 24-well plates and switched to serum-free medium 2 h before the experiment. Just before the experiment, the CHO cells were washed and then incubated in Hanks balanced salt solution (HBSS) containing CellTracker Green (Life Technologies)–labeled HL-60 cells (1.5 × 106 cells/dish) in the presence of 100 ng/ml phorbol myristate acetate (PMA) for 30 min. Cells were gently washed in warm PBS containing CaCl2 and MgCl2 to remove any unbound cells before being fixed with 3.7% formaldehyde in the same buffer for 20 min. Five random fields of view from three separate coverslips per condition were analyzed and quantified as cells per field of view. Images were captured using a Zeiss Axiovert 200M microscope equipped with a 20× objective lens and a Hamamatsu ORCA-ERAG digital camera. The experiment was conducted three times, and representative results from one experiment are shown.
CHO cell adhesion to fc-chimeric proteins
Black-walled 96-well microtiter (Corning) or E-16 RTCA plates were coated with LFA-1/fc chimera (20 μg/ml; R&D Biosystems) or JAM-A/fc (20 μg/ml) in 10 mM Tris, pH 9.0, at 4°C overnight. For some experiments JAM-A/fc was digested with 0.1 U of PNGaseF (Sigma- Aldrich) for 1 h at 37°C to remove N-glycans. Wells were then blocked with 0.5% heat- denatured BSA in PBS for 1 h at room temperature. CHO cells were labeled with CellTracker Green (and allowed to adhere for 30 min at 37°C in HBSS. Wells were gently washed with warm PBS with 1 mM CaCl2 and MgCl2, and fluorescence of input and adherent cells was determined with a fluorescence plate reader (Tecan). Background binding to BSA-coated wells was negligible and subtracted.
Bead adhesion assay
Assays were performed as previously described (Lessey-Morillon et al., 2014). Briefly, 4.5-μm tosylactivated paramagnetic beads (Invitrogen) were coated with LFA-1/fc according to the manufacturer. Beads were added to confluent cells in a 1:1 ratio and allowed to adhere for 15 min. Cells were washed five times with warm PBS to remove any unattached beads. Five fields of view from two separate experiments were acquired, and bound beads were determined.
Surface biotinylation assay
CHO cells were grown to confluence and washed three times with warm PBS containing 1 mM CaCl2 and 1 mM MgCl2 before being incubated with the same buffer containing 0.5 mM sulfo-NHSbiotin (Pierce). After a 15-min incubation at room temperature, the reaction was quenched by the addition of 25 mM glycine for 10 min. Cells were again washed with PBS and lysed (25 mM Tris, pH 7.6, 150 mM NaCl, 0.1% SDS, 0.5% NP-40, protease inhibitors). Biotin-tagged proteins were enriched on streptavidin agarose for 1 h and released by boiling in sample buffer. Precipitated proteins and reserved input were analyzed for JAM-A by Western blot analysis.
BS3 cross-linking: detection of JAM-A dimerization
Cells were washed three times with ice-cold PBS containing CaCl2 and MgCl2 (1 mM each) before being incubated with 5 μM BS3 (Pierce) for 30 min at 4°C. The reaction was quenched by the addition of 25 μM glycine for 10 min at room temperature. Cells were washed three times and lysed directly in sample buffer for Western blot analysis.
Barrier function analysis
For FITC-dextran flux, cells were seeded (5 × 104 cells/well) onto FN-coated (10 μg/ml) 0.4-μm polycarbonate Transwell membranes (Corning) and cultured for 48 h. FITC-dextran (10 kDa) (Sigma-Aldrich) at a final concentration of 1 mg/ml was added to the upper chamber, and after 2 h, medium from the bottom chamber was collected and transferred to a black-walled, 96-well microtiter plate (Corning) for analysis. Fluorescence intensity was analyzed using a plate reader (Tecan; excitation 485 nm, emission 520 nm). For RTCA experiments, 5 × 104 cells were plated in each well of an E-Plate 16 (Roche Applied Science), and cell index was assessed for 30 h.
Supplementary Material
Acknowledgments
This work was funded by National Institutes of Health Grants 5T32CA009156 (D.W.S.) and HL114388 and GM029860 (K.B.) and American Heart Association Postdoctoral Fellowship 15POST24470070 (D.W.S.).
Abbreviations used:
- EV
empty vector
- FN
fibronectin
- HUVEC
human umbilical vein endothelial cell
- JAM-A
junctional adhesion molecule-A
- N185Q
JAM-A point mutant with asparagine 185 mutated to glutamine
- wt
wild-type human JAM-A.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.3205) on July 29, 2015.
REFERENCES
- Aurrand-Lions M, Duncan L, Ballestrem C, Imhof BA. JAM-2, a novel immunoglobulin superfamily molecule, expressed by endothelial and lymphatic cells. J Biol Chem. 2001;276:2733–2741. doi: 10.1074/jbc.M005458200. [DOI] [PubMed] [Google Scholar]
- Azari BM, Marmur JD, Salifu MO, Cavusoglu E, Ehrlich YH, Kornecki E, Babinska A. Silencing of the F11R gene reveals a role for F11R/JAM-A in the migration of inflamed vascular smooth muscle cells and in atherosclerosis. Atherosclerosis. 2010;212:197–205. doi: 10.1016/j.atherosclerosis.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Babinska A, Azari BM, Salifu MO, Liu R, Jiang XC, Sobocka MB, Boo D, Al Khoury G, Deitch JS, Marmur JD, et al. The F11 receptor (F11R/JAM-A) in atherothrombosis: overexpression of F11R in atherosclerotic plaques. Thromb Haemost. 2007;97:272–281. [PubMed] [Google Scholar]
- Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, Wooster R. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91:355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. Junction adhesion molecule is a receptor for reovirus. Cell. 2001;104:441–451. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Tonetti P, Manzi L, Cera MR, Balconi G, Dejana E. Expression of junctional adhesion molecule-A prevents spontaneous and random motility. J Cell Sci. 2005;118:623–632. doi: 10.1242/jcs.01661. [DOI] [PubMed] [Google Scholar]
- Breitling J, Aebi M. N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2013;5:a013359. doi: 10.1101/cshperspect.a013359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Nie W, Li J, Zhang Y, Yan X, Guan X, Chen X, Zen K, Zhang CY, Jiang X, Hou D. MicroRNA-495 induces breast cancer cell migration by targeting JAM-A. Protein Cell. 2014;5:862–872. doi: 10.1007/s13238-014-0088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cera MR, Fabbri M, Molendini C, Corada M, Orsenigo F, Rehberg M, Reichel CA, Krombach F, Pardi R, Dejana E. JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J Cell Sci. 2009;122:268–277. doi: 10.1242/jcs.037127. [DOI] [PubMed] [Google Scholar]
- Chacko BK, Scott DW, Chandler RT, Patel RP. Endothelial surface N-glycans mediate monocyte adhesion and are targets for anti-inflammatory effects of peroxisome proliferator-activated receptor gamma ligands. J Biol Chem. 2011;286:38738–38747. doi: 10.1074/jbc.M111.247981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Jiang X, Sun D, Han G, Wang F, Ye M, Wang L, Zou H. Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. J Proteome Res. 2009;8:651–661. doi: 10.1021/pr8008012. [DOI] [PubMed] [Google Scholar]
- Couldrey C, Green JE. Metastases: the glycan connection. Breast Cancer Res. 2000;2:321–323. doi: 10.1186/bcr75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Staunton DE, Marlin SD, Springer TA. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991;65:961–971. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- Ebnet K, Schulz CU, Meyer Zu Brickwedde MK, Pendl GG, Vestweber D. Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO-1. J Biol Chem. 2000;275:27979–27988. doi: 10.1074/jbc.M002363200. [DOI] [PubMed] [Google Scholar]
- Ebnet K, Suzuki A, Horikoshi Y, Hirose T, Meyer Zu Brickwedde MK, Ohno S, Vestweber D. The cell polarity protein ASIP/PAR-3 directly associates with junctional adhesion molecule (JAM) EMBO J. 2001;20:3738–3748. doi: 10.1093/emboj/20.14.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong D, Spizzo G, Mitterer M, Seeber A, Steurer M, Gastl G, Brosch I, Moser P. Low expression of junctional adhesion molecule A is associated with metastasis and poor survival in pancreatic cancer. Ann Surg Oncol. 2012;19:4330–4336. doi: 10.1245/s10434-012-2381-8. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. The Catalogue of Somatic Mutations in Cancer (COSMIC) 2008 doi: 10.1002/0471142905.hg1011s57. Curr Protoc Hum Genet Chapter 10:Unit 10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraemohs L, Koenen RR, Ostermann G, Heinemann B, Weber C. The functional interaction of the beta 2 integrin lymphocyte function-associated antigen-1 with junctional adhesion molecule-A is mediated by the I domain. J Immunol. 2004;173:6259–6264. doi: 10.4049/jimmunol.173.10.6259. [DOI] [PubMed] [Google Scholar]
- Geyer H, Geyer R, Odenthal-Schnittler M, Schnittler HJ. Characterization of human vascular endothelial cadherin glycans. Glycobiology. 1999;9:915–925. doi: 10.1093/glycob/9.9.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotte M, Mohr C, Koo CY, Stock C, Vaske AK, Viola M, Ibrahim SA, Peddibhotla S, Teng YH, Low JY, et al. miR-145-dependent targeting of junctional adhesion molecule A and modulation of fascin expression are associated with reduced breast cancer cell motility and invasiveness. Oncogene. 2010;29:6569–6580. doi: 10.1038/onc.2010.386. [DOI] [PubMed] [Google Scholar]
- Guo HB, Johnson H, Randolph M, Pierce M. Regulation of homotypic cell-cell adhesion by branched N-glycosylation of N-cadherin extracellular EC2 and EC3 domains. J Biol Chem. 2009;284:34986–34997. doi: 10.1074/jbc.M109.060806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Nairn A, dela Rosa M, Nagy T, Zhao S, Moremen K, Pierce M. Transcriptional regulation of the protocadherin beta cluster during Her-2 protein-induced mammary tumorigenesis results from altered N-glycan branching. J Biol Chem. 2012;287:24941–24954. doi: 10.1074/jbc.M112.369355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Srikrishna G, Freeze HH. N-glycosylation deficiency reduces ICAM-1 induction and impairs inflammatory response. Glycobiology. 2014;24:392–398. doi: 10.1093/glycob/cwu006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Cruz F, Bazzoni G. Junctional adhesion molecule-A regulates cell migration and resistance to shear stress. J Cell Physiol. 2006;209:122–130. doi: 10.1002/jcp.20712. [DOI] [PubMed] [Google Scholar]
- Kitazume S, Imamaki R, Kurimoto A, Ogawa K, Kato M, Yamaguchi Y, Tanaka K, Ishida H, Ando H, Kiso M, et al. Interaction of platelet endothelial cell adhesion molecule (PECAM) with alpha2,6-sialylated glycan regulates its cell surface residency and anti-apoptotic role. J Biol Chem. 2014;289:27604–27613. doi: 10.1074/jbc.M114.563585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazume S, Imamaki R, Ogawa K, Komi Y, Futakawa S, Kojima S, Hashimoto Y, Marth JD, Paulson JC, Taniguchi N. Alpha2,6-sialic acid on platelet endothelial cell adhesion molecule (PECAM) regulates its homophilic interactions and downstream antiapoptotic signaling. J Biol Chem. 2010;285:6515–6521. doi: 10.1074/jbc.M109.073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmi SP, Reddy AT, Naik MU, Naik UP, Reddy RC. Effects of JAM-A deficiency or blocking antibodies on neutrophil migration and lung injury in a murine model of ALI. Am J Physiol Lung Cell Mol Physiol. 2012;303:L758–L766. doi: 10.1152/ajplung.00107.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer MD, Guo H, Shashikanth N, Pierce JM, Leckband DE. N-glycosylation alters cadherin-mediated intercellular binding kinetics. J Cell Sci. 2012;125:2478–2485. doi: 10.1242/jcs.101147. [DOI] [PubMed] [Google Scholar]
- Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EU, Roth J, Paulson JC. Alteration of terminal glycosylation sequences on N-linked oligosaccharides of Chinese hamster ovary cells by expression of beta-galactoside alpha 2,6-sialyltransferase. J Biol Chem. 1989;264:13848–13855. [PubMed] [Google Scholar]
- Lessey-Morillon EC, Osborne LD, Monaghan-Benson E, Guilluy C, O’Brien ET, Superfine R, Burridge K. The RhoA guanine nucleotide exchange factor, LARG, mediates ICAM-1-dependent mechanotransduction in endothelial cells to stimulate transendothelial migration. J Immunol. 2014;192:3390–3398. doi: 10.4049/jimmunol.1302525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Nie H, Zhang Y, Yao Y, Maitikabili A, Qu Y, Shi S, Chen C, Li Y. Cell surface-specific N-glycan profiling in breast cancer. PLoS One. 2013;8:e72704. doi: 10.1371/journal.pone.0072704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell KJ, McCall IC, Parkos CA. Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J Biol Chem. 2004;279:16254–16262. doi: 10.1074/jbc.M309483200. [DOI] [PubMed] [Google Scholar]
- Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSherry EA, Brennan K, Hudson L, Hill AD, Hopkins AM. Breast cancer cell migration is regulated through junctional adhesion molecule-A-mediated activation of Rap1 GTPase. Breast Cancer Res. 2011;13:R31. doi: 10.1186/bcr2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSherry EA, McGee SF, Jirstrom K, Doyle EM, Brennan DJ, Landberg G, Dervan PA, Hopkins AM, Gallagher WM. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int J Cancer. 2009;125:1343–1351. doi: 10.1002/ijc.24498. [DOI] [PubMed] [Google Scholar]
- Monteiro AC, Luissint AC, Sumagin R, Lai C, Vielmuth F, Wolf MF, Laur O, Reiss K, Spindler V, Stehle T, et al. Trans-dimerization of JAM-A regulates Rap2 and is mediated by a domain that is distinct from the cis-dimerization interface. Mol Biol Cell. 2014;25:1574–1585. doi: 10.1091/mbc.E14-01-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro AC, Parkos CA. Intracellular mediators of JAM-A-dependent epithelial barrier function. Ann NY Acad Sci. 2012;1257:115–124. doi: 10.1111/j.1749-6632.2012.06521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev. Mol Cell Biol. 2012;13:448–462. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Giampietro C, Giannotta M, Corada M, Torselli I, Orsenigo F, Cocito A, d’Ario G, Mazzarol G, Confalonieri S, et al. Abrogation of junctional adhesion molecule-A expression induces cell apoptosis and reduces breast cancer progression. PLoS One. 2011;6:e21242. doi: 10.1371/journal.pone.0021242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik UP, Ehrlich YH, Kornecki E. Mechanisms of platelet activation by a stimulatory antibody: cross-linking of a novel platelet receptor for monoclonal antibody F11 with the Fc gamma RII receptor. Biochem J. 1995;310:155–162. doi: 10.1042/bj3100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik MU, Naik UP. Junctional adhesion molecule-A-induced endothelial cell migration on vitronectin is integrin alpha v beta 3 specific. J Cell Sci. 2006;119:490–499. doi: 10.1242/jcs.02771. [DOI] [PubMed] [Google Scholar]
- Naik UP, Naik MU, Eckfeld K, Martin-DeLeon P, Spychala J. Characterization and chromosomal localization of JAM-1, a platelet receptor for a stimulatory monoclonal antibody. J Cell Sci. 2001;114:539–547. doi: 10.1242/jcs.114.3.539. [DOI] [PubMed] [Google Scholar]
- Naik MU, Naik TU, Suckow AT, Duncan MK, Naik UP. Attenuation of junctional adhesion molecule-A is a contributing factor for breast cancer cell invasion. Cancer Res. 2008;68:2194–2203. doi: 10.1158/0008-5472.CAN-07-3057. [DOI] [PubMed] [Google Scholar]
- Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol. 2002;3:151–158. doi: 10.1038/ni755. [DOI] [PubMed] [Google Scholar]
- Peddibhotla SS, Brinkmann BF, Kummer D, Tuncay H, Nakayama M, Adams RH, Gerke V, Ebnet K. Tetraspanin CD9 links junctional adhesion molecule-A to alphavbeta3 integrin to mediate basic fibroblast growth factor-specific angiogenic signaling. Mol Biol Cell. 2013;24:933–944. doi: 10.1091/mbc.E12-06-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho SS, Seruca R, Gartner F, Yamaguchi Y, Gu J, Taniguchi N, Reis CA. Modulation of E-cadherin function and dysfunction by N-glycosylation. Cell Mol Life Sci. 2011;68:1011–1020. doi: 10.1007/s00018-010-0595-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt MM, Megens RT, Zernecke A, Bidzhekov K, van den Akker NM, Rademakers T, van Zandvoort MA, Hackeng TM, Koenen RR, Weber C. Endothelial junctional adhesion molecule-a guides monocytes into flow-dependent predilection sites of atherosclerosis. Circulation. 2014;129:66–76. doi: 10.1161/CIRCULATIONAHA.113.004149. [DOI] [PubMed] [Google Scholar]
- Scott DW, Chen J, Chacko BK, Traylor JG, Jr, Orr AW, Patel RP. Role of endothelial N-glycan mannose residues in monocyte recruitment during atherogenesis. Arterioscler Thromb Vasc Biol. 2012;32:e51–e59. doi: 10.1161/ATVBAHA.112.253203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DW, Dunn TS, Ballestas ME, Litovsky SH, Patel RP. Identification of a high-mannose ICAM-1 glycoform: effects of ICAM-1 hypoglycosylation on monocyte adhesion and outside in signaling. Am J Physiol Cell Physiol. 2013;305:C228–C237. doi: 10.1152/ajpcell.00116.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DW, Patel RP. Endothelial heterogeneity and adhesion molecules N-glycosylation: implications in leukocyte trafficking in inflammation. Glycobiology. 2013;23:622–633. doi: 10.1093/glycob/cwt014. [DOI] [PubMed] [Google Scholar]
- Severson EA, Jiang L, Ivanov AI, Mandell KJ, Nusrat A, Parkos CA. Cis-dimerization mediates function of junctional adhesion molecule A. Mol Biol Cell. 2008;19:1862–1872. doi: 10.1091/mbc.E07-09-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson EA, Lee WY, Capaldo CT, Nusrat A, Parkos CA. Junctional adhesion molecule A interacts with Afadin and PDZ-GEF2 to activate Rap1A, regulate beta1 integrin levels, and enhance cell migration. Mol Biol Cell. 2009;20:1916–1925. doi: 10.1091/mbc.E08-10-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson EA, Parkos CA. Structural determinants of Junctional Adhesion Molecule A (JAM-A) function and mechanisms of intracellular signaling. Curr Opin Cell Biol. 2009;21:701–707. doi: 10.1016/j.ceb.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw SK, Ma S, Kim MB, Rao RM, Hartman CU, Froio RM, Yang L, Jones T, Liu Y, Nusrat A, et al. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med. 2004;200:1571–1580. doi: 10.1084/jem.20040965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriramarao P, Berger E, Chambers JD, Arfors KE, Gehlsen KR. High mannose type N-linked oligosaccharides on endothelial cells may influence beta 2 integrin mediated neutrophil adherence in vitro. J Cell Biochem. 1993;51:360–368. doi: 10.1002/jcb.240510316. [DOI] [PubMed] [Google Scholar]
- Stanley P, Schachter H, Taniguchi N. N-glycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. NY: Cold Spring Harbor Laboratory Press, 101–114: Cold Spring Harbor; 2009. [Google Scholar]
- Vetrano S, Danese S. The role of JAM-A in inflammatory bowel disease: unrevealing the ties that bind. Ann NY Acad Sci. 2009;1165:308–313. doi: 10.1111/j.1749-6632.2009.04045.x. [DOI] [PubMed] [Google Scholar]
- Wittchen ES, Aghajanian A, Burridge K. Isoform-specific differences between Rap1A and Rap1B GTPases in the formation of endothelial cell junctions. Small GTPases. 2011;2:65–76. doi: 10.4161/sgtp.2.2.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittchen ES, Worthylake RA, Kelly P, Casey PJ, Quilliam LA, Burridge K. Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. J Biol Chem. 2005;280:11675–11682. doi: 10.1074/jbc.M412595200. [DOI] [PubMed] [Google Scholar]
- Wojcikiewicz EP, Koenen RR, Fraemohs L, Minkiewicz J, Azad H, Weber C, Moy VT. LFA-1 binding destabilizes the JAM-A homophilic interaction during leukocyte transmigration. Biophys J. 2009;96:285–293. doi: 10.1529/biophysj.108.135491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Nagarajan H, Lewis NE, Pan S, Cai Z, Liu X, Chen W, Xie M, Wang W, Hammond S, et al. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat Biotechnol. 2011;29:735–741. doi: 10.1038/nbt.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Luo W, Huang B, Liu Z, Sun L, Zhang Q, Qiu X, Xu K, Wang E. Overexpression of JAM-A in non-small cell lung cancer correlates with tumor progression. PLoS One. 2013;8:e79173. doi: 10.1371/journal.pone.0079173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.