

Abstract
Background
Roxadustat (FG-4592) is an oral hypoxia-inducible factor prolyl hydroxylase inhibitor that stimulates erythropoiesis. This Phase 2a study tested efficacy (Hb response) and safety of roxadustat in anemic nondialysis-dependent chronic kidney disease (NDD-CKD) subjects.
Methods
NDD-CKD subjects with hemoglobin (Hb) ≤11.0 g/dL were sequentially enrolled into four dose cohorts and randomized to roxadustat or placebo two times weekly (BIW) or three times weekly (TIW) for 4 weeks, in an approximate roxadustat:placebo ratio of 3:1. Efficacy was assessed by (i) mean Hb change (ΔHb) from baseline (BL) and (ii) proportion of Hb responders (ΔHb ≥ 1.0 g/dL). Pharmacodynamic evaluation was performed in a subset of subjects. Safety was evaluated by adverse event frequency/severity.
Results
Of 116 subjects receiving treatment, 104 completed 4 weeks of dosing and 96 were evaluable for efficacy. BL characteristics for roxadustat and placebo groups were comparable. In roxadustat-treated subjects, Hb levels increased from BL in a dose-related manner in the 0.7, 1.0, 1.5 and 2.0 mg/kg groups. Maximum ΔHb within the first 6 weeks was significantly higher in the 1.5 and 2.0 mg/kg groups than in the placebo subjects. Hb responder rates were dose dependent and ranged from 30% in the 0.7 mg/kg BIW group to 100% in the 2.0 mg/kg BIW and TIW groups versus 13% in placebo.
Conclusions
Roxadustat transiently and moderately increased endogenous erythropoietin and reduced hepcidin. Adverse events were similar in the roxadustat and placebo groups. Roxadustat produced dose-dependent increases in blood Hb among anemic NDD-CKD patients in a placebo-controlled trial.
Clinical Trials Registration
Clintrials.gov #NCT00761657.
Keywords: anemia, chronic kidney disease, erythropoietin, hepcidin, HIF-PHI
INTRODUCTION
Anemia is a common complication of chronic kidney disease (CKD) and is associated with significant morbidity and mortality in dialysis-dependent (DD-) and nondialysis-dependent (NDD-) CKD populations [1, 2]. Erythropoietin (EPO) analogues reduce the need for red blood cell (RBC) transfusion and have improved clinical outcomes and quality of life in dialysis [1, 3, 4] and nondialysis [5, 6] settings. Anemia remains largely undertreated in NDD-CKD, in part from delayed referral to nephrologists and because EPO analogues require either intravenous (IV) or subcutaneous administration and are only available in nephrology settings [7, 8]. Further, 40–45% of patients in the USA initiate dialysis without prior contact with a nephrologist, and as of 2011, only 28% of newly initiated dialysis patients in the USA had received one or more doses of EPO analogue prior to initiation of dialysis [9]. Similarly, in two European settings, 26.5% [10] and 41% [11] received EPO analogues prior to dialysis.
The safety of using EPO analogues to treat CKD-associated anemia has been questioned. In controlled trials, greater risks for death, serious adverse cardiovascular (CV) reactions and stroke occurred when EPO analogues were administered targeting hemoglobin (Hb) levels >13 g/dL [12, 13]. These findings prompted the US Food and Drug Administration (FDA) to issue multiple warnings in 2007 to reduce EPO analogue utilization and limit target Hb levels, further lowering targets in 2011 [14]. The resulting downward trend in Hb in US CKD patients newly initiated on dialysis [7] may have contributed to the doubling of the blood transfusion rate in such patients [15]. Since June 2011, FDA guidance for NDD-CKD patients has emphasized ‘use the lowest dose of erythropoietin analogue to avoid transfusions’ and to limit initiation of EPO analogues to those patients with Hb <10 g/dL. Untreated or delayed treatment of CKD-associated anemia with Hb <10 g/dL is associated with a higher likelihood of hospitalization, RBC transfusion [16] and mortality rates [17, 18]. A higher RBC transfusion rate reduces a CKD patient's suitability for transplant because of allosensitization from transfusion [19, 20]. These consequences of untreated anemia and the limitations of EPO analogues call for exploration of safer, effective and accessible treatments for CKD anemia based on alternative erythropoietic mechanisms [21].
Roxadustat, also known as FG-4592, is a first in class, potent hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHI). With respect to erythropoiesis, HIF stabilization upon hypoxia or by HIF-PHIs induces activation of a group of ‘early response’ target genes, including those for EPO, EPO receptor, proteins promoting iron absorption, iron transport and heme synthesis [22]. Selective stabilization of HIF with small-molecule HIF-PHIs may be an innovative therapeutic approach to anemia treatment. Herein, we report the first Phase 2 clinical trial of roxadustat in anemic NDD-CKD patients.
MATERIALS AND METHODS
Study design
This was a multicenter, randomized study [single-blind (subjects), placebo-controlled, with sequential dose escalation and evaluation of frequency of administration] of roxadustat in subjects with anemia and Stage 3 or 4 CKD. The study consisted of a 28-day screening period, a 4-week treatment period [Day 1 to Day 29 (two times weekly, BIW) or 26 (three times weekly, TIW)] and up to a 12-week follow-up period. The trial was registered at Clinicaltrials.gov (NCT00761657), approved by all appropriate institutional review boards, was conducted in accordance with the Declaration of Helsinki and subjects provided written informed consent.
Initially, there were pharmacokinetic/pharmacodynamic (PK/PD) cohorts (at Phase 1 units) and treatment cohorts (mostly nephrology clinics but some from the PK/PD sites); at the latter, the focus was evaluation of dose–response of roxadustat in raising Hb in anemic NDD-CKD patients. For the PK/PD cohort, the major goal was to measure roxadustat levels and endogenous EPO (eEPO) levels in blood over time, which involves more frequent phlebotomies. Thus, subjects in the PK/PD cohort were permitted to have an entry Hb up to 13 g/dL to avoid declines in Hb from repeated blood sampling and were not eligible for efficacy evaluation. In contrast, subjects in the treatment cohorts had to be anemic with baseline (BL) Hb ≤ 11.0 g/dL to be included in efficacy analyses. Because of these differences between PK/PD cohorts and treatment cohorts, some entry criteria and analyses differed from those at treatment sites (see Supplementary Information).
After 35 patients had been enrolled, 18 into the PK/PD cohort (11 to roxadustat, 6 to placebo and 1 untreated) and 17 into the treatment cohort (13 on roxadustat and 4 to placebo), the study was interrupted and then resumed under an amended protocol with removal of PK-only cohorts that were initially planned to be tested (2.5, 3, 3.5 and 4.0 mg/kg) to avoid excessive rate of rise in Hb levels, so subjects enrolled subsequent to this protocol amendment were all ‘treatment subjects’ (screening Hb ≤ 11 g/dL), while a small subset of these patients at selected study sites could also participate in the PK/PD substudy. An additional 82 treatment subjects were enrolled (64 to roxadustat and 18 to placebo). Thus, there were a total of 99 treatment subjects randomized to one of four dose cohorts (1, 1.5, 2.0 or 0.7 mg/kg, BIW versus TIW or to placebo) as detailed in Supplementary Information and Supplementary Figure 1. Treatment subjects are included in the efficacy analyses, and all subjects from the PK and treatment cohorts are included in the safety analyses.
PK data from this trial will be presented together with those of subsequent Phase 2 studies in a different manuscript.
Subjects and treatment
Eligible subjects were 18−80 years old with Stage 3 or 4 CKD, defined by an estimated glomerular filtration rate using the modification of diet in renal disease (eGFR-MDRD) formula [23] of 15−59 mL/min/1.73 m2 and not receiving dialysis. Subjects were enrolled at 27 treatment and 2 PK sites. BL Hb was defined as the mean of the three most recent Hb measurements prior to the first dose of study drug. Subjects with severe hypertension [diastolic blood pressure (BP) > 109 mmHg or systolic BP > 170 mmHg at screening] were excluded. A comprehensive list of eligibility criteria including those for iron indices is provided in the Supplementary Information. If the subject was on oral iron at BL during enrollment, they had to remain on a stable dose of oral iron throughout the study. Decisions relating to need for oral iron were otherwise left to the investigator's discretion.
Eligible subjects were sequentially enrolled to one of four dose cohorts: 1.0, 1.5, 2.0 and then 0.7 mg/kg. Because of robust Hb responses in the 2.0 mg/kg dose cohorts, planned higher dose cohorts (2.5 and 3.0 mg/kg) were not evaluated. The first 35 subjects were randomized in a 7:7:2:2 ratio at treatment sites, and a 2:2:1:1 ratio at PK sites, to roxadustat BIW, roxadustat TIW, placebo BIW or placebo TIW, respectively. The remaining 82 subjects were randomized 10:10:3:3 at treatment sites only (see also legend to Figure 1). During the 4-week treatment period, oral roxadustat or placebo was administered as capsules (FibroGen, Inc., San Francisco, CA) according to dose group and frequency. Weekly Hb was obtained. Treatment with EPO analogues, IV iron, androgens or RBC transfusions was prohibited during the treatment period and for the first 4 weeks postdrug treatment but allowed during the remainder of the follow-up period. Oral iron was permitted at all times.
FIGURE 1:

Patient disposition. *The AEs in the roxadustat arm were acute prostatitis (in the 1.0 mg/kg TIW group) and elevated liver enzymes (in the 2.0 mg/kg BIW group). One placebo patient was discontinued because of SAEs of acute pericarditis and renal failure.
Dose modifications
Excessive erythropoiesis was defined as ≥2.0 g/dL increase in Hb within any 2-week period or ΔHb ≥3.0 g/dL at any assessment during the treatment period. In cases of the former, study drug doses were lowered by 50%. In confirmed cases of the latter, dosing was discontinued, and the subject was followed up.
Assessments
Hb response was defined as a change from BL Hb (ΔHb) of ≥1 g/dL at any time from Day 1 of treatment through 2 weeks of follow-up, the 2-week period beyond the dosing period permitting assessment of ongoing erythropoiesis. The magnitude of response was also assessed by the mean ΔHb at the end of Week 6, two weeks beyond study drug, to capture those with delayed response and the mean maximum ΔHb during the 6-week study period (ΔHbmax).
Safety assessments were based on physical examinations, electrocardiograms, clinical laboratory tests and the incidence and severity of adverse events (AEs) and assessed by each study investigator from the onset of treatment through to the end of follow-up. Optional automated blood pressure monitoring (ABPM) was performed in some patients.
Exploratory biologic correlates of erythropoiesis and iron utilization included serum iron, transferrin, transferrin saturation (TSAT), ferritin and hepcidin. Levels of plasma eEPO for up to 72 h following dosing on the first and last days of treatment were measured in the 17 subjects enrolled at the PK/PD sites. Subjects with any missing values were excluded from analysis.
Statistical analysis
Sample size of this exploratory study was not determined by statistical power, the goal being exploration of trends in Hb at the doses selected in order to determine dose levels for future studies. Efficacy analyses were conducted on those subjects who had an Hb ≤11 g/dL at BL and who received study treatment for ≥2.5 weeks [efficacy-evaluable (EE) population]. ΔHb was compared between roxadustat-treated and placebo subjects using analysis of variance. The proportion of roxadustat subjects who achieved Hb response was compared with those of placebo subjects using Fisher's exact test. Time to response was estimated using the Kaplan–Meier method. All analyses were performed using SAS v9.1.3 or higher, and statistical significance was based on P ≤0.05 unadjusted for multiple comparisons. Results are presented as mean ± SD (text or tables) and mean ± SEM in figures. PD data for eEPO are presented as median.
RESULTS
Patient disposition
Of 293 subjects screened, 117 were randomized to receive roxadustat (n = 88) or placebo (n = 29) at 29 US study sites. Patient screening and disposition are summarized in Figure 1. The safety population of 116 subjects included 88 roxadustat-treated subjects and 28 placebo subjects. Of these, 17 (11 randomized to roxadustat and 6 randomized to placebo) were enrolled at PK/PD sites and comprised the PK/PD population. Seventy-eight (88.6%) roxadustat subjects and 26 (92.2%) placebo subjects completed the study through the end of the treatment period. Reasons for drop out are itemized in Figure 1. The EE population consisted of 73 (83.0%) roxadustat subjects and 23 (82.1%) placebo subjects.
Patient characteristics
BL characteristics were representative of subjects with Stage 3–4 CKD and generally comparable across treatment groups, except for some gender and race differences (Table 1). Oral iron was taken by 53.4% of subjects at least once during study.
Table 1.
Patient demographics and BL characteristics (safety population)
| Characteristics | Placebo (n = 28) | 0.7 mg/kg |
1.0 mg/kg |
1.5 mg/kg |
2.0 mg/kg |
Pooled roxadustat (n = 88) | Total (N = 116) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| BIW (n = 10) | TIW (n = 13) | BIW (n = 12) | TIW (n = 9) | BIW (n = 10) | TIW (n = 11) | BIW (n = 11) | TIW (n = 12) | ||||
| Sex, n (%) | |||||||||||
| Male | 16 (57.1) | 6 (60.0) | 6 (46.2) | 4 (33.3) | 6 (66.7) | 4 (40.0) | 1 (9.1) | 3 (27.3) | 3 (25.0) | 33 (37.5) | 49 (42.2) |
| Female | 12 (42.9) | 4 (40.0) | 7 (53.8) | 8 (66.7) | 3 (33.3) | 6 (60.0) | 10 (90.9) | 8 (72.7) | 9 (75.0) | 55 (62.5) | 67 (57.8) |
| Race, n (%) | |||||||||||
| White | 15 (53.6) | 4 (40.0) | 9 (69.2) | 6 (50.0) | 5 (55.6) | 7 (70.0) | 4 (36.4) | 8 (72.7) | 6 (50.0) | 49 (55.7) | 64 (55.2) |
| Black | 11 (39.3) | 6 (60.0) | 4 (30.8) | 3 (25.0) | 3 (33.3) | 3 (30.0) | 7 (63.5) | 3 (27.3) | 5 (41.7) | 34 (38.6) | 45 (38.8) |
| Asian | 2 (7.1) | 0 | 0 | 1 (8.3) | 0 | 0 | 0 | 0 | 1 (8.3) | 2 (2.3) | 4 (3.4) |
| Other | 0 | 0 | 0 | 2 (16.7) | 1 (11.1) | 0 | 0 | 0 | 0 | 3 (3.4) | 3 (2.6) |
| Age in years | |||||||||||
| Mean | 68.6 | 64.6 | 60.6 | 69.5 | 67.0 | 63.8 | 63.5 | 64.3 | 66.8 | 64.0 | 65.8 |
| Range | 56–79 | 57–73 | 47–75 | 52–80 | 54–79 | 52–77 | 49–72 | 53–82 | 49–76 | 47–82 | 47–82 |
| eGFR (mL/min/1.73 m2), mean (SD) | 31.4 (12.4) | 32.1 (14.2) | 33.0 (11.1) | 38.0 (15.5) | 35.2 (9.7) | 27.9 (8.2) | 40.1 (15.3) | 34.7 (15.1) | 32.7 (9.9) | 34.3 (12.7) | 33.6 (12.6) |
| Hb (g/dL),mean (SD) | 10.3 (0.9) | 10.3 (0.7) | 9.9 (0.8) | 10.4 (1.5) | 10.6 (0.9) | 10.3 (0.6) | 10.1 (0.7) | 10.3 (1.0) | 10.1 (1.1) | 10.3 (0.9) | 10.3 (0.9) |
| TSAT (%), mean (SD) | 28.3 (6.8) | 30.1 (6.4) | 30.1 (11.3) | 24.0 (9.4) | 23.5 (5.2) | 31.1 (8.1) | 25.8 (6.5) | 30.0 (9.3) | 31.6 (11.0) | 28.4 (9.0) | 28.4 (8.5) |
| >20%, n (%) | 26 (92.9) | 10 (100) | 12 (92.3) | 8 (66.7) | 7 (77.8) | 9 (90.0) | 9 (81.8) | 11 (100) | 11 (91.7) | 77 (87.5) | 103 (88.8) |
| ≤20%, n (%) | 2 (7.1) | 0 | 1 (7.7) | 4 (33.3) | 2 (22.2) | 1 (10) | 2 (18.2) | 0 | 1 (8.3) | 11 (12.5) | 13 (11.2) |
| Ferritin (ng/mL), mean (SD) | 228 (193) | 164 (68.1) | 231 (143) | 174 (181) | 167 (178) | 228 (184) | 184 (101) | 242 (218) | 190 (89.4) | 199 (150) | 206 (161) |
| >100 ng/mL, n (%) | 21 (75.0) | 7 (70.0) | 13 (100) | 8 (66.7) | 4 (44.4) | 9 (90.0) | 11 (100) | 6 (54.5) | 10 (83.3) | 68 (77.3) | 89 (76.7) |
| ≤100, ng/mL n (%) | 7 (25.0) | 3 (30.0) | 0 | 4 (33.3) | 5 (55.6) | 1 (10.0) | 0 | 5 (45.5) | 2 (16.7) | 20 (22.7) | 27 (23.3) |
| Cuff BP (mmHg), mean (SD) | 92.4 (10.5) | 93.3 (7.0) | 93.7 (12.1) | 90.5 (13.2) | 96.6 (14.8) | 92.5 (14.2) | 91.0 (7.0) | 88.6 (7.7) | 88.7 (5.4) | 91.7 (10.7) | 91.9 (10.6) |
eGFR, estimated glomerular filtration rated (MDRD formula); Hb, hemoglobin; TSAT, transferrin saturation.
Hb response and other biologic activity
Roxadustat increased Hb in a dose-dependent manner, ΔHbmax during the 6-week study period ranged from 0.8 ± 0.9 to 2.2 ± 0.8 g/dL (Figure 2). With study medication having been administered TIW during the first 4 weeks, ΔHb at 6 weeks in the 1.5 and 2.0 mg/kg treatment groups was both significantly greater than that of the placebo group: +1.2 and +1.8 versus −0.1 g/dL (both at P < 0.01) (Figure 3). Three subjects had a ΔHbmax > 3.0 g/dL.
FIGURE 2:

Mean maximum change from BL in Hb (ΔHbmax) and % subjects achieved Hb response, defined as Hb increase by ≥1 g/dL (EE population). Mean (SD) BL Hb was 10.1 (0.7) g/dL for roxadustat subjects and 10.1 (0.6) g/dL for placebo subjects. Pooled placebo data used. Time to response was estimated using the Kaplan–Meier method, estimable for groups with >50% response. Nonresponders were censored at Day 42. *From an intergroup t-test compared with placebo; n.s.: not significant.
FIGURE 3:
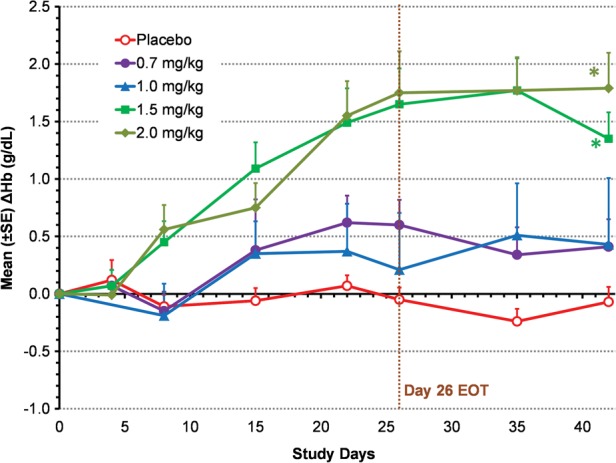
Mean change from BL in Hb (ΔHb) in TIW cohorts (EE population). Mean (SD) BL Hb was 10.1 (0.7) g/dL for roxadustat TIW subjects and 10.1 (0.6) g/dL for placebo subjects. Last-observation-carried-forward (LOCF) method was used to impute missing values. *P < 0.01 intergroup two-sample t-tests comparing roxadustat change from BL with placebo change from BL. End of treatment (EOT) for TIW was Day 26.
Hb response rates were 13% in the pooled placebo group, 30 and 58% in the roxadustat 0.7 mg/kg BIW and TIW groups, 60 and 40% in the 1.0 mg/kg BIW and TIW groups, 80 and 91% in the 1.5 mg/kg BIW and TIW groups and 100% in both 2.0 mg/kg groups, respectively. Subjects treated with 1.5 or 2.0 mg/kg roxadustat TIW (4.5–6.0 mg/kg per week) achieved a response at a median of 14 days compared with 25 or 21 days in the 1.5 or 2.0 mg/kg BIW groups (3–4 mg/kg per week), respectively.
Plasma eEPO levels over time on Day 1 and Day 29 are presented in Figure 4 for six subjects in the placebo group and five subjects in the 1.0 mg/kg BIW group. In placebo subjects, eEPO levels postdose remained essentially unchanged from BL. For subjects in the 1.0 mg/kg BIW group, eEPO began to rise ∼4 h after dosing on Day 1, peaked at ∼10 h, median of 113 IU/L and returned to BL within 24−48 h. On Day 29, the time course of eEPO rise and fall was essentially the same as on Day 1, with median peak eEPO levels reaching 121 IU/L.
FIGURE 4:

Changes in median plasma EPO on first and final days of treatment. Data are for the PK/PD population (subjects with complete dataset only).
Fewer observations (n = 1−3) were available for the other dose regimens. Peak eEPO increased with dose but not frequency. At 2 mg/kg dose, the median peak level was 397 IU/L with a maximal observed value of 705 IU/L. Levels invariably returned to BL within 48 h.
Iron indices were evaluated in the EE subjects, most of whom were iron-replete at BL. Of the 116 subjects, 45% were already on oral iron prior to study (10–626 days), which was then continued. For the EE population, there was no significant difference in the fraction receiving oral iron between placebo (39%) and roxadustat (47%) groups. Daily iron dose pre- and poststudy ranged from 39 to 300 mg/day with a median of 130 mg/day without a difference between placebo and pooled roxadustat-treated groups. During the active study period, oral iron was begun in two roxadustat subjects only. No IV iron was permitted on study.
During 6 weeks of roxadustat-induced erythropoiesis, TSAT fell while total iron-binding capacity (TIBC) rose significantly, consistent with increased iron utilization. No significant changes occurred in the placebo group (Table 2).
Table 2.
Change from BL in mean serum iron, TIBC, ferritin and TSAT levels
| Mean (SD) levels |
|||
|---|---|---|---|
| BL | EOT | Change from BL | |
| Roxadustat | (n = 73) | (n = 67) | |
| Serum iron (μg/dL) | 69.1 (17.5) | 58.3 (21.3) | −11.0 (23.3) |
| TIBC (μg/dL) | 246.3 (43.5) | 287.8 (61.7) | +41.8 (45.4)* |
| Ferritin (ng/mL) | 203.5 (150) | 141.4 (128.5)a | −68.8 (70.1) |
| TSAT (%) | 28.8 (9.4) | 20.8 (8.1) | −8.1 (9.3)** |
| Placebo | (n = 23) | (n = 18) | |
| Serum iron (μg/dL) | 71.1 (19.7) | 64.1 (19.4) | −9.5 (19.3) |
| TIBC (μg/dL) | 248.5 (51.6) | 248.3 (52.1) | −7.6 (26.6) |
| Ferritin (ng/mL) | 234.9 (198.0) | 193.2 (170.2) | −37.8 (40.3) |
| TSAT (%) | 29.0 (6.9) | 25.9 (6.3) | −3.1 (7.8) |
Data are for the EE population. BIW and TIW placebo groups and all roxadustat treatment groups were combined. P-values are from intergroup two-sample t-tests comparing roxadustat change from BL with placebo change from BL. EOT was Day 29 (BIW) or Day 26 (TIW).
an = 66.
*P < 0.0001, **P = 0.036.
Serum hepcidin levels decreased significantly during treatment with roxadustat (Figure 5). By 4 weeks, mean (±SD) hepcidin levels in the 1.5 and 2.0 mg/kg dose groups were both significantly decreased from BL (−150 ± 89.5, P = 0.048, and −225 ± 192 ng/mL, P = 0.0013, respectively) compared with the placebo group (−17.8 ± 114 ng/mL).
FIGURE 5:
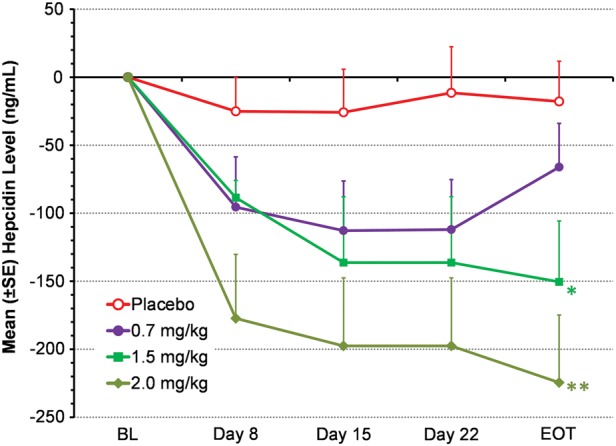
Mean change from BL in serum hepcidin (EE population). Data from BIW and TIW groups were pooled for each dose level. Serum hepcidin was not measured in the roxadustat 1.0 mg/kg dose group. LOCF method was used to impute missing data. *P = 0.048, **P = 0.0013, intergroup two-sample t-tests comparing roxadustat change from BL with placebo change from BL. EOT was Day 29 (BIW) or Day 26 (TIW).
Safety
AEs were reported by 52 (59%) roxadustat-treated and 13 (46%) placebo subjects; the most common AEs were expected in CKD (Table 3) and did not differ clinically differ between groups. All AEs are reported in Supplementary Table 1. Serious AEs (SAEs) were reported by four (5%) roxadustat-treated subjects and one (4%) placebo patient; the SAEs in roxadustat-treated subjects included vascular access complications, femoral neck fracture, noncardiac chest pain and dyspnea. The vascular access complication was reported in a patient with graft infection 4 days after arteriovenous (AV) graft placement whose BL eGFR was 18.4 mL/min. Initial Hb was 9.3 g/dL. Despite a good Hb response for 6 weeks, a left arm AV graft was placed to prepare for future dialysis. Four days later, the patient presented with signs and symptoms compatible with ‘graft’ infection and was treated with clindamycin and vancomycin.
Table 3.
AEs reported in three or more subjects
| Number of subjects with TEAEa, n (%) |
||
|---|---|---|
| Roxadustat (n = 88) | Placebo (n = 28) | |
| Any TEAE | 52 (59.1) | 13 (46.4) |
| Diarrhea | 8 (9.1) | 2 (7.1) |
| Headache | 6 (6.8) | 1 (3.6) |
| Back pain | 4 (4.5) | 1 (3.6) |
| Fatigue | 4 (4.5) | 0 |
| Hyperkalemia | 4 (4.5) | 0 |
| Peripheral edema | 3 (3.4) | 0 |
| Dizziness | 2 (2.3) | 2 (7.1) |
| Insomnia | 2 (2.3) | 1 (3.6) |
| Seasonal allergy | 1 (1.1) | 2 (7.1) |
| Urinary tract infection (UTI)b | 1 (1.1) | 3 (10.7) |
SAEs with roxadustat included a Grade 3 [National Cancer Institute's Common Terminology Criteria for Adverse Events (CTCAE), see legend to Supplementary Table 1] AV fistula site reaction, hip fracture, noncardiac chest pain and dyspnea. All these occurred in the 0.7 mg/kg TIW dose group and were considered to be unrelated to the study drug. Two AEs of moderate exacerbation of hypertension occurred in the same patient receiving 0.7 mg/kg BIW. BL weight at entry was 100 kg for this 165 cm tall individual. BL BP was 131/83 mmHg. This patient rapidly gained 8.6 and 5.4 kg above his BL weight over periods of 12 and 8 days, respectively. On both occasions, this was accompanied by worsening lower extremity edema, worsening heart failure symptoms and increases in systolic pressure from BL of 131–184 and 173 mmHg, respectively. Diastolic pressure increased only with the first larger weight gain to 99 mmHg. Aggressive diuresis was needed to remove 8.6 and 5.5 kg of fluid with decreases of BP reduction to 138/89 mmHg after first episode and 137/87 mmHg after second episode. At 6 weeks (2 weeks postdosing) and end of study (16 weeks), BP was 136/87 and 138/84 mmHg with weight below BL at 97.3–98.2 kg. One patient with a history of heavy alcohol intake had study treatment discontinued due to elevated ALT, AST and bilirubin levels following the first dose of study drug, which then normalized within a week. Although variable transient heart rate increases were noted, no clinically significant changes or trends in vital signs or laboratory values were observed.
aMedDRA version 11 (Part 1) or 13.1 (Part 2) preferred term.
bPost hoc analysis roxadustat versus placebo using Fischer's exact test showed a difference for UTI only, P = 0.043.
No CV SAEs and no death occurred during study.
The incidences of seizure, thromboembolic and CV events during roxadustat treatment are of special interest in this population; no such AEs were reported. Two episodes of moderate exacerbation of hypertension were reported by one site investigator as AEs in the same patient who had a prior history of hypertension, received the lowest roxadustat dose 0.7 mg/kg BIW and gained excessive fluid weight. Details are given in Table 3. These events were deemed unrelated to study medication. No hypertension exacerbation or other AEs of special interest were reported in the higher dose groups, and overall, no safety signal was detected from the ABPM (data not shown).
Liver function was monitored. One patient with a history of heavy alcohol intake had study treatment discontinued due to elevated alanine aminotransferase (ALT), aspartate aminotransferase (AST) and bilirubin levels following the first dose of study drug, all of which normalized within a week. No evidence of liver toxicity or sustained increases of liver enzymes or serum bilirubin were reported in this study.
Excessive erythropoiesis occurred in eight (9.1%) roxadustat-treated subjects during the dosing period of 4 weeks, all but one with the 1.5 and 2.0 mg/kg doses. Two of these had their last dose held because of ΔHb ≥3.0 g/dL. In total, 11 (12.5%) roxadustat-treated subjects and 1 (3.6%) placebo subject experienced an Hb level >13 g/dL on at least one occasion by the end of Week 6, all of which decreased to <12 g/dL within 4–8 weeks. None of the above episodes was associated with hypertension, seizure or CV event.
One (1.1%) roxadustat patient (0.7 mg/kg TIW) received a transfusion of three units of RBC 2 days after first roxadustat dose (for Hb 8.0 g/dL during treatment for an SAE of dyspnea secondary to worsening congestive heart failure). During the follow-up period of 12 weeks, five (17.9%) placebo subjects and eight (9.1%) roxadustat subjects received EPO analogues.
DISCUSSION
To our knowledge, the data from the present clinical trial are the first to demonstrate the ability of an orally available HIF-PHI to treat CKD-associated anemia in man. Roxadustat doses of 0.7−2.0 mg/kg administered orally BIW or TIW increased Hb concentrations in NDD-CKD subjects who received no IV iron supplementation. Hb responses were prompt. The overall median time to increase Hb ≥1 g/dL was 21 days, shorter than the range of 29–43 days reported in similar patient populations treated with EPO analogues [24].
Concomitant changes in eEPO, hepcidin and iron indices indicated coordinated erythropoiesis from both eEPO as well as improved iron delivery to the erythron from hepcidin reduction and increased TIBC. Peak eEPO levels were within the physiological range seen with acute bleeding [25, 26] whereas the 48-h time-averaged concentration is comparable with that of high-altitude acclimation [27]. Roxadustat was well tolerated.
Limitations of this first proof of concept study of roxadustat in CKD patients include short treatment duration of 4 weeks and modest sample size. However, other completed Phase 2 roxadustat trials have demonstrated durability of Hb response for 24 weeks (FibroGen Inc., data on file).
Current EPO analogue therapies rely upon supraphysiologic levels of recombinant variants of EPO to induce erythropoiesis [28, 29]. Use of greater doses in CKD patients is associated with worsening hypertension and increased risk for CV events (myocardial infarction, hospitalization for congestive heart failure and stroke) [4, 30, 31]. The basis for poorer CV outcomes with greater EPO analogue doses is controversial; underlying comorbidities, iron status, rate of Hb rise and direct stimulation of nonerythroid tissue have all been proposed [29, 30, 32, 33]. Induction of erythropoiesis by HIF stabilization through roxadustat, in a coordinated manner, is characterized by transient increases in eEPO to near physiologic levels and improved iron transport and potentially enables effective anemia therapy without the adverse effects associated with EPO analogues.
Functional iron deficiency is a common cause of suboptimal Hb responses to EPO analogue therapy. Supplemental IV iron improves Hb response to recombinant EPO [34] and is frequently provided as a necessary adjuvant therapy to EPO analogues [35–37]. Roxadustat stimulated erythropoiesis and increased Hb concentration with the use of oral rather than supplemental IV iron. As ferritin and TSAT tended to decrease during 4 weeks of treatment, HIF stabilization by HIF-PHI appears to support erythropoiesis when biomarkers of iron metabolism indicate relative iron deficiency, possibly via direct impact on iron metabolism. This will be explored and confirmed in subsequent Phase 3 studies. Improvement in TIBC and suppression of hepcidin, permitting the efflux of stored iron from macrophages and influx of oral iron onto transferrin, can prevent the development of functional iron deficiency. Hepcidin, a key regulator of iron absorption and remobilization, is normally downregulated by erythropoiesis, anemia and hypoxia [38]. Results of numerous investigations demonstrate that HIF acts as an iron sensor [39] and that HIF stabilization is associated with hepcidin suppression, increased intestinal iron absorption and increases in iron transport enzymes [40]. Recent studies in HIF-2α knockout mice have shown that intestinal HIF-2α is critical not only for the erythropoietic induction of iron absorption genes in the small intestine but also for the increase in serum iron, which is necessary for efficient erythropoiesis [41].
In summary, oral roxadustat administered BIW or TIW in NDD-CKD patients increased Hb in the absence of IV iron supplementation. The balance of benefits and risks with roxadustat is being evaluated in ongoing large, controlled trials in both nondialysis and dialysis populations.
SUPPLEMENTARY DATA
Supplementary data are available online at http://ndt.oxfordjournals.org.
AUTHORS' CONTRIBUTIONS
FibroGen Inc. was the study sponsor that designed the study in consultation with the Principal Investigator (A.B.). All authors except those of the sponsor but including A.B. contributed patients to the study. FibroGen was responsible for data collection and analysis. All authors had full access to the study data and the analyses. All authors reviewed the manuscript and signed off on its accuracy.
CONFLICT OF INTEREST STATEMENT
A.B., T.L., R.L., S.H., K.-H.P.Y. and T.B.N. are employees of FibroGen and hold stock and/or stock options in FibroGen. S.J.K. is a former employee of FibroGen. There are no other disclosures. At the time the study was performed, A.B. was affiliated with the Henry Ford Health System and with Wayne State University School of Medicine.
Supplementary Material
REFERENCES
- 1.Collins AJ, Ma JZ, Xia A et al. Trends in anemia treatment with erythropoietin usage and patient outcomes. Am J Kidney Dis 1998; 32: S133–S141 [DOI] [PubMed] [Google Scholar]
- 2.Coresh J, Selvin E, Stevens LA et al. Prevalence of chronic kidney disease in the United States. JAMA 2007; 298: 2038–2047 [DOI] [PubMed] [Google Scholar]
- 3.Eschbach JW, Egrie JC, Downing MR et al. Correction of the anemia of end-stage renal disease with recombinant human erythropoietin. Results of a combined phase I and II clinical trial. N Engl J Med 1987; 316: 73–78 [DOI] [PubMed] [Google Scholar]
- 4.Regidor DL, Kopple JD, Kovesdy CP et al. Associations between changes in hemoglobin and administered erythropoiesis-stimulating agent and survival in hemodialysis patients. J Am Soc Nephrol 2006; 17: 1181–1191 [DOI] [PubMed] [Google Scholar]
- 5.Revicki DA, Brown RE, Feeny DH et al. Health-related quality of life associated with recombinant human erythropoietin therapy for predialysis chronic renal disease patients. Am J Kidney Dis 1995; 25: 548–554 [DOI] [PubMed] [Google Scholar]
- 6.Benz R, Schmidt R, Kelly K et al. Epoetin alfa once every 2 weeks is effective for initiation of treatment of anemia of chronic kidney disease. Clin J Am Soc Nephrol 2007; 2: 215–221 [DOI] [PubMed] [Google Scholar]
- 7.U.S. Renal Data System. 2013 Annual Data Report: Atlas of End-Stage Renal Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; http://www.usrds.org/atlas.htm (1 October 2014, date last accessed) [Google Scholar]
- 8.Minutolo R, Locatelli F, Gallieni M et al. Anaemia management in non-dialysis chronic kidney disease (CKD) patients: a multicentre prospective study in renal clinics. Nephrol Dial Transplant 2013; 28: 3035–3045 [DOI] [PubMed] [Google Scholar]
- 9.Yan G, Cheung AK, Ma JZ et al. The associations between race and geographic area and quality-of-care indicators in patients approaching ESRD. Clin J Am Soc Nephrol 2013; 8: 610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valderrabano F, Horl WH, Macdougall IC et al. PRE-dialysis survey on anaemia management. Nephrol Dial Transplant 2003; 18: 89–100 [DOI] [PubMed] [Google Scholar]
- 11.Perez-Garcia R, Martin-Malo A, Fort J et al. Baseline characteristics of an incident haemodialysis population in Spain: results from ANSWER—a multicentre, prospective, observational cohort study. Nephrol Dial Transplant 2009; 24: 578–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfeffer MA, Burdmann EA, Chen CY et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361: 2019–2032 [DOI] [PubMed] [Google Scholar]
- 13.Singh AK, Szczech L, Tang KL et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355: 2085–2098 [DOI] [PubMed] [Google Scholar]
- 14.Epogen® Package Insert, 2009. http://pi.amgen.com/united_states/epogen/epogen_pi_hcp_english.pdf (20 March 2015, date last accessed)
- 15.Gill KS, Muntner P, Lafayette RA et al. Red blood cell transfusion use in patients with chronic kidney disease. Nephrol Dial Transplant 2013; 28: 1504–1515 [DOI] [PubMed] [Google Scholar]
- 16.Seliger S, Fox KM, Gandra SR et al. Timing of erythropoiesis-stimulating agent initiation and adverse outcomes in nondialysis CKD: a propensity-matched observational study. Clin J Am Soc Nephrol 2010; 5: 882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levin A. The treatment of anemia in chronic kidney disease: understandings in 2006. Curr Opin Nephrol Hypertens 2007; 16: 267–271 [DOI] [PubMed] [Google Scholar]
- 18.Mohanram A, Zhang Z, Shahinfar S et al. Anemia and end-stage renal disease in patients with type 2 diabetes and nephropathy. Kidney Int 2004; 66: 1131–1138 [DOI] [PubMed] [Google Scholar]
- 19.Karpinski M, Pochinco D, Dembinski I et al. Leukocyte reduction of red blood cell transfusions does not decrease allosensitization rates in potential kidney transplant candidates. J Am Soc Nephrol 2004; 15: 818–824 [DOI] [PubMed] [Google Scholar]
- 20.Obrador GT, Macdougall IC. Effect of red cell transfusions on future kidney transplantation. Clin J Am Soc Nephrol 2013; 8: 852–860 [DOI] [PubMed] [Google Scholar]
- 21.Macdougall IC. Recent advances in erythropoietic agents in renal anemia. Semin Nephrol 2006; 26: 313–318 [DOI] [PubMed] [Google Scholar]
- 22.Haase VH. Hypoxic regulation of erythropoiesis and iron metabolism. Am J Physiol Renal Physiol 2010; 299: F1–F13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levey AS, Bosch JP, Lewis JB et al. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med 1999; 130: 461–470 [DOI] [PubMed] [Google Scholar]
- 24.Macdougall IC, Walker R, Provenzano R et al. C.E.R.A. corrects anemia in patients with chronic kidney disease not on dialysis: results of a randomized clinical trial. Clin J Am Soc Nephrol 2008; 3: 337–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross SP, McCrea JB, Besarab A. Erythropoietin response to blood loss in hemodialysis patients is blunted but preserved. ASAIO J 1994; 40: M880–M885 [DOI] [PubMed] [Google Scholar]
- 26.Kato A, Hishida A, Kumagai H et al. Erythropoietin production in patients with chronic renal failure. Ren Fail 1994; 16: 645–651 [DOI] [PubMed] [Google Scholar]
- 27.Milledge JS, Cotes PM. Serum erythropoietin in humans at high altitude and its relation to plasma renin. J Appl Physiol 1985; 59: 360–364 [DOI] [PubMed] [Google Scholar]
- 28.Cheung WK, Goon BL, Guilfoyle MC et al. Pharmacokinetics and pharmacodynamics of recombinant human erythropoietin after single and multiple subcutaneous doses to healthy subjects. Clin Pharmacol Ther 1998; 64: 412–423 [DOI] [PubMed] [Google Scholar]
- 29.Fishbane S, Besarab A. Mechanism of increased mortality risk with erythropoietin treatment to higher hemoglobin targets. Clin J Am Soc Nephrol 2007; 2: 1274–1282 [DOI] [PubMed] [Google Scholar]
- 30.Szczech LA, Barnhart HX, Inrig JK et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int 2008; 74: 791–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Thamer M, Stefanik K et al. Epoetin requirements predict mortality in hemodialysis patients. Am J Kidney Dis 2004; 44: 866–876 [PubMed] [Google Scholar]
- 32.Erslev AJ. Erythropoietin. N Engl J Med 1991; 324: 1339–1344 [DOI] [PubMed] [Google Scholar]
- 33.Arcasoy MO. The non-haematopoietic biological effects of erythropoietin. Br J Haematol 2008; 141: 14–31 [DOI] [PubMed] [Google Scholar]
- 34.Spinowitz BS, Kausz AT, Baptista J et al. Ferumoxytol for treating iron deficiency anemia in CKD. J Am Soc Nephrol 2008; 19: 1599–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Locatelli F, Olivares J, Walker R et al. Novel erythropoiesis stimulating protein for treatment of anemia in chronic renal insufficiency. Kidney Int 2001; 60: 741–747 [DOI] [PubMed] [Google Scholar]
- 36.Macdougall IC, Wiecek A, Tucker B et al. Dose-finding study of peginesatide for anemia correction in chronic kidney disease patients. Clin J Am Soc Nephrol 2011; 6: 2579–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roger SD, Locatelli F, Woitas RP et al. C.E.R.A. once every 4 weeks corrects anaemia and maintains haemoglobin in patients with chronic kidney disease not on dialysis. Nephrol Dial Transplant 2011; 26: 3980–3986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nemeth E, Ganz T. Regulation of iron metabolism by hepcidin. Annu Rev Nutr 2006; 26: 323–342 [DOI] [PubMed] [Google Scholar]
- 39.Peyssonnaux C, Zinkernagel AS, Schuepbach RA et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest 2007; 117: 1926–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah YM, Matsubara T, Ito S et al. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab 2009; 9: 152–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson ER, Xue X, Shah YM. Intestinal hypoxia-inducible factor-2α (HIF-2α) is critical for efficient erythropoiesis. J Biol Chem 2011; 286: 19533–19540 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.