

Abstract
Aflatoxins are toxic and potent carcinogenic metabolites produced from the fungi Aspergillus flavus and A. parasiticus. Aflatoxins can contaminate cottonseed under conducive preharvest and postharvest conditions. United States federal regulations restrict the use of aflatoxin contaminated cottonseed at >20 ppb for animal feed. Several strategies have been proposed for controlling aflatoxin contamination, and much success has been achieved by the application of an atoxigenic strain of A. flavus in cotton, peanut and maize fields. Development of cultivars resistant to aflatoxin through overexpression of resistance associated genes and/or knocking down aflatoxin biosynthesis of A. flavus will be an effective strategy for controlling aflatoxin contamination in cotton. In this study, genome-wide transcriptome profiling was performed to identify differentially expressed genes in response to infection with both toxigenic and atoxigenic strains of A. flavus on cotton (Gossypium hirsutum L.) pericarp and seed. The genes involved in antifungal response, oxidative burst, transcription factors, defense signaling pathways and stress response were highly differentially expressed in pericarp and seed tissues in response to A. flavus infection. The cell-wall modifying genes and genes involved in the production of antimicrobial substances were more active in pericarp as compared to seed. The genes involved in auxin and cytokinin signaling were also induced. Most of the genes involved in defense response in cotton were highly induced in pericarp than in seed. The global gene expression analysis in response to fungal invasion in cotton will serve as a source for identifying biomarkers for breeding, potential candidate genes for transgenic manipulation, and will help in understanding complex plant-fungal interaction for future downstream research.
Introduction
Aflatoxins represent the group of four polyketide-derived mycotoxins (B1, B2, G1 and G2) that are highly toxic and carcinogenic chemicals produced as secondary metabolites from toxigenic isolates of the saprophytic fungi Aspergillus flavus and A. parasiticus [1–6]. Aflatoxin B1 is the most widely occurring structure that is carcinogenic to humans and animals [2–4]. Aflatoxins cause suppression of the immune system, cancer, retardation in growth, and in extreme cases death of both humans and animals. Aflatoxins have the ability to contaminate variety of crops such as corn, cotton, peanut and tree nuts during their growth and development, accounting to an estimated economic loss of ~$270 M annually worldwide [4], [5], [7]. The occurrence of aflatoxin in agricultural products is highly prohibited. U.S. Food and Drug Administration (FDA) has imposed strict regulations on the levels of aflatoxin contamination in foods and feeds; the permitted aflatoxin levels in human food and milk is 20 parts per billion (ppb) and 0.5 ppb, respectively [8], but for the cereals, nuts and dried fruits, aflatoxin standards are more stringent, which is 4 ppb for total aflatoxin content and 2 ppb for aflatoxin B1 [8], [9]. The cottonseeds alone contribute ~15% of the income of the farmers from cotton. The contamination of cottonseed with aflatoxin is of high concern to the cotton industry because cottonseeds are used as a preferred meal to dairy cows due to their high protein content, and cottonseeds are also used for oil production. Further, cows fed with contaminated cottonseeds can metabolize the aflatoxin B1 to M1 (hydroxylated derivative of metabolized aflatoxin B1), which in their milk will ultimately affect humans [8]. The prices of cottonseeds are largely determined by the content of aflatoxin present. Aflatoxin contamination is a major problem in the arid cotton growing regions of Arizona, the Imperial Valley of California, South Texas, and to extent in Louisiana in the U.S., and accounts to high annual economic losses.
Considering the declining economy of the cottonseed industry due to the infection of cotton by A. flavus, it is highly important to take necessary steps to manage aflatoxin contamination in cotton. Both pre- and post-harvest strategies have been used to lessen the aflatoxin contamination in cotton and other crops. Pre-harvest strategies include control of insect pests and proper irrigation to manage aflatoxin contamination. The post-harvest strategies include control of storage conditions that are less favorable to fungal growth, and detoxification of aflatoxin from contaminated seeds and grains [10]. Some plant metabolites, such as linoleic acid derivative 13(S)-hydroperoxide, are known to inhibit aflatoxin synthesis [8], [11]. Further, the bio-competition by application of atoxigenic strains A. flavus and/or A. parasiticus to outcompete toxigenic strains in the fields has been shown to be an effective strategy to reduce the aflatoxin contamination [5], [8], [12]. Atoxigenic strains of Aspergillus were reported to reduce the contamination of aflatoxin by ~70–90% in cotton and peanut [13–15]. This bio-competition strategy is of utmost importance in cotton because cotton has limited genetic diversity, and to date, no aflatoxin-resistant genotype is available in cotton [4], [8].
The defense responses in plants depend on the type of pathogen [6], [16]. Among different mechanisms, defense responses in plants are known to be regulated by the phytohormones, such as salicylic acid (SA), jasmonic acid (JA), ethylene (ET), cytokinin (CK) and auxins [6], [16], [17]. As a general rule, plant resistance to biotrophic pathogens is controlled by SA. In contrast, the resistance to necrotrophic pathogens is controlled by JA- and ET-dependent signaling pathways [6], [16], [17]. Moreover, resistance to necrotrophic fungal pathogens is known to be quantitative in nature and regulated by multiple genes [6], [18]. Toxigenic strain of A. flavus is characterized with the features of a necrotrophic fungal pathogen [6]. It is essential to develop germplasm that can resist the fungal invasion and/or shut down toxin production for long-term control of A. flavus infections [4], [19]. However, conventional breeding for resistance to A. flavus in cotton has been handicapped due to the unavailability of the genetic resistance in the available cotton gene pool. Genetic engineering of cotton with genes induced or upregulated in response to A. flavus infection will provide a promising approach to develop cotton varieties resistant to A. flavus. This necessitates detail investigation into the host-pathogen interaction to identify genes that are induced in cotton in response to A. flavus invasion or by toxin production. Further, understanding the largely unknown molecular basis of bio-competition strategy in controlling toxigenic A. flavus infection using atoxigenic strain of A. flavus could lead to identification of candidate genes for their use in manipulation of A. flavus resistance in cotton. Strategies such as small-scale expressed sequence tag (EST) library sequencing and oligonucleotide microarray have been used for the identification of genes induced or regulated in response to A. flavus infection in crops, such as maize, peanuts and cotton [4], [20], [21]. These small-scale targeted strategies based on the identification one or a few genes are not sufficient to understand the complex host-pathogen interaction responses [22]. Therefore, to identify key regulators in the interaction of A. flavus infection with cotton, it is necessary to discover genes on a global scale using high-coverage transcriptome analysis approach. We report here the identification of differentially expressed/regulated genes in the pericarp and seed tissues of cotton in response to A. flavus infection with an objective to understand the complex genetics involved in defense response of cotton to both toxigenic and atoxigenic strains of A. flavus infection.
Materials and Methods
1. Fungal Culture Preparation and Cotton Boll Inoculation
Fungal cultures of toxigenic (AF13) and atoxigenic (AF36) strains of A. flavus were prepared as described earlier [4]. Briefly, the strains were grown on maltose extract agar medium at 30°C for a week. Conidia were harvested by scrapping the mycelium in nine ml of potato dextrose broth, and the suspension was adjusted to a concentration of 104 conidia/ml.
Cotton variety ‘Coker 312’ was grown in the greenhouse for the present study as described earlier [4]. A hole to a depth of 5–10 mm was made in the center of one of the locules (L1) of cotton bolls (28–30 dpa) with the help of a 3 mm dia cork borer. Ten L of the conidia suspension was applied into the hole with the help of a Pasteur pipet. Bolls inoculated with only potato dextrose broth (PDB) without conidia served as the control. Pericarp and fiber-free seeds from non-inoculated and inoculated locule (L1) and adjacent/distal (Adj) locules of cotton bolls were harvested in liquid nitrogen at 6, 24, 48, and 72 h after inoculation, and stored at -80°C for RNA isolation. Three bolls each from two different plants (biological replicates) were used for each treatment.
2. RNA Extraction, Library Preparation and Sequencing
The total RNA was separately extracted from seed and pericarp tissues from L1 and Adj locules collected at each time point by using Spectrum total RNA isolation kit (Sigma-Aldrich, St. Louis, MO). RNA quantity and integrity were assessed as described earlier [4].
For library preparation, 2 μg of RNA from each different time points and replications were mixed for each tissue and experimental condition in order to minimize the cost of library preparation. Altogether, six libraries–non-inoculated pericarp (NIP) and seed (NIS), Pericarp (NTP) and seed (NTS) inoculated with atoxigenic strain, and pericarp (TP) and seed (TS) inoculated with toxigenic strain were prepared as per the manual of Illumina RNA-library construction kit. The libraries were single-end sequenced using the Illumina HiSeq-2000 platform at the sequencing facilities of the Iowa State University, Johnston, IA.
3. Read Filtering and Sequence Assembly
The single-end raw short Illumina sequencing reads (100 bp) were subjected to filtering to obtain high quality reads for downstream analysis. The raw reads were filtered and trimmed for adapter contamination and low quality, ambiguous and uncalled nucleotide bases. The reads containing more than 5% of uncalled bases and of average quality < = 20 over a window size of 5 bp in 5’ to 3’ direction were discarded. The filtering and trimming of the raw reads were performed by an in-house pipeline developed with Perl and Python programming. Subsequently, the high quality reads were assembled de novo using Trinity (release 2013-02-25) [23] with parameters of k-mer size 25, minimum contig length 200 bp and min_kmer_cov 2. The assembly was performed individually for reads of each of the six libraries. The overlapping k-mers were assembled into linear transcripts followed by clusters of overlapping transcripts. The transcripts for alternative spliced form and paralogous genes from these overlapping transcripts were obtained. All bioinformatics data analysis was performed using the Louisiana State University High Performance Computing resource SuperMike-II configured with 16 CPUs. After the assembly was performed for each library, exactly duplicate (100% similar) transcripts were removed to determine the total unigenes. We have used the term “transcript” here to describe individual sequence assembly and “unigene” to denote the longest transcript from a particular alternatively spliced isoforms cluster.
4. Functional Annotation
The assembled transcripts were subjected to functional annotation using homology search against publicly available databases, such as G. raimondii protein database (http://phytozome.jgi.doe.gov/pz), G. arboreum protein database (http://cgp.genomics.org.cn/page/species/index.jsp), NCBI's non-redundant (nr) plant nucleotide sequence database (http://www.ncbi.nlm.nih.gov), and UniProtKB (SwissProt and Tr-EMBL plant sequences) database (http://www.uniprot.org). The homology search was performed using BLASTx algorithm [24] against the databases at an E-value cut-off of 1e-05. If the annotation from different databases conflicted with each other, priority was given to the match with G. raimondii protein database, NR and UniprotKB, in that order. The Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg) database was utilized for assigning biological pathways to the transcripts. Further, based on the sequence similarity with G. raimondii protein database, GO annotations for biological process, molecular function and cellular component were assigned to the assembled transcripts. The GO enrichment analysis was performed with agriGO analysis toolkit [25] with default P-value and false discovery rate (FDR). For identification of enrichment of metabolic pathways, the PathExpress analysis tool with criteria of P< 0.05 was used [26].
5. Mapping Reads to Reference Sequence
The cleaned reads from each library were mapped individually to the D genome G. raimondii (http://www.phytozome.net/) and the A genome G. arboreum (http://cgp.genomics.org.cn/page/species/index.jsp) [27], [28] using Tophat (version 2.0.9) spliced aligner [29] and Bowtie2 aligner [30] with number of threads set to 10. The unaligned reads were mapped by Bowtie2 that split these into smaller segments for realigning and finding potential spiced sites. Mapped and unmapped reads were reported as BAM files, which were used for downstream analysis.
6. Differential Gene Expression Analysis
Differentially expressed genes from the six RNA-Seq libraries were identified by using respective BAM files for alignment with Tophat-Cufflink pipeline (version 2.1.1) that produced the transcript assembly [31], [32]. The read counts were normalized as Fragments per Kilobase of Transcripts per Million mapped fragments (FPKM). The assembly files created across infected and uninfected control conditions were pooled in a single file using Cuffmerge for differential expression analysis. Finally, the pooled file from Cuffmerge was fetched to Cuffdiff for calculating expression level and statistical significance of genes across control and inoculated conditions. Cuffdiff employed a blind dispersion model, which conservatively treated all conditions as a replicate of each other in the absence of transcripts from biological replicates as is the case in the present study [31], [32]. The codes used in Cuffdiff for identifying DEGs under NIP vs TP were as follows: cuffdiff-o cuffdiff_out_NIP_TP-b GraimondiiGenome.fa-p 10-L NIP,TP—max-bundle-frags 10000000—FDR-u merged.gtf accepted_hits_NIP.bam accepted_hits_TP.bam (Generalized code: cuffdiff-o cuffdiff_out_NIP_TP-b genome.fa-p 10-L NIP,TP-u merged.gtf accepted_hits_NIP.bam accepted_hits_TP.bam). The same code was used for other experimental conditions. The heatmaps for gene expression analysis (log2 fold change) were plotted using heatmap.2 function within R package (version 3.1.2). Three-way comparisons–NI vs NT, NI vs T, and NT vs T were performed to understand the modulation of gene expression between different experimental conditions for both pericarp and seed. The (digital) expression of 10 selected genes showing maximum log2 fold change under A. flavus infection in comparison to non-inoculated control was validated by reverse-transcription PCR using gene specific primers (S1 Table; sheet 1) following the method described earlier [4].
Results and Discussion
1. Illumina Sequencing, Quality Control and Alignment to the Reference Genome
Sequencing of the six libraries generated 911,040,814 reads of 100 bp long resulting in a total of 91.1 Gbp sequence data (Table 1). The average quality of the reads in the libraries after filtering was >33. There was negligible amount of adapter/primer contamination in the reads. Altogether, 907,357,848 high quality reads were obtained from all six libraries, which totaled to 90.73 Gbp sequencing data (Table 1). The raw sequences have been deposited in the NCBI SRA database (http://www.ncbi.nlm.nih.gov/sra; Biopoject accession PRJNA275482). Out of high quality filtered reads, 59.6% mapped to the D genome of cotton (Gossypium raimondii), whereas 75.6% mapped to the A genome (G. arboreum) across all six libraries. The higher percentage of alignment of the reads to the A genome could be due to its higher genome size as compared to the D genome.
Table 1. Sequence and assembly statistics of six cotton libraries with/without A. flavus infection.
| Tissue/library | Pericarp | Seed | |||||
|---|---|---|---|---|---|---|---|
| Parameter | NIP* | NTP | TP | NIS | NTS | TS | Total |
| # Raw single-end reads | 163,090,907 | 138,340,968 | 196,958,025 | 193,242,949 | 137,348,154 | 82,059,811 | 911,040,814 |
| Bases sequenced (Gbp) | 16.30 | 13.83 | 19.69 | 19.32 | 13.73 | 8.205 | 91.1 |
| Sequence coverage | 65.24X | 55.34X | 78.78X | 77.3X | 54.94X | 32.82X | 364.42X |
| # Cleaned reads used in assembly | 162,591,684 | 137,657,801 | 196,080,070 | 192,530,847 | 136,780,468 | 81,716,978 | 907,357,848 |
| # Assembled unique transcripts | 99,772 | 107,294 | 122,657 | 100,510 | 82,896 | 73,440 | 586,569 |
| # unigenes | 31,425 | 35,831 | 40,010 | 31,635 | 28,019 | 23,638 | 190,558 |
| Transcript N50 (bp) | 1887 | 1842 | 1976 | 1927 | 1703 | 1787 | 1864 |
| Transcript N90 (bp) | 701 | 647 | 699 | 705 | 634 | 658 | 675 |
| Maximum transcript size (bp) | 15441 | 14102 | 15414 | 15383 | 10154 | 14933 | 15441 |
| Minimum transcript size (bp) | 201 | 201 | 201 | 201 | 201 | 201 | 201 |
| Average transcript Size (bp) | 1339.58 | 1275.85 | 1351.4 | 1353.22 | 1217.53 | 1278.19 | 1307.8 |
| Transcriptome size (Mbp) | 133.65 | 136.89 | 165.75 | 136.01 | 100.92 | 93.87 | 767.11 |
*NIP = non-inoculated pericarp
NTP = atoxigenic pericarp; TP = toxigenic pericarp; NIS = non-inoculated seed; NTS = atoxigenic seed; TS = toxigenic seed.
2. De Novo Sequence Assembly
The high quality reads from each of the six libraries from different experimental conditions were assembled independently into transcripts with a length more than 200 bp. The lengthwise distribution of transcripts of six independent libraries of cotton under different experimental conditions is shown in Fig 1. Trinity has a better resolving power than others in identifying alternative spliced transcript, and thus produces less duplicates and chimeric transcripts [33]. However, redundancy was encountered in the assembled transcriptome due to high sequencing depth, duplication and assembly process. Therefore, exactly duplicate transcripts were removed from all six libraries to obtain 586,569 unique transcripts (Table 1). Further, total transcripts from six libraries clustered into 190,558 unigenes (Table 1). The size of the transcripts ranged from 201 to 15,441 bp with a mean of 1307.80 bp. Similarly, unigenes size ranged from 201 to 15,441 bp with a mean of 841.88 bp (Table 1). Of the total reads, 87% aligned to the assembled transcripts indicating good coverage of the transcriptome. The high N50 and N90 values for unique transcriptome were 1864 bp and 675 bp, respectively, further suggesting a good quality assembly. Furthermore, complete alignment of the longest transcript comp54729_c0_seq1_NIP (15,441 bp) from NIP library to a gene coding for auxin transport protein of Theobroma cacao (TCM_019010) in the NCBI database demonstrated that the transcript was not chimeric which could have occurred due to repetitive regions in the genes. These results strongly supported a high quality transcriptome assembly of cotton.
Fig 1. Lengthwise (in bp) distribution of transcripts in RNA-Seq libraries from pericarp and seed tissues of cotton with and without Aspergillus flavus infection.
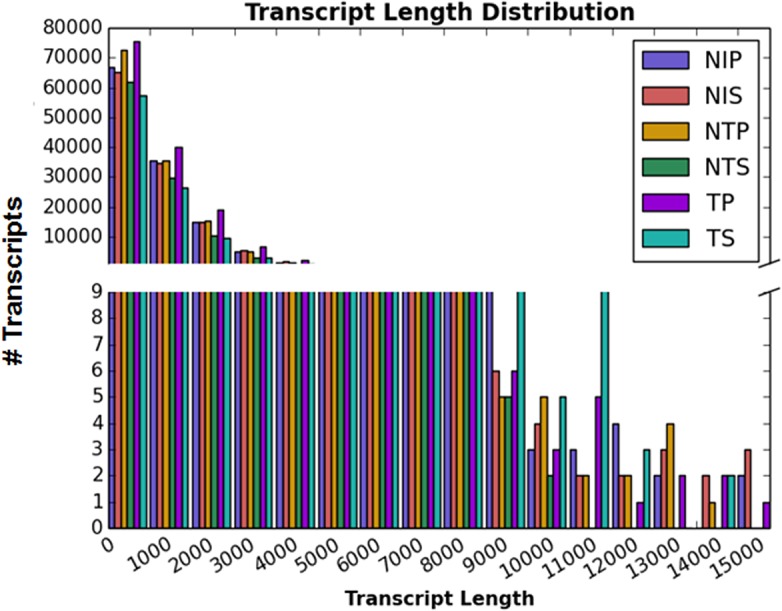
NIP = non-inoculated pericarp, NTP = non-toxigenic pericarp, TP = toxigenic pericarp, NIS = non-inoculated seed, NTS = non-toxigenic seed, TS = toxigenic seed.
3. Functional Annotation
Out of the 586,569 total unique transcripts, 466,054 (79.45%) were assigned functions based on their similarity to cotton protein database. The remaining un-annotated sequences were searched against NCBI nr and UniProtKB plant databases. Longer sequences produced significant blast hits as compared to the shorter sequences. Out of the total annotated transcripts, 405,652 (87.04%) transcripts with more than 500 bp length showed similarity to proteins in the cotton database. Of the remaining unmapped transcripts, 8,755 transcripts mapped to plant NCBI nr/nt and UniProtKB database. Further, the cotton unique transcriptome was mapped to the protein sequences of G. arboreum. In total, 19,750 un-annotated unique transcripts matched with G. arboreum protein sequences. Thus, we annotated total 494,559 transcripts (84.31%) of the cotton transcriptome. The transcripts which did not match to known genes may represent novel genes or genes that may have diverged from their homologs or noncoding RNAs [34]. The homology search showed 79.45% and 82.89% unique transcripts matching to G. raimondii and G. arboreum proteins, respectively.
4. Identification of Differentially Expressed Genes (DEGs) in Response to Aspergillus flavus Infection
Statistically significant differentially expressed genes (DEGs) in terms of FPKM (fragments per kb per million mapped reads) were calculated using combination of log2FC and P-value criteria based on mapping of the cotton reads against the G. raimondii genome as reference. In pericarp tissue, 1265, 832 and 396 genes were up-regulated (log2FC ≥2, P < 0.05) under NIP vs NTP, NIP vs TP and NTP vs TP conditions, respectively. On the other hand, 247, 123 and 869 genes were down-regulated (log2FC ≤-2, P < 0.05) under same experimental conditions, respectively. Similarly, in the seed tissue, 680, 492 and 369 genes were up-regulated under NIS vs NTS, NIS vs TS and NTS vs TS conditions, respectively, whereas, 321, 80 and 302 genes were down-regulated under same experimental conditions, respectively (Fig 2A, 2B and 2C). Principal component analysis (PCA) showed distinct response of pericarp and seed tissues to toxigenic and atoxigenic strains of A. flavus (Fig 3). The total variance contributed by three principal components was 72% (Fig 3). The results further showed significant differences in the expression profile of genes in response to atoxigenic and toxigenic infection in pericarp, whereas, in seed, the difference in response was not significant with the infection by both strains of A. flavus. This suggested that the pericarp tissue, being the primary tissue for inoculation, exhibited higher level of differential response of genes as compared to the seed tissue. Thus, identification of specific category of highly up-regulated genes from different clusters, and characterization of their biochemical response would provide potential candidates for functional characterization through genetic manipulation toward improvement of resistance to A. flavus infection. The distributions of DEGs under different conditions are shown in three-way Venn diagrams for pericarp (Fig 2B) and seed (Fig 2C). The DEGs were further characterized into different groups based on their putative functional significance as described below. Because A. flavus is a necrotrophic fungus, JA and ET dependent signaling pathways presumably function in defense response and regulate the expression of defense related genes, genes involved in oxidative burst, synthesis of antimicrobial compounds, regulation of transcription factors and localized programmed cell death in cotton in response to the fungal infection.
Fig 2. Gene expression profile of cotton in pericarp and seed tissue in response to A. flavus infection.
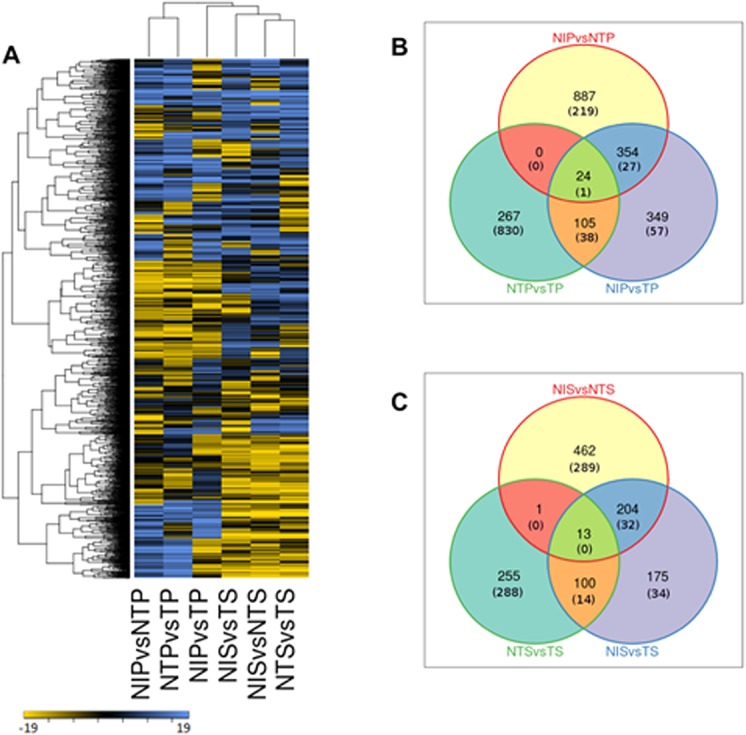
A) Heatmap showing differentially expressed genes (DEGs) of cotton in response to infection by atoxigenic and toxigenic strains of A. flavus. The up-regulated genes (log2FC> = 2 and P<0.05) and down-regulated genes (log2FC< = -2 and P<0.05) are represented by blue and yellow color, respectively. Genes with similar expression profiles were clustered together by hierarchical clustering. For description of the gene names represented in the heatmaps please refer to the S1 Table, sheet 2. Venn diagram shows the unique and common DEGs in pericarp (B) and seed (C) tissues under different experimental conditions.
Fig 3. Principal component analysis (PCA) showing the variability (72% variance) of DEGs of cotton in pericarp and seed tissue in response to infection by toxigenic and atoxiganic strains of A. flavus.
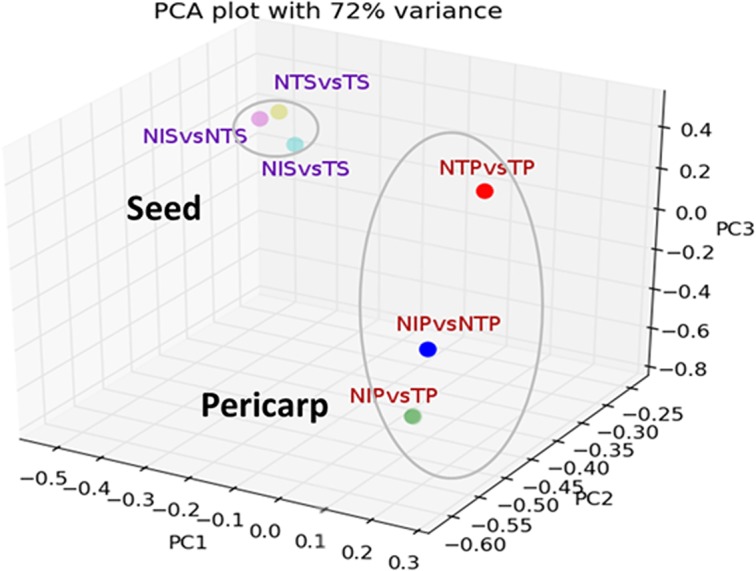
Expression of genes under different experimental conditions in seed (small oval) and pericarp (large oval) were distinct with the variability of expression higher in pericarp compared to seed. PC1, PC2 and PC3 explained 32%, 24% and 16% of the total variance.
Genes interfering with fungal virulence and growth
Eleven transcripts encoding chitinases were differentially expressed under infection by both atoxigenic and toxigenic strains in pericarp and seed. The transcripts encoding B-CHI and CHIV were induced by infection with both atoxigenic and toxigenic strains in pericarp and seed. But, CTL2 was up-regulated specifically in pericarp by both strains, whereas, CHIA was specifically induced in seed by both strains. Among the transcripts encoding CTL2, Gorai.006G078900 and Gorai.011G198500 were induced under atoxigenic and toxigenic strain infection, respectively. The three genes encoding β-1,3-glucanases (BG) were up-regulated specifically in seed. BG3 (Gorai.006G134600) and BG1 (Gorai.006G134700) were highly induced by the atoxigenic strain, whereas, another BG3 transcript (Gorai.010G003600) was up-regulated specifically by the toxigenic strain. Plant pathogenic fungi infect the plants through wounds or release of hydrolytic enzymes, such as pectinases, proteases and amylases for successful colonization [22]. Therefore, identification and characterization of plant genes, which interfere with invasion of fungus in plants, can be useful to reduce fungal pathogenicity. The hydrolytic enzymes chitinase and β-1,3-glucanase genes possess antifungal activity by degrading the fungal cell wall containing chitins [22], [35], [36]. Plant chitinases possess lysozyme activity and are highly active in inhibiting fungal growth [37]. Moreover, over-expression of chitinase genes has conferred resistance to fungal infection in plants, such as tobacco, peanuts and rice [38–40].
Five transcripts encoding trypsin and protease inhibitor proteins (TPI) were induced in pericarp, and only one TPI was induced in seed (Fig 4A). The two transcripts, Gorai.011G254400 and Gorai.012G027700, were up-regulated under infections by both the strains in pericarp, but the TPI (Gorai.011G254900) was specifically induced under the atoxigenic infection in pericarp. Among the TPI genes induced in pericarp, Gorai.011G254500 and Gorai.011G254600 were highly up-regulated by the toxigenic strain and down-regulated by the atoxigenic strain (Fig 4A). For example, Gorai.011G254500 and Gorai.011G254600 were up-regulated by 2.9- and 6.5-fold, and 9.8- and 12.7-fold higher by the toxigenic strain in comparison with the non-inoculated control and the atoxigenic strain, respectively (S1 Table, sheet 3). In seed, the TPI (Gorai.012G027600) was induced under the toxigenic strain infection only. Reduced growth of A. flavus has direct impact on aflatoxin production [41]. Trypsin inhibitors are known to possess antifungal activity [22], [41] and inhibit conidia germination and hyphal growth of A. flavus [41]. Four genes encoding serine protease inhibitor (SPI) were also differentially expressed in pericarp and seed. Gorai.012G105800 and Gorai.007G143500 were up-regulated specifically under the atoxigenic strain infection in pericarp and the toxigenic strain infection in seed, respectively. Serine protease inhibitor gene has been shown to be induced in response to infection with A. flavus in peanut [20].
Fig 4. Heatmaps showing DEGs involved in interference of fungal virulence and growth (A) and DEGs involved in defense signaling (B).
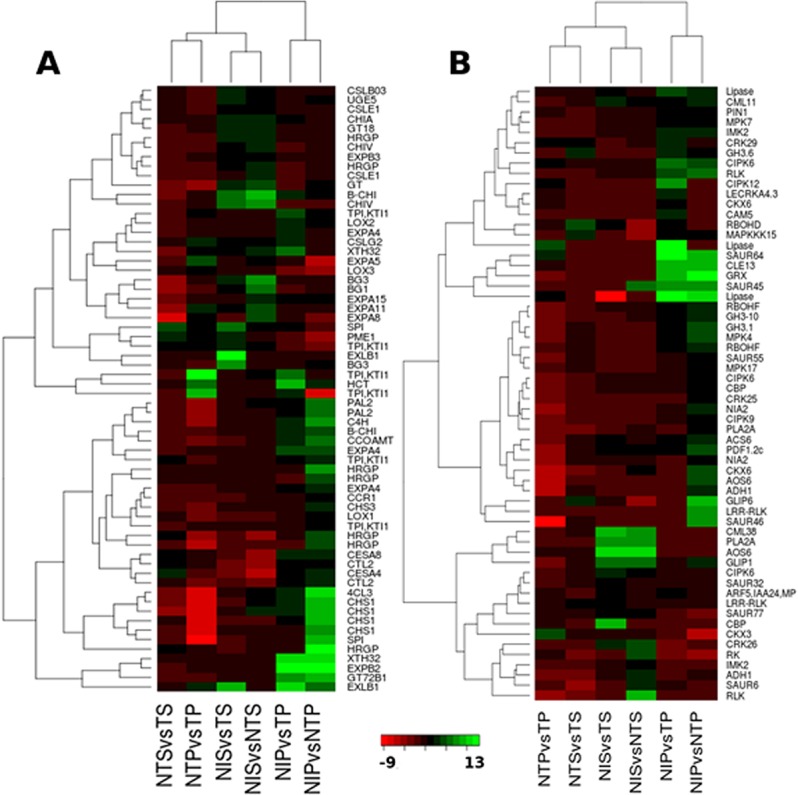
The green color represents up-regulated (log2FC≥2) genes and red color represents down-regulated (log2FC≤2) genes. For description of the gene names represented in the heatmaps please refer to the S1 Table, sheet 3.
Plants accumulate hydroxyproline-rich glycoproteins (HRGPs), phytoalexins and lignin-like substances as a resistance mechanism in response to fungal pathogen infection [42]-[44]. The HRGPs and lignification are involved in fortifying the cell-wall structure of plants and contribute to the resistance to pathogen invasion [45]. Among 20 DEGs encoding HRGP, 10 and two genes were specifically induced in pericarp and seed, respectively (Fig 4A). In pericarp, most of these genes were induced under the atoxigenic strain infection. Similarly, in seed the two genes were induced by the atoxigenic strain infection. The increase in the level of HRGP transcripts has been observed in several plants in response to wound or pathogen infection, and is associated with higher resistance to pathogen [37]. The genes involved in the metabolism of phenylpropanoids, which serve as a source for furanocoumarin and isoflavonoid phytoalexins and lignins, were differentially expressed in the pericarp and seed tissues. The phenylpropanoid pathway precursors are also involved in the synthesis of lignin and phenolic substances, and possess antifungal activities [37], [42], [43]. The increase in cell wall lignification as a structural modification has been observed in plants for defense in response to fungal pathogen [37]. The enzymes phenylalanine ammonia-lyase (PAL), 4-coumarate CoA ligase (4CL) and chalcone synthase (CHS) are involved in the phenylpropanoid pathway [37], [42], [43]. Two genes encoding each PAL2 and 4CL1 were highly up-regulated in pericarp under the atoxigenic strain infection (Fig 4A). The transcript encoding 4CL3 was up-regulated in both tissues, but had the higher fold change in gene expression in pericarp specifically under the atoxigenic strain infection as compared to the toxigenic strain infection. The enzyme chalcone synthase (CHS) was highly induced in pericarp (Fig 4A). The three genes encoding CHS (Gorai.005G035100, Gorai.006G000200 and Gorai.009G339300) were specific to pericarp and showed higher expression under the atoxigenic strain infection. The two CHS genes (Gorai.011G161200 and Gorai.011G161300) were up-regulated in both tissues under the atoxigenic infection, but only in pericarp under the toxigenic infection, The fold change for these two CHS genes were higher in pericarp as compared to seed (Fig 4A). Other genes involved in the lignin biosynthesis, such as cinnamate-4-hydroxylase (C4H), cinnamoyl-CoA reductase 1 (CCR), hydroxycinnamoyl-CoA shikimate/quinate hydroxycinnamoyl transferase (HCT) and caffeoyl-CoA 3-O-methyltransferase (CCOAMT), were specifically up-regulated in the pericarp tissue under fungal infection (Fig 4A). Two transcripts encoding C4H, one transcript encoding CCR1 and CCOAMT each were highly up-regulated under the atoxigenic strain infection, while HCT was highly induced under the toxigenic strain infection in pericarp. Simultaneous up-regulation of PAL, 4CL and CHS genes of the phenylpropanoid pathway suggested that these genes are coordinately regulated in response to fungal infection and that wounding and fungal infection at pericarp induced and accumulated these transcripts at a much higher level in pericarp as a part of an induced plant defense response to the invading fungus. The genes involved in the phenylpropanoid pathway are also induced in response to wounding [46]. The production of phytoalexins and accumulation of PAL, CHS and HRGP upon infection indicates hypersensitivity response, thus establishing immunity of uninfected distant cells [37], [42]. The increase in lignification was also shown to be associated with hypersensitive resistant reaction in wheat in response to Puccinia graminis f.sp. tritici infection [37], [47].
Lipoxygenase (LOX) genes of the LOX biosynthesis pathway were differentially expressed in pericarp tissue under A. flavus infection. The LOX2 gene was up-regulated under infection by both the strains in pericarp, whereas, the LOX1 (Gorai.004G241400) was specifically up-regulated under the atoxigenic strain infection in pericarp. The LOX3 gene was down-regulated in the pericarp tissue under infection by both strains (Fig 4A). However, the LOX genes did not show any change in response to A. flavus infection in seed. Most volatile compounds and hydroperoxy fatty acids are the products of the LOX biosynthesis pathway [22]. These results suggested that LOX genes expression was more abundant in pericarp as a possible mechanism of resistance against A. flavus infection. Plant-derived volatile compounds have the capability to inhibit A. flavus growth and aflatoxin biosynthesis [22]. Previous studies have reported antifungal role of LOX genes in peanut, corn and soybean [20]. In human too, LOX genes were shown to be involved in degradation of aflatoxin B1 by oxidative metabolism [20].
Wounding and pathogen infection have also been shown to regulate the genes involved in cell wall biosynthesis and modifications, such as pectins, cellulose and hemicellulose [46]. The transcripts encoding xyloglucan endotransglucosylase (XTH), cellulose synthase (CS), UDP-D-galactose 4-epimerase (UGE), pectin methylesterase (PME), expansin (EXP) and glycosyltransferase (GT), which are involved in the cell wall modification, were regulated by A. flavus infection in both pericarp and seed (Fig4A). The results also suggested that cell-wall modifying genes and genes involved in the production of antimicrobial substances were more active in pericarp as compared to seed. It is thus evident that the atoxigenic strain of A. flavus played a major role in activation of the antifungal and cell-wall modifying genes in the pericarp and seed tissues. Further, the characteristic response of these genes under specific fungal strain infection and tissue will help elucidate the mechanism of defense.
The cross-talk between JA/ET and SA signaling pathway
Salicylic acid (SA), jasmonic acid (JA), ethylene (ET) and abscisic acid (ABA) are known to be involved in defense signaling pathways in response to pathogen infections and wounding [16], [17], [46], [48], [49]. SA-induced defense response generally involves resistance to biotrophic and hemibiotrophic pathogens, whereas JA and ET initiate the defense reaction in response to necrotrophic pathogens [16], [17]. The activation of SA and JA/ET pathways are pathogen dependent that involves mutually antagonistic activities [16]. JA and ET work synergistically in response to pathogen infection in plants [16]. In the present study, the transcripts encoding phospholipase, GDSL lipase, allene oxide synthase (AOS) and alcohol dehydrogenase (ADH), which are involved in the JA biosynthesis [46], [49], were induced in both pericarp and seed tissues (Fig 4B). Similarly, the gene 1-aminocyclopropane-1-carboxylic acid synthase (ACS), involved in ET biosynthesis [46], was up-regulated in both pericarp and seed tissues (Fig 4B). A higher number of lipase-encoding transcripts were induced in pericarp as compared to seed (Fig 4B). In pericarp, the lipases genes showed similar expression pattern under both atoxigenic and toxigenic strains infection, whereas in seed the genes were more active under the toxigenic strain infection (Fig 4B). AOS and ACS6 genes were up-regulated under the atoxigenic strain infection in pericarp, while in seed AOS was up-regulated by both strains, and ACS6 was specific to toxigenic strain infection (Fig 4B). The jasmonate zim-domain protein (JAZ) inhibits the JA signaling in plants by interacting with JIN1/MYC2 gene and represses the expression of JA responsive genes [16]. The transcript encoding JAZ8 (Gorai.006G092400) was down-regulated in both pericarp and seed tissues under A. flavus infection (Fig 4B). Further, SA mediated signaling pathway was inhibited by JA/ET signaling pathway in both pericarp and seed tissue in response to the fungal infection. The non-expresser of PR genes (NPR) which are a vital component of SA signaling pathway [16] was down-regulated in both pericarp and seed in response to A. flavus infection (S1 Table). The mitogen activated protein kinase gene, MPK4, acts as a positive regulator of JA and a negative regulator of SA signaling pathway in plants [16]. MPK4 was up-regulated under the atoxigenic strain infection in pericarp. Interestingly, the glutaredoxin (GRX), which is identified as a negative regulator of JA/ET signaling pathway [16], was up-regulated in pericarp, but down-regulated in seed. The enhanced expression of JA-responsive marker gene plant defensin 1.2 (PDF1.2) is known to be associated with resistance to necrotrophic pathogens [16]. In the present study, the transcript encoding PDF1.2c was specifically up-regulated under the atoxigenic A. flavus strain infection in pericarp tissue (Fig 4B). The transcription factor ERF1 that acts as a positive regulator of JA and ET signaling pathway in Arabidopsis [16] was highly induced in seed by the infection with both strains of A. flavus. These results indicated that JA/ET signaling pathway may be a component in the resistance mechanism of cotton to necrotrophic A. flavus.
Genes involved in defense signaling pathways
Plant receptor protein kinases (RPK) represent PRRs that are involved in the perception of pathogen signal and trigger inducible defense [50]. The transcripts similar to receptor-like protein kinase (RLK), leucine-rich repeat receptor-like protein kinase (LRR-RLK), cysteine-rich RLK (CRK), lectin receptor kinase, inflorescence meristem receptor-like kinase (IMK) and receptor kinase (RK) were differentially expressed in both tissues under the atoxigenic and the toxigenic A. flavus strains infection (Fig 4B). In seed, the transcripts encoding RLK were highly induced by the atoxigenic strain. The transcripts for LRR-RLK were specifically induced by the atoxigenic strain in pericarp and the toxigenic strain in seed. The CRK genes were induced in both tissues by both atoxigenic and toxigenic strains (Fig 4B). Plants produce elicitors that are perceived by RPK to amplify immunity and resistance to fungal infection [51], [52]. The elicitor CLAVATA3 (CLV3), secreted by shoot apical meristem, binds to the LRR-RLK and activates it [51]. In pericarp, the transcript encoding CLAVATA3 (CLV3) was up-regulated under both atoxigenic and toxigenic strains infection (Fig 4B).
The increase in the level of Ca2+ is indicative of the activation of plant’s innate immunity. The increase in Ca2+ levels is the result of release of pathogen elicitors after the infection in plants. The elevated levels of calcium under stress conditions are recognized by calcium binding proteins such as calcium dependent kinases (CDPKs), calmodulins (CaMs) and calcineurin B-like proteins (CBL), which in turn induce downstream target gene expression [46], [51] [53]. The up-regulation of a large number of transcripts encoding calcium binding proteins including CaMs, CBL and calcium binding EF-hand family proteins (CBP) in the present study suggested that there were elevated levels of Ca2+ after fungal infection in both pericarp and seed tissues (Fig 4B). In pericarp, most of the transcripts encoding CBP were induced under the atoxigenic strain infection, whereas in seed these were under the toxigenic strain infection (Fig 4B). Recent studies on plant-pathogen interactions reported that Ca2+-mediated activation of CaMs, CBL interacting protein kinases (CIPKs) and CDPKs are involved in plant’s immunity responses [51]. The transcripts similar to CIPK9, CIPK12, CIPK6 and CIPK21 were differentially expressed in both tissues under fungal infection (Fig 4B). CIPK9 and CIPK6, and CIPK12 and CIPK6 genes were induced under the atoxigenic and the toxigenic infection in pericarp, respectively (Fig 4B). However in seed, only CIPK6 was induced under the toxigenic infection. The CIPK genes are involved in late immune responses (3–24 h after infection) and promote accumulation of phytoalexin, and expression of cell death and PR genes in response to fungal infection [51].
The activation of MAPK pathway is also one of the defense responses that contribute to resistance to fungal infection in plants starting as early as 1 min after infection. Wounding and pathogen elicitors induce fast activation of MAPK cascade signaling [51]. The MAPK cascade signaling involves three components: MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK) and MAPK. In MAPK signaling cascade, the MAPKKK activates MAPKK, which in turn activates MAPK. The four transcripts encoding MAPKKK15, MPK17, MPK7 and MPK4 were highly up-regulated specifically under the atoxigenic strain infection in pericarp (Fig 4B). MAPK cascade signaling was not induced in seed tissue in response to the fungal infection. The activation of MAPK signaling cascade regulates the downstream transcription factors, which further induce the expression of defense related genes leading to enhanced long-term defense response and resistance to fungal infection by regulating the synthesis of antimicrobial peptides and chemicals, programmed cell death, stress hormones (JA and ET), nitric oxide (NO) and reactive oxygen species (ROS) [51], [52]. The NO synthesis in plants is catalyzed by the enzyme nitrate reductase (NIA), which plays an important role in plant defense responses [54], [55]. Under the atoxigenic strain infection, the two transcripts encoding NIA2 were up-regulated in pericarp (Fig 4B). In tobacco cells, the fungal elicitors contributed to prolonged activation of MAPKs, which regulate the expression of NO and ROS [51], [56]. The ROS, which play an important role in plant defense responses together with NO, are synthesized by the phagocyte enzyme complex of NADPH oxidase [54], [56]. The respiratory burst oxidase homolog (RBOH) which is plant NADPH oxidase [56], [57] was differentially expressed in both pericarp and seed tissues of cotton under fungal infection (Fig 4B). The RBOH is regulated by MAPK signaling cascade and its increased expression is associated with resistance to pathogens [51], [56]. The three homologs of RBOH including RBOHF (Gorai.003G085100, Gorai.008G199100) and RBOHD (Gorai.009G202500) were highly induced specifically in pericarp (Fig 4B). The RBOHF transcripts were induced under both atoxigenic and toxigenic strains infection in pericarp, whereas RBOHD was specifically up-regulated under the toxigenic strain infection (Fig 4B). The production of NO and ROS together are necessary for inducing the hypersensitive response (HR) and cell death in plants [54].
The phytohormone auxin (Aux), besides regulating growth and developments of plants, plays an important role in the defense responses to pathogens, [16], [58], [59]. Aux regulates the expression of Aux/IAA, Gretchenhagen-3 (GH3) family, Auxin response factor (ARF) and small auxin-up RNA (SAUR) genes [16], [58], [60]. Over-expression of OsGH3.1 in rice enhanced the resistance to fungal pathogen by reducing the auxin level and suppressing the expression of expansin genes [16], [59]. In this study, the transcripts similar to GH3.1 and GH3.10 were specifically induced in pericarp by both strains ns (Fig 4B). The GH3.6 (Gorai.005G208000) was specifically induced under the toxigenic strain infection in pericarp. Similarly, SAUR family genes were differentially expressed in pericarp and seed tissues under A. flavus infection (Fig 4B). SAUR genes have inhibitory activity on auxin biosynthesis and transport [60]. The over-expression of SAUR39 transcript in rice showed reduced free IAA level and auxin transport [60].
The role of phytohormone cytokinin has also been elucidated in disease resistance reaction in Arabidopsis [16], [61]. Arabidopsis lines overexpressing cytokinin oxidase/dehydrogenase (CKX) showed enhanced resistance to clubroot disease [61]. The transcripts similar to CKX6 (Gorai.012G081000 and Gorai.011G295400) were induced under atoxigenic and toxigenic strains infection only in pericarp. Contrastingly, CKX3 was down-regulated in pericarp under the atoxigenic strain infection (Fig 4B). The detailed characterization of these genes is necessary to understand the role and cross-talk of auxin- and cytokinin-signaling pathways in A. flavus-mediated defense response in cotton.
Genes encoding transcription factors (TFs)
Transcription factors control the transcriptional regulation by activating or suppressing the expression of downstream genes in response to pathogens infection [62]. The transcription factor GhWRKY3 is known to be induced under wounding and fungal infection in cotton [62]. Mutation in WRKY70 and WRKY33 enhanced the susceptibility of Arabidopsis to necrotrophic B. cinerea fungal infection [17]. WRKY40, WRKY33, WRKY53, WRKY22, WRKY11, WRKY15 and WRKY60 are known to be induced under wounding in Arabidopsis [46]. In the present study, 28 WRKY-related transcripts were differentially expressed under fungal infection in pericarp and seed of cotton (Fig 5A). The WRKY75 (Gorai.001G057600 and Gorai.005G164300) and WRKY72 were induced in both pericarp and seed in response to A. flavus infection. Most of the WRKY TFs were up-regulated under the atoxigenic strain infection in pericarp and the toxigenic strain infection in seed (Fig 5A). WRKY75 (Gorai.006G043200) and WRKY40 (Gorai.009G124000) were specifically up-regulated under the atoxigenic strain infection in both pericarp and seed. WRKY6 (Gorai.001G214800), WRKY53 (Gorai.008G253300), WRKY50 (Gorai.001G021500) and WRKY41 (Gorai.007G014600) were down-regulated in pericarp. WRKY50 is reported to negatively regulate the plant-fungus interaction, and mutation in WRKY50 has been associated with enhanced resistance to pathogen [17]. It was also reported that the WRKY TFs regulate the expression of RLK genes, which are induced in response to pathogen infection [46], [63].
Fig 5. Heatmaps showing DEGs involved in transcriptional regulation (A), involved in oxidative burst (B) and stress response (C).
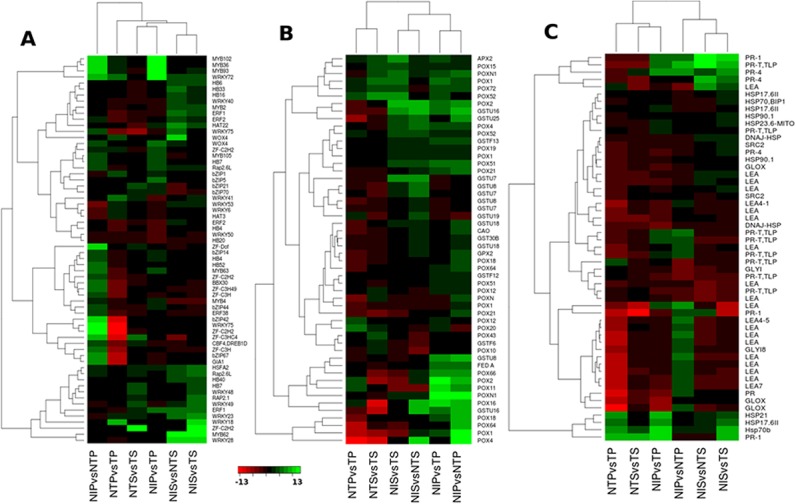
The green color represents up-regulated (log2FC≥2) genes and red color represents down-regulated (log2FC≤2) genes. For gene names represented in the heatmaps please refer to the S1 Table.
The APETALA2 (AP2)/Ethylene responsive factor (ERF) family of TFs are known to be involved in the activation of defense-related genes in response to pathogen infection through ET and JA pathways [17], [64]. The members of AP2/ERF family were differentially expressed in response to the necrotrophic A. flavus infection in pericarp and seed (Fig 5A). The transcripts similar to RAP2.6L (Gorai.012G125700) and ERF38 were up-regulated specifically in pericarp (Fig 5A). RAP2.6L was induced under both atoxigenic and toxigenic strains infection, whereas, ERF38 expression was specific to the atoxigenic strain infection. The number of AP2/ERF TFs up-regulated in seed was higher as compared to pericarp. ERF1 (Gorai.005G049300) and RAP2.6L (Gorai.005G197100) were induced specifically in seed under both strain infections. In seed, ERF2 (Gorai.009G165500) and RAP2.1 were up-regulated under the atoxigenic and the toxigenic strain infection, respectively. However, ERF2 (Gorai.010G156600) was down-regulated under the toxigenic strain infection in pericarp. The AP2/ERF TF family genes are known to be highly induced in response to wounding in Arabidopsis [46]. Overexpression of an ERF TF in Arabidopsis conferred enhanced resistance to necrotrophic fungus B. cinerea [17]. A transcript encoding DREB1D/CBF4 gene was induced under the atoxigenic strain infection in pericarp (Fig 5A). DREB/CBF TFs belong to AP2/ERF TF family and have been reported to be induced in response to wounding, in addition to abiotic stresses, such as cold, heat, salt and drought in Arabidopsis [46].
The role of bZIP family TFs in biotic stress responses of plants is well established [65]. The bZIP-family TFs were induced in pericarp of cotton by A. flavus infection. The five bZIP TFs, bZIP14, GIA1, bZIP67, bZIP44 and bZIP42, were specifically induced under the atoxigenic strain infection, whereas bZIP1 and bZIP5 were induced under the toxigenic strain infection (Fig 5A). The bZIP TFs were not induced by the fungus in seed; bZIP70 and bZIP21 were down-regulated under the atoxigenic strain infection in seed.
Many MYB TFs were also differentially expressed under A. flavus infection in cotton. More MYB genes were up-regulated in pericarp as compared to seed (Fig 5A). MYB93, MYB63, MYB102, MYB105, MYB102 and MYB36 were induced under both the atoxigenic and the toxigenic strains infection in pericarp, whereas MYB62 and MYB2 were induced in seed (Fig 5A). The gene expression pattern of MYB TFs has been studied in Arabidopsis in response to wounding and pathogen infection [46]. The MYB TFs regulate the expression of flavonoid genes, PR genes and genes involved in secondary metabolism [46]. In the present study, MYB4, which is a repressor of phenylpropanoid pathway [49], was down-regulated under both strains infection in seed (Fig 5A).
The gene expression profiles of zinc finger (ZF), heat shock factors (HSF) and homeobox (HB) type TFs were altered in response to wounding in Arabidopsis [46]. In cotton, a number of transcripts encoding ZF TFs were up-regulated in response to the atoxigenic strain infection in pericarp, whereas in seed, only one transcript similar to ZF-C2H2 (Gorai.002G223300) was induced under the toxigenic strain infection (Fig 5A). The HB type TFs showed high activity in seed than pericarp, which was evident from the up-regulation of a large number of HB genes in seed as compared to pericarp (Fig 5A). The HB TFs, HAT3, HB20 and HB4, were down-regulated in pericarp under A. flavus infection. The HSFs did not show any activity in pericarp, and were specifically induced in seed (Fig 5A). The transcript showing similarity with HSFA2 was highly up-regulated in seed under both strains infections. Further characterization of these TFs is necessary to understand their regulatory mechanisms in controlling expression of downstream genes and their roles in defense response of cotton to A. flavus infection.
Genes involved in oxidative burst
Oxidative burst is one of the earliest defense responses of plants against pathogen infection and wounding, and is considered as the hallmark of pathogen recognition [46], [56], [57], [66]. ROS production is observed in both plant triggered immunity (PTI) and effector triggered immunity ETI [57]. In addition to their involvement in direct defense reaction by killing the pathogens, ROS are also involved in the activation of defense-related genes through signaling mechanism [57]. ROS can regulate the TFs and produce antimicrobial phytoalexins and other secondary metabolites, which have inhibitory activity on pathogen growth [57]. As discussed earlier, the MAPK pathway activated the expression of RBOH genes in response to A. flavus infection, which could trigger the plant apoplastic oxidative burst. The transcripts encoding glutathione S-transferase (GST), ascorbate peroxidase (APX), copper amine oxidase (CAO), ferredoxin (FED) and peroxidase (POX), which are involved in ROS processing and scavenging, showed increased activity under A. flavus infection in cotton (Fig 5B). Most of these genes were induced in pericarp, and their expression patterns were different under the atoxigenic and the toxigenic strains infection in both pericarp and seed tissues. The transcripts similar to copper amine oxidase (CAO) under atoxigenic strain infection and ferredoxin under both strains infections were specifically induced in pericarp tissue (Fig 5B). Most of the transcripts encoding glutathione S-transferase (GST) were induced under the atoxigenic strain infection in both pericarp and seed (Fig 5B). The expression patterns for peroxidase genes (POX) were similar under the atoxigenic and the toxigenic strain infection in pericarp, but in seed POX genes were highly induced under the toxigenic strain infection (Fig 5B). The peroxidase 2 (POX2) genes were specifically induced in pericarp tissue under both the atoxigenic and the toxigenic strains infection. The transcript similar to glutathione peroxidase (GPX2) was induced under the atoxigenic strain infection in pericarp (Fig 5B). Hydrogen peroxide can also act as a secondary messenger and initiate regulation of defense related genes [46] These results suggested that wounding and subsequent A. flavus infection activated the ROS-regulated defense response in both pericarp and seed tissues of cotton.
Genes involved in stress response
Many stress responsive genes have been implicated in plant’s response to fungal infection [4], [22]. The late embryogenesis abundant (LEA) storage protein was shown to be induced in response to A. flavus infection in cotton [4]. In this study, 12 transcripts similar to LEA genes were specifically up-regulated in response to the atoxigenic A. flavus infection in pericarp (Fig 5C). In seed, only 4 LEA4 transcripts were differentially induced under the atoxigenic and the toxigenic strains infection. The heat shock proteins (HSP) play a major protective role in biotic and abiotic stresses by controlling chaperone activity and other cellular processes [22], [46]. The expression of HSPs is under the regulation of HSFs [46]. There are several HSPs and HSFs that have been reported to be induced by pathogen infection and wounding [22], [46]. Most of the transcripts similar to HSPs, in this study, were induced in seed as compared to pericarp tissue (Fig 5C). HSP70B was specifically induced under the atoxigenic strain infection in pericarp. HSP23.6-MITO, HSP90.1, HSP21 and HSP17.6II were all induced in both pericarp and seed tissues by both the strains. HSP90.1 and DNAJ-HSP were up-regulated byboth the strains specifically in seed, whereas HSP70 was up-regulated under the toxigenic strain infection. The DNAJ-HSP (Gorai.008G099300) was down-regulated in pericarp tissue. The up-regulation of a large number of HSPs in seed as compared to pericarp could be correlated to the induction of HSFs specifically in seed tissue, as discussed earlier. Stress-inducible cold regulated gene (SRC2) was specifically up-regulated in the seed tissue (Fig 5C). Two transcripts encoding glyoxal oxidase-related protein were specifically induced under the toxigenic strain infection in seed (Fig 5C). The overexpression of glyoxal oxidase gene was shown to enhance the resistance of grape plant to fungal infection [67]. The stress responsive gene glyoxalase I was also known to be induced in response to abiotic and biotic stresses in plants [22], [68]. Glyoxalase I was up-regulated in response to necrotrophic hemibiotroph fungus T. basicola in G. hirsutum [68]. The expression of a transcript coding for a glyoxalase I family protein, GLYI8, was specific to pericarp and up-regulated in response to the atoxigenic A. flavus infection (Fig 5C). The pathogenesis related genes (PR) that are associated with the resistance reactions [22], [68], [69] were also differentially expressed in the pericarp and seed tissues (Fig 5C). Among the 15 differentially expressed transcripts that were similar to PR genes, seven and three were specific to pericarp and seed, respectively. In pericarp, PR and PR-1 (Gorai.006G115900) were up-regulated specifically under the atoxigenic strain infection, and PR-T (Gorai.007G193600) was up-regulated under the toxigenic strain infection. Two PR genes (PR-4 and PR-1) were equally induced in seed, while PR-T (Gorai.009G194000) was down-regulated under both strains infection in seed. Most of these stress responsive genes have also been known to be induced under abiotic stress conditions.
5. GO and KEGG Enrichment Analysis of DEGs
In this study, GO enrichment analysis of the DEGs identified the functional categories, such as biological process, molecular function and cellular component that were distinctly represented by the atoxigenic and the toxigenic strain infection in pericarp and seed tissues of cotton. The GO categories under biological process, such as response to stimulus (GO:0050896), response to wounding (GO:0009611), response to external stimulus (GO:0009605), response to chemical stimulus (GO:0042221) and flavonoid biosynthesis (GO:0009813) were highly represented under the atoxigenic strain infection in pericarp. On the other hand, response to biotic stimulus (GO:0009607), response to stress (GO:0006950), regulation of defense response (GO:0031347), response to chitin (GO:0010200), defense response to fungus (GO:0050832) and signal transduction (GO:0007165) were enriched under the toxigenic strain infection in seed (Fig 6). Under molecular function category, most of the responses were highly enriched in pericarp as compared to seed. In pericarp, transcription regulator activity (GO:0030528), transcription factor activity (GO:0003700), transmembrane transporter activity (GO:0022857) and enzyme inhibitor activity (GO:0004857) were enriched under the atoxigenic strain infection in comparison with the toxigenic strain infection (Fig 6). Peroxidase activity (GO:0004601), pectate lyase activity (GO:0030570), antioxidant activity (GO: 0016209) and hydrolase activity (GO:0004553) were enriched under the toxigenic strain infection in pericarp as compared to the atoxigenic strain infection (Fig 6). Most of the cellular components were enriched in pericarp as compared to seed tissue (Fig 6). Component of cell wall, membrane and vacuoles were differentially enriched under the atoxigenic and the toxigenic strain infection in pericarp and seed.
Fig 6. Gene ontology enrichment analysis of DEGs in pericarp and seed tissues of cotton in response to infection with atoxigenic and toxigenic strains of A. flavus.
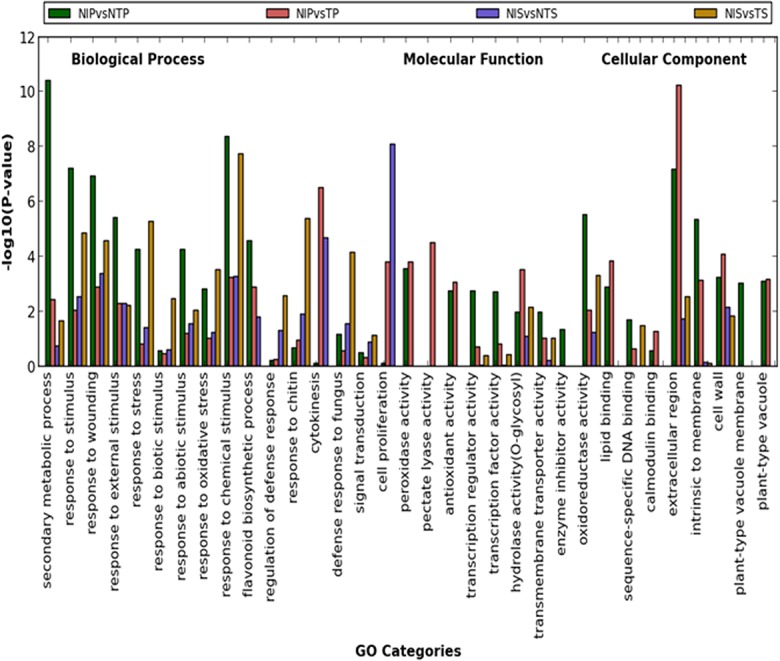
The X-axis represents the GO categories and Y-axis represents enrichment in terms of P-value.
Analysis of the biochemical pathways represented by the DEGs showed that 94, 77, 59, and 63 KEGG pathways were represented under the atoxigenic and the toxigenic strains infection in pericarp and seed, respectively. Highly enriched pathways (P < 0.05) in pericarp and seed are shown in Fig 7. The phenylpropanoid pathway, which is involved in the production of antimicrobial phytoalexins, lignins and phenolic substances [37], [42], was enriched in the toxigenic strain infection in pericarp followed by the atoxigenic infection in pericarp and seed. The flavonoid biosynthesis pathway was the most highly enriched under the atoxigenic strain infection in seed followed by pericarp. Genes in the flavonoid pathway are involved in the production of antifungal compounds and are associated with defense reactions [70]. The alkaloid biosynthesis pathway was highly enriched under both atoxigenic and toxigenic strains infection in pericarp as compared to seed (Fig 7). In tobacco plants, alkaloid biosynthesis is induced in response to insect damage and application of jasmonate [71]. This suggests that JA-regulated defense response was activated in cotton in response to A. flavus infection. Further, enrichment of arachidonic acid (AA) metabolism was observed under the toxigenic strain infection in seed followed by the atoxigenic infection in pericarp (Fig 7). AA acts as a signaling molecule, and activates plant’s defense responses through fatty acids. AA is a potent elicitor present in the pathogen, which activates plant innate immunity leading to programmed cell death and defense responses [72]. The alpha-linolenic acid metabolism pathway was enriched in pericarp in comparison to seed under both atoxigenic and toxigenic strains infection (Fig 7). JA and its derivatives, which are key regulators of plant defense responses to necrotrophic pathogens, are synthesized from the alpha-linolenic acid pathway [73], [74]. The primary metabolic pathways, such as starch and sucrose metabolism and glycerolipid metabolism, were also highly enriched under the atoxigenic strain infection in pericarp (Fig 7). The up-regulation of carbohydrate, amino acids and lipid metabolisms was suggested to regulate the signal transduction cascade during plant defense responses [75]. The biochemical pathways involved in response to the atoxigenic strain of A. flavus infection in pericarp and seed tissue of cotton can be manipulated for stress tolerance in cotton.
Fig 7. Highly represented KEGG metabolic pathways in pericarp and seed tissues of cotton under A. flavus infection.

The X-axis represents the enrichment in terms of P-value and Y-axis represents the biochemical pathways.
Validity of the next generation sequence data was confirmed by reverse-transcription PCR of 10 genes belonging to different functional categories with fold change expression of 5-fold or above (from sequence data) under experimental conditions relative to non-inoculated control. The results showed significant up-regulation of their mRNA accumulation under infection by the atoxigenic or the toxigenic strain infection in a tissue-dependent manner (S1 Fig).
Conclusions
Little is known about the molecular mechanisms of A. flavus resistance in cotton, largely due to the lack of a resistant cotton germplasm. However, understanding the expression profile of the genes, especially in response to the atoxigenic strain infection, could provide clues to the molecular mechanism of resistance, in addition to the physical barrier, conferred by the atoxigenic strains against the toxigenic strain. Comparative analysis of the genes involved in specific GO categories of the atoxigenic vis-à-vis the toxigenic strain infection will lead to the identification of promising candidates for genetic manipulation toward development of cotton varieties resistant to A. flavus. For example, genes with transcriptional regulation involved in response to stress stimulus, involved in flavonoid biosynthesis and lipid biding in extracellular regions (Fig 6) could be considered promising candidates for further validation through functional characterization. In addition, our ongoing comparative analysis of the cotton transcriptome with available corn and peanut transcriptome, induced under A. flavus infection, will provide a better understanding of the genetic and biochemical basis of A. flavus-cotton interaction and also identify conserved orthologous genes in cotton for their functional translation in conferring resistance to A. flavus through genetic manipulation in cotton.
Supporting Information
Gene expression was normalized using cotton elongation factor [4] as the internal reference against the expression in non-inoculated control, which was set to 1.
(TIF)
Sheet 1, Nomenclature of genes and primer sequences used for expression analysis through RT-PCR; sheet 2, All differentially expressed genes discussed in the manuscript and used for heatmap in Fig 2A; sheet 3, Genes from different classes used in the generation of heatmaps for Figs 4 and 5.
(XLSX)
Acknowledgments
The authors thank Greg Ford, SRRC-USDA, for his help with the fungal inoculation of cotton bolls. The research was funded by a grant through Specific Cooperative Agreement (SCA# 58-6435-0-529) from the USDA-AFRI. This manuscript has been approved for publication by the Louisiana Agricultural Experiment Station as MS# 2015-306-22458.
Data Availability
All data are available at http://www.ncbi.nlm.nih.gov/sra; Submission ID: SUB828139.
Funding Statement
Funded by United States Department of Agriculture—National Institute of Food and Agriculture (SCA# 58-6435-0-529).
References
- 1. Cleveland TE, Dowd PF, Desjardins AE, Bhatnagar D, Cotty PJ. United States Department of Agriculture—Agricultural Research Service research on pre-harvest prevention of mycotoxins and mycotoxigenic fungi in US crops. Pest Manag Sci. 2003;59(6–7):629–42. [DOI] [PubMed] [Google Scholar]
- 2. Wogan GN. Impacts of chemicals on liver cancer risk. Semin Cancer Biol. 2000;10(3):201–10. [DOI] [PubMed] [Google Scholar]
- 3. International Agency for Research on Cancer (IARC). IARC Monographs on the evaluation of the carcinogenic risks to humans: some naturally occurring substances: food items and constituents, heterocyclic aromatic amines and mycotoxins IARC, Lyon, France: 1993;56:245–540. [Google Scholar]
- 4. Lee S, Rajasekaran K, Ramanarao MV, Bedre R, Bhatnagar D, Baisakh N. Identifying cotton (Gossypium hirsutum L.) genes induced in response to Aspergillus flavus infection. Physiol Mol Plant Pathol. 2012;80:35–40. [Google Scholar]
- 5. Yin YN, Yan LY, Jiang JH, Ma ZH. Biological control of aflatoxin contamination of crops. J Zhejiang Univ-Sc B. 2008;9(10):787–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kelley RY, Williams WP, Mylroie JE, Boykin DL, Harper JW, Windham GL, et al. Identification of Maize Genes Associated with Host Plant Resistance or Susceptibility to Aspergillus flavus Infection and Aflatoxin Accumulation. Plos One. 2012;7(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richard JL, Payne GA. Mycotoxins: risks in plant, animal and human systems Ames, IA: Council for Agricultural Science and Technology; 2003. [Google Scholar]
- 8. Yu JJ. Current Understanding on Aflatoxin Biosynthesis and Future Perspective in Reducing Aflatoxin Contamination. Toxins. 2012;4(11):1024–57. 10.3390/toxins4111024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van Egmond HP, Jonker MA, Abbas HK. Worldwide regulations on Aflatoxins. Aflatoxin and food safety. 2005;77–93. [Google Scholar]
- 10.Lillehoj EB, Wall JH. Decontamination of aflatoxin-contaminated maize grain. In Proceedings of US Universities-CIMMYT Maize Aflatoxin Workshop, El Batan, Mexico.1987.
- 11. Wright MS, Greene-McDowelle DM, Zeringue HJ, Bhatnagar D, Cleveland TE. Effects of volatile aldehydes from Aspergillus-resistant varieties of corn on Aspergillus parasiticus growth and aflatoxin biosynthesis. Toxicon. 2000;38(9):1215–23. [DOI] [PubMed] [Google Scholar]
- 12. Cotty PJ. Influence of field application of an atoxigenic strain of Aspergillus flavus on the populations of A. flavus infecting cotton bolls and on the aflatoxin content of cottonseed. Phytopathology. 1994;84:1270–1277. [Google Scholar]
- 13. Dorner JW. Biological control of aflatoxin contamination of crops. J Toxicol-Toxin Rev. 2004;23(2–3):425–50. [Google Scholar]
- 14. Pitt JI, Hocking AD. Mycotoxins in Australia: biocontrol of aflatoxin in peanuts. Mycopathologia. 2006;162(3):233–43. [DOI] [PubMed] [Google Scholar]
- 15. Dorner JW. Management and prevention of mycotoxins in peanuts. Food Addit Contam. 2008;25(2):203–8. [DOI] [PubMed] [Google Scholar]
- 16. Bari R, Jones J. Role of plant hormones in plant defense responses. Plant Mol Biol. 2009;69(4):473–88. 10.1007/s11103-008-9435-0 [DOI] [PubMed] [Google Scholar]
- 17. Birkenbihl RP, Somssich IE. Transcriptional plant responses critical for resistance towards necrotrophic pathogens. Front Plant Sci. 2011;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. St Clair DA. Quantitative Disease Resistance and Quantitative Resistance Loci in Breeding. Annu Rev Phytopathol. 2010;48:247–68. 10.1146/annurev-phyto-080508-081904 [DOI] [PubMed] [Google Scholar]
- 19. Yu J, Bhatnagar D, Cleveland TE, Payne G, Nierman WC, Bennett JW. Aspergillus flavus genetics and genomics in solving mycotoxin contamination of food and feed. OMICs technologies: Tools for Food Science; 2012. pp 367–402. [Google Scholar]
- 20. Guo BZ, Fedorova ND, Chen XP, Wan CH, Wang W, Nierman WC, et al. Gene Expression Profiling and Identification of Resistance Genes to Aspergillus flavus Infection in Peanut through EST and Microarray Strategies. Toxins. 2011;3(7):737–5 10.3390/toxins3070737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luo M, Brown RL, Chen ZY, Menkir A, Yu JJ, Bhatnagar D. Transcriptional Profiles Uncover Aspergillus flavus-Induced Resistance in Maize Kernels. Toxins. 2011;3(7):766–86. 10.3390/toxins3070766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cleveland TE, Yu JJ, Bhatnagar D, Chen ZY, Brown RL, Chang PK, et al. Progress in elucidating the molecular basis of the host plant—Aspergillus flavus interaction, a basis for devising strategies to reduce aflatoxin contamination in crops. J Toxicol-Toxin Rev. 2004;23(2–3):345–80. [Google Scholar]
- 23. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–U130. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Du Z, Zhou X, Ling Y, Zhang ZH, Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010;38:W64–W70. 10.1093/nar/gkq310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goffard N, Weiller G. PathExpress: a web-based tool to identify relevant pathways in gene expression data. Nucleic Acids Res. 2007;35:W176–W81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang KB, Wang ZW, Li FG, Ye WW, Wang JY, Song GL, et al. The draft genome of a diploid cotton Gossypium raimondii . Nat Genet. 2012;44(10):1098–1103. 10.1038/ng.2371 [DOI] [PubMed] [Google Scholar]
- 28. Li FG, Fan GY, Wang KB, Sun FM, Yuan YL, Song GL, et al. Genome sequence of the cultivated cotton Gossypium arboreum . Nat Genet. 2014;46(6):567–72. 10.1038/ng.2987 [DOI] [PubMed] [Google Scholar]
- 29. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11. 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–U54. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–U174. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. 10.1038/nbt.2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Drew DP, Dueholm B, Weitzel C, Zhang Y, Sensen CW, Simonsen HT. Transcriptome Analysis of Thapsia laciniata Rouy Provides Insights into Terpenoid Biosynthesis and Diversity in Apiaceae. Int J Mol Sci. 2013;14(5):9080–98. 10.3390/ijms14059080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cardoso-Silva CB, Costa EA, Mancini MC, Balsalobre TWA, Canesin LEC, Pinto LR, et al. De Novo Assembly and Transcriptome Analysis of Contrasting Sugarcane Varieties. Plos One. 2014;9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tohidfar M, Mohammadi M, Ghareyazie B. Agrobacterium-mediated transformation of cotton (Gossypium hirsutum) using a heterologous bean chitinase gene. Plant Cell Tiss Org Cult. 2005;83(1):83–96. [Google Scholar]
- 36. Wang SY, Ye XY, Chen J, Rao PF. A novel chitinase isolated from Vicia faba and its antifungal activity. Food Res Int. 2012;45(1):116–22. [Google Scholar]
- 37. Collinge DB, Slusarenko AJ. Plant Gene-Expression in response to pathogens. Plant Mol Biol. 1987;9(4):389–410. 10.1007/BF00014913 [DOI] [PubMed] [Google Scholar]
- 38. Prasad K, Bhatnagar-Mathur P, Waliyar F, Sharma KK. Overexpression of a chitinase gene in transgenic peanut confers enhanced resistance to major soil borne and foliar fungal pathogens. J Plant Biochem Biotechnol. 2013;22(2):222–33. [Google Scholar]
- 39. Rohini VK, Rao KS. Transformation of peanut (Arachis hypogaea L.) with tobacco chitinase gene: variable response of transformants to leaf spot disease. Plant Sci. 2001;160(5):889–98. [DOI] [PubMed] [Google Scholar]
- 40. Baisakh N, Datta K, Oliva N, Ona I, Rao GJN, Mew TW, et al. Rapid development of homozygous transgenic rice using anther culture harboring rice chitinase gene for enhanced sheath blight resistance. Plant Biotechnol. 2001;18:101–8. [Google Scholar]
- 41. Chen ZY, Brown RL, Russin JS, Lax AR. Cleveland TE. A corn trypsin inhibitor with antifungal activity inhibits Aspergillus flavus α-amylase. Phytopathology. 1999;89:902–907. 10.1094/PHYTO.1999.89.10.902 [DOI] [PubMed] [Google Scholar]
- 42. Lawton MA, Lamb CJ. Transcriptional Activation of Plant Defense Genes by Fungal Elicitor, Wounding, and Infection. Mol Cell Biol. 1987;7(1):335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chappell J, Hahlbrock K. Transcription of plant defense genes in response to UV-Light or fungal elicitor. Nature. 1984;311(5981):76–8. [Google Scholar]
- 44. Ecker JR, Davis RW. Plant Defense Genes Are Regulated by Ethylene. P Natl Acad Sci USA. 1987;84(15):5202–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. HammondKosack KE, Jones JDG. Resistance gene-dependent plant defense responses. Plant Cell. 1996;8(10):1773–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheong YH, Chang HS, Gupta R, Wang X, Zhu T, Luan S. Transcriptional profiling reveals novel interactions between wounding, pathogen, abiotic stress, and hormonal responses in Arabidopsis. Plant Physiol. 2002;129(2):661–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Beardmore J, Ride JP, Granger JW. Cellular Lignification as a Factor in the Hypersensitive Resistance of Wheat to Stem Rust. Physiol Plant Pathol. 1983;22(2):209. [Google Scholar]
- 48. Sanchez-Vallet A, Lopez G, Ramos B, Delgado-Cerezo M, Riviere MP, Llorente F, et al. Disruption of Abscisic Acid Signaling Constitutively Activates Arabidopsis Resistance to the Necrotrophic Fungus Plectosphaerella cucumerina . Plant Physiol. 2012;160(4):2109–24. 10.1104/pp.112.200154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dolezal AL, Shu XM, OBrian GR, Nielsen DM, Woloshuk CP, Boston RS, et al. Aspergillus flavus infection induces transcriptional and physical changes in developing maize kernels. Front Microbiol. 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Garcia-Brugger A, Lamotte O, Vandelle E, Bourque S, Lecourieux D, Poinssot B, et al. Early signaling events induced by elicitors of plant Defenses. Mol Plant Microbe In. 2006;19(7):711–24. [DOI] [PubMed] [Google Scholar]
- 51. Tena G, Boudsocq M, Sheen J. Protein kinase signaling networks in plant innate immunity. Curr Opin Plant Biol. 2011;14(5):519–29. 10.1016/j.pbi.2011.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dodds PN, Rathjen JP. Plant immunity: towards an integrated view of plant-pathogen interactions. Nat Rev Genet. 2010;11(8):539–48. 10.1038/nrg2812 [DOI] [PubMed] [Google Scholar]
- 53. Asano T, Hayashi N, Kikuchi S, Ohsugi R. CDPK-mediated abiotic stress signaling. Plant Signaling & Behavior; 2012;7:817–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoshioka H, Mase K, Yoshioka M, Kobayashi M, Asai S. Regulatory mechanisms of nitric oxide and reactive oxygen species generation and their role in plant immunity. Nitric Oxide-Biol Ch. 2011;25(2):216–21. [DOI] [PubMed] [Google Scholar]
- 55. Wang P, Du Y, Li Y, Ren D, Song CP. Hydrogen peroxide–mediated activation of MAP kinase 6 modulates nitric oxide biosynthesis and signal transduction in Arabidopsis . The Plant Cell Online. 2010;22:2981–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Asai S, Ohta K, Yoshioka H. MAPK signaling regulates nitric oxide and NADPH oxidase-dependent oxidative bursts in Nicotiana benthamiana . Plant Cell. 2008;20(5):1390–406. 10.1105/tpc.107.055855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Torres MA. ROS in biotic interactions. Physiol Plant. 2010;138(4):414–29. 10.1111/j.1399-3054.2009.01326.x [DOI] [PubMed] [Google Scholar]
- 58. Ghanashyam C, Jain M. Role of auxin-responsive genes in biotic stress responses. Plant Signal Behav. 2009;4:846–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Domingo C, Andres F, Tharreau D, Iglesias DJ, Talon M. Constitutive Expression of OsGH3.1 Reduces Auxin Content and Enhances Defense Response and Resistance to a Fungal Pathogen in Rice. Mol Plant Microbe In. 2009;22(2):201–10. [DOI] [PubMed] [Google Scholar]
- 60. Kant S, Bi YM, Zhu T, Rothstein SJ. SAUR39, a Small Auxin-Up RNA Gene, Acts as a Negative Regulator of Auxin Synthesis and Transport in Rice. Plant Physiol. 2009;151(2):691–701. 10.1104/pp.109.143875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Siemens J, Keller I, Sarx J, Kunz S, Schuller A, Nagel W, et al. Transcriptome analysis of Arabidopsis clubroots indicate a key role for cytokinins in disease development. Mol Plant Microbe In. 2006;19(5):480–94. [DOI] [PubMed] [Google Scholar]
- 62. Guo RY, Yu FF, Gao Z, An HL, Cao XC, Guo XQ. GhWRKY3, a novel cotton (Gossypium hirsutum L.) WRKY gene, is involved in diverse stress responses. Mol Biol Rep. 2011;38(1):49–58. 10.1007/s11033-010-0076-4 [DOI] [PubMed] [Google Scholar]
- 63. Du L, Chen Z. Identification of genes encoding receptor‐like protein kinases as possible targets of pathogen‐and salicylic acid‐induced WRKY DNA‐binding proteins in Arabidopsis . Plant J. 2000;24:837–47. [DOI] [PubMed] [Google Scholar]
- 64. Lorenzo O, Piqueras R, Sanchez-Serrano JJ, Solano R. Ethylene Response Factor1 Integrates Signals from Ethylene and Jasmonate Pathways in Plant Defense. Plant Cell. 2003;15(1):165–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Singh KB, Foley RC, Onate-Sanchez L. Transcription factors in plant defense and stress responses. Curr Opin Plant Biol. 2002;5(5):430–6. [DOI] [PubMed] [Google Scholar]
- 66. Bolwell GP, Wojtaszek P. Mechanisms for the generation of reactive oxygen species in plant defense—a broad perspective. Physiol Mol Plant P. 1997;51(6):347–66. [Google Scholar]
- 67. Guan X, Zhao HQ, Xu Y, Wang YJ. Transient expression of glyoxal oxidase from the Chinese wild grape Vitis pseudoreticulata can suppress powdery mildew in a susceptible genotype. Protoplasma. 2011;248(2):415–23. 10.1007/s00709-010-0162-4 [DOI] [PubMed] [Google Scholar]
- 68. Coumans JVF, Polijak A, Raftery MJ, Backhouse D, Pereg-Gerk L. Analysis of cotton (Gossypium hirsutum) root proteomes during a compatible interaction with the black root rot fungus Thielaviopsis basicola . Proteomics. 2009;9(2):335–49. 10.1002/pmic.200800251 [DOI] [PubMed] [Google Scholar]
- 69. Pritsch C, Muehlbauer GJ, Bushnell WR, Somers DA, Vance CP. Fungal development and induction of defense response genes during early infection of wheat spikes by Fusarium graminearum . Mol Plant Microbe In. 2000;13(2):159–69. [DOI] [PubMed] [Google Scholar]
- 70. Treutter D. Significance of flavonoids in plant resistance and enhancement of their biosynthesis. Plant Biology. 2005;7(6):581–91. [DOI] [PubMed] [Google Scholar]
- 71. Todd AT, Liu EW, Polvi SL, Pammett RT, Page JE. A functional genomics screen identifies diverse transcription factors that regulate alkaloid biosynthesis in Nicotiana benthamiana . Plant J. 2010;62(4):589–600. 10.1111/j.1365-313X.2010.04186.x [DOI] [PubMed] [Google Scholar]
- 72. Savchenko T, Walley JW, Chehab EW, Xiao YM, Kaspi R, Pye MF, et al. Arachidonic Acid: An Evolutionarily Conserved Signaling Molecule Modulates Plant Stress Signaling Networks. Plant Cell. 2010;22(10):3193–205. 10.1105/tpc.110.073858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wasternack C. Action of jasmonates in plant stress responses and development applied aspects. Biotechnol Adv. 2014;32(1):31–9. 10.1016/j.biotechadv.2013.09.009 [DOI] [PubMed] [Google Scholar]
- 74. Robert-Seilaniantz A, Grant M, Jones JDG. Hormone Crosstalk in Plant Disease and Defense: More than just JASMONATE-SALICYLATE antagonism. Annu. Rev. Phytopathol., 2011;49:317–43. 10.1146/annurev-phyto-073009-114447 [DOI] [PubMed] [Google Scholar]
- 75. Rojas CM, Senthil-Kumar M, Tzin V, Mysore KS. Regulation of primary plant metabolism during plant-pathogen interactions and its contribution to plant defense. Front Plant Sci. 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene expression was normalized using cotton elongation factor [4] as the internal reference against the expression in non-inoculated control, which was set to 1.
(TIF)
Sheet 1, Nomenclature of genes and primer sequences used for expression analysis through RT-PCR; sheet 2, All differentially expressed genes discussed in the manuscript and used for heatmap in Fig 2A; sheet 3, Genes from different classes used in the generation of heatmaps for Figs 4 and 5.
(XLSX)
Data Availability Statement
All data are available at http://www.ncbi.nlm.nih.gov/sra; Submission ID: SUB828139.