

Abstract

The thiazolidinediones (TZD) typified by rosiglitazone are the only approved therapeutics targeting PPARγ for the treatment of type-2 diabetes (T2DM). Unfortunately, despite robust insulin sensitizing properties, they are accompanied by a number of severe side effects including congestive heart failure, edema, weight gain, and osteoporosis. We recently identified PPARγ antagonists that bind reversibly with high affinity but do not induce transactivation of the receptor, yet they act as insulin sensitizers in mouse models of diabetes (SR1664).1 This Letter details our synthetic exploration around this novel series of PPARγ antagonists based on an N-biphenylmethylindole scaffold. Structure–activity relationship studies led to the identification of compound 46 as a high affinity PPARγ antagonist that exhibits antidiabetic properties following oral administration in diet-induced obese mice.
Keywords: PPARγ, nuclear receptor, indole, diabetes
The peroxisome proliferator-activated receptors (PPARs) are ligand-dependent transcription factors and members of the nuclear receptor superfamily that regulate gene expression patterns of diverse biological processes.2 The PPARs play a key role in transcriptional regulation of genes involved in lipid and glucose metabolism and inflammation. The PPAR family consists of three genes, PPARα, PPARγ, and PPARβ/δ with each having different tissue distribution, selectivity, and responsiveness to ligands. PPARγ has been implicated in several disorders including atherosclerosis, diabetes, obesity, and cancer. Thus, the search for potent and selective modulators of PPARγ to be used as therapies to treat these pathologies is well justified.3 In fact PPARγ agonists have for many years represented a promising approach to treat insulin resistance associated with type 2 diabetes.4,5 PPARγ is the molecular target of the thiazolidinedione (TZD) class of antidiabetic drugs rosiglitazone and pioglitazone.6 These drugs were developed prior to their molecular mechanism of action being determined. Both compounds are high affinity full agonists of PPARγ. While the TZD class of drugs exhibits robust effects on glucose metabolism and insulin sensitivity, it is associated with a myriad of undesirable side effects (adverse events; AEs) including fluid retention with increased risk of heart failure, weight gain, loss of bone mineral density, and cancer.7−9 In efforts to improve the therapeutic index of PPARγ modulators, partial agonists were developed that proved to be equally efficacious as insulin sensitizers as full agonists yet with significantly improved AE profiles.10−15 This disconnect between agonism and efficacy raised questions about the mechanism of action of such compounds. Recently it was reported that efficacy of both full and partial agonists correlated with their ability to block the obesity-linked phosphorylation of PPARγ at serine 273 (pS273), and counter AEs correlate with the magnitude of agonism.16 Importantly, blockage of pS273 was shown to be mediated via a conformational change in the ligand binding domain of PPARγ and not by kinase inhibition. Subsequently, compounds were developed (e.g., SR1664) that bind PPARγ with high affinity and block pS273, yet are devoid of agonism but still afford robust antidiabetic activity in rodent models of diabetes.1 These findings clearly suggest that it is possible to separate agonism of the receptor from efficacy, and such efforts should yield compounds with improved therapeutic index. In this Letter, we report the discovery and structure–activity relationship (SAR) of a novel series of indole-based carboxylic acids related to SR1664.
This project initiated with the prospect of identifying or synthesizing noncovalent antagonists of PPARγ.17 Covalent antagonists have been identified but are unlikely to be developed as therapeutics.18,19 It was not clear if this proposal was even feasible at the onset, but it entailed a challenge that would shed light on the hypothesis that one could separate agonism of PPARγ from its efficacy as a potential diabetic therapeutic. Without access to a high-throughput screen, we investigated several alternative approaches to identify antagonists of PPARγ. One approach involved starting with known PPARγ partial agonists from both the primary and patent literature and designing out the agonism. With a plethora of potential starting points in the public domain and with little guidance on where to start, we chose a handful of different scaffolds and began synthesizing some analogures. Structures are provided in the Supporting Information (Figure S1).
Partials agonists are defined as weak activators of PPARγ that elicit the same activation pattern as full agonists, but with a lower maximal activity. The antihypertensive agent Telmisartan is an angiotensin II receptor antagonist, but it has also been reported to function as a weak PPARγ agonist.20,21 Scientists at GlaxoSmithKline optimized the telmisartan scaffold to afford GSK 7b, as a potent PPARγ partial agonist (Figure 1A).22 A group at Amgen developed AMG-131 as a potent and highly selective PPARγ partial agonist that advanced into Phase 2 clinical trials.23 FK614 from Astellas Pharma is reported as a selective partial agonist for PPARγ in a GAL4-PPAR transactivation assay.11,24 MBX-102 (metaglidasen) is a weak PPARγ ligand that exhibits both partial agonist and antagonist activity.25,26 Lastly, Merck identified MRL-24 as a potent partial agonist of PPARγ, which was ultimately optimized and advanced into clinical trials as MK-0533.12,27
Figure 1.

(A) Identification of SR1664 from GSK 7b (SR9034). (B) Effect of ligands on PPARγ GAL-4 transactivation.
Roughly two dozen analogues in each series were synthesized and screened in both a PPARγ binding assay using a competitive Lanthascreen assay format and a fluorescently labeled probe as well as in a transactivation assay using PPARγ-GAL4 and a UAS-Luciferase reporter assay system in an effort to help identify which series held the most promise. Compounds that exhibited minimum transactivation in GAL-4 were profiled using full-length PPARγ and a 5xPPRE-Luciferase reporter construct. Our definition of an antagonist of PPARγ is one wherein the transactivation efficiency is ≤10% of that of rosiglitazone at 10 μM concentration, while still retaining good affinity for the receptor (i.e., IC50 ≤ 250 nM). Most analogues synthesized in the different scaffolds in Figure S1 afforded partial agonists, but several compounds in the GSK 7b series had minimal PPARγ transactivation and looked promising as PPARγ antagonists. Initial SAR simply focused on different indole amides and led to the discovery of SR1664 (Figure 1).1 Remarkably, SR1664 has similar affinity for PPARγ as rosiglitazone, but virtually no activation of the receptor as is inherent to GSK7b.
While this was proof-of-principle that one could in fact identify compounds that bound PPARγ with high affinity with little to no transactivation of the receptor, very little was known about the SAR around this scaffold. Additionally, SR1664 had poor oral in vivo exposure in rodents, and the nitro group needed to be replaced for potential toxicity concerns. In an effort to expand the SAR around SR1664 and design orally active PPARγ antagonists, considerable optimization was undertaken. We envisioned modifications of all parts of the molecule as shown in Figure 2. First, we focused our efforts on modification of the substituents on the phenyl ring as well as at the stereogenic center. In the context of expanding the SAR and improving in vivo properties, we sought to make modifications to the indole ring as well as to the biphenyl carboxylic acid. The synthesis of these compounds 15–69 is outlined in Scheme 1 and Schemes S1–S4 in the Supporting Information.
Figure 2.

SAR optimization of SR1664.
Scheme 1. Synthesis of Biphenyl Analogues.

Reagents and conditions: (a) 2-butanone, HCl, dioxane, reflux; (b) allyl bromide, K2CO3, DMAC, rt; (c) tert-butyl 4′-(bromomethyl)-[1,1′-biphenyl]-2-carboxylate, NaH, DMF; (d) Pd(PPh3)4, THF, rt; (e) R-PhCH(R′)NH2, HATU, DCM, rt; (f) TFA, DCM, rt.
Indole acid (2) was obtained through the condensation of 4-hydrazine benzoic acid 1 and 2-butanone in high yield followed by esterification with allyl bromide to give 3. Alkylation of the indole with tert-butyl 4′-(bromomethyl)-[1,1′-biphenyl]-2-carboxylate provided 4. Cleavage of the allyl group with tetrakis(triphenylphosphine)palladium(0) gave acid 5. Lastly, amide coupling with an amine of interest followed by hydrolysis of the tert-butyl ester gave final products (Tables 1–4).
Table 1. SAR Modification of the Benzamide.

| Cmpd | R | Lantha (IC50, nM)a | GAL-4 (EC50, nM)b | Cmpd | R | Lantha (IC50, nM)a | GAL-4 (EC50, nM)b |
|---|---|---|---|---|---|---|---|
| 9034 | α-ethyl-benzylamine | 0.37 | 13(40%) | 22 | 4-Me-benzylamine | 4 | 14 (30%) |
| 15 | benzylamine | 1 | 300 (40%) | 23 | 4-Br-benzylamine | 14 | 516 (25%) |
| 16 | 2-Me-benzylamine | 6 | 632 (10%) | 24 | 4-NO2-benzylamine | 38 | 1344 (20%) |
| 17 | 2-NH2-benzylamine | 32 | 2532 (10%) | 25 | 4-t-Bu-benzylamine | 667 | 124 (10%) |
| 18 | 2-NO2-benzylamine | 24 | 831 (10%) | 26 | thiophen-2-ylmethanamine | 1 | 196 (35%) |
| 19 | 3-Me-benzylamine | 5 | 363 (30%) | 27 | diphenylmethanamine | 26 | 1 (15%) |
| 20 | 3-OMe-benzylamine | 4 | 188 (20%) | 28 | cyclobutanamine | 32 | 5665 (40%) |
| 21 | 3-NO2-benzylamine | 20 | 822 (18%) | 39 | cyclohexylmethanamine | 78 | 179(40%) |
Values are means of at least two experiments. All standard deviations are <20%.
Transactivation at 10 μM.
Table 4. Substitutions at the Stereogenic Center.

| Cmpd | R | stereochemistry | Lantha (IC50, nM)a | GAL-4 (EC50, nM)b |
|---|---|---|---|---|
| 53 | H | 5 | 133 (30%) | |
| 54 | CF3 | S | 17 | 100(10%) |
| 45 | Me | S | 30 | 852 (5%) |
| 55 | Et | S | 9 | 100(15%) |
| 56 | iPr | S | 80 | 133(10%) |
| 57 | cyclopropyl | S | 8 | 60(18%) |
| 58 | nBu | S | 81 | 116(15%) |
| 59 | iBu | S | 1222 | 226(18%) |
| 60 | Bn | S | 97 | 150 (20%) |
Values are means of at least two experiments. All standard deviations are <20%.
Transactivation at 10 μM.
For compounds wherein the enantiomerically pure amine was not commercially available, these were synthesized using Ellman chemistry28,29 as shown in Scheme S1 in the Supporting Information. A few modifications to the indole core were also investigated. The synthesis of these analogues is outlined in Scheme S2 in the Supporting Information. Finally, we maintained the biphenylmethyl substitution and explored acid bioisosteres (Schemes S3 and S4 in the Supporting Information).
We first explored SAR of the left-hand side of the molecule (Table 1). Initial exploration of the amide group seemed simplest by first removing the stereogenic center found in GSK 7b (SR9034). Removing the α-ethyl substituent (15) led to a slight loss in affinity; however, the compound retained its partial agonism of the receptor. In general, most types and positions of substitutions on the phenyl ring were well tolerated and yielded potent PPARγ partial agonists, which were of little interest given we were searching for antagonists.
Substitution was tolerated at the ortho-, meta-, and para-positions with only slight variations in activity. Indeed if we compare ortho- (18), meta- (21), and para-nitro (24) analogues, these compounds have similar affinities and transactivation potentials for PPARγ. The benzyl group was not required as heterocyclic replacements such as 26 (IC50 = 1 nM, EC50 = 196 nM) were also potent. Surprisingly, the sterically encumbering benzhydryl amide (27) with an IC50 = 26 nM and an EC50 = 1 nM is also well tolerated, perhaps giving an indication of the size of the binding pocket. Lastly, aryl groups are not required for potency, as simple cycloalkyl groups (28, 29) afford potent partial agonists as well.
We next examined the effect of the α-substituent in the benzyl amine. The parent molecule, SR9034, contains a racemic ethyl group and is a potent PPARγ partial agonist (Table 2). Separation of the enantiomers led to (S)-30 (IC50 = 3 nM and EC50 = 53 nM) and (R)-31 (IC50 = 0.13 nM and EC50 = 3 nM), both of which are potent partial agonists; however, the (R)-enantiomer was an order of magnitude more potent both in binding and transactivation. We switched from α-ethyl to α-methyl given the increased access to commercially available starting materials. This did not seem to affect in vitro activity as 4-substituted α-methylbenzyl amine analogues were equipotent to the lead SR9034. Introduction of a p-substituent of the appropriate size led to an unexpected breakthrough. Smaller groups such as fluorine (S)-32 and (R)-33 or methoxy groups (S)-34 and (R)-35 did not seem to have much effect on in vitro activity, with the same general trend observed in that the (R)-enantiomer was the more potent of the pair. However, the (S)-4-nitro analogue SR1664, despite good affinity for PPARγ, exhibited minimal transactivation in GAL-4. The different effects on transactivation between SR1664 and its enantiomer 36, are quite striking (Figure S2 in the Supporting Information). SR1664 was also profiled using the full length PPARγ-reporter assay and exhibited no transactivation at 10 μM. It is possible to observe no transactivation for compounds that do not get into cells. Therefore, we screened rosiglitazone in the presence of increasing concentrations of SR1664. This resulted in a right-fold shift in the rosiglitazone transactivation curve indicating competitive binding by SR1664 alleviating any concerns of lack of cell penetrance. Thus, SR1664 was identified as our first PPARγ antagonist from this series. From this small subset of analogues, we concluded that not only was the stereochemistry at the amine alpha-center important but so was the size of the phenyl substituent. To fully test this, however, we would need to go beyond what was commercially available, and synthesize additional amines, and for this, we turned to Ellman chemistry as highlighted in Scheme S1 (Table 3).
Table 2. SAR of the Benzyl Amine.
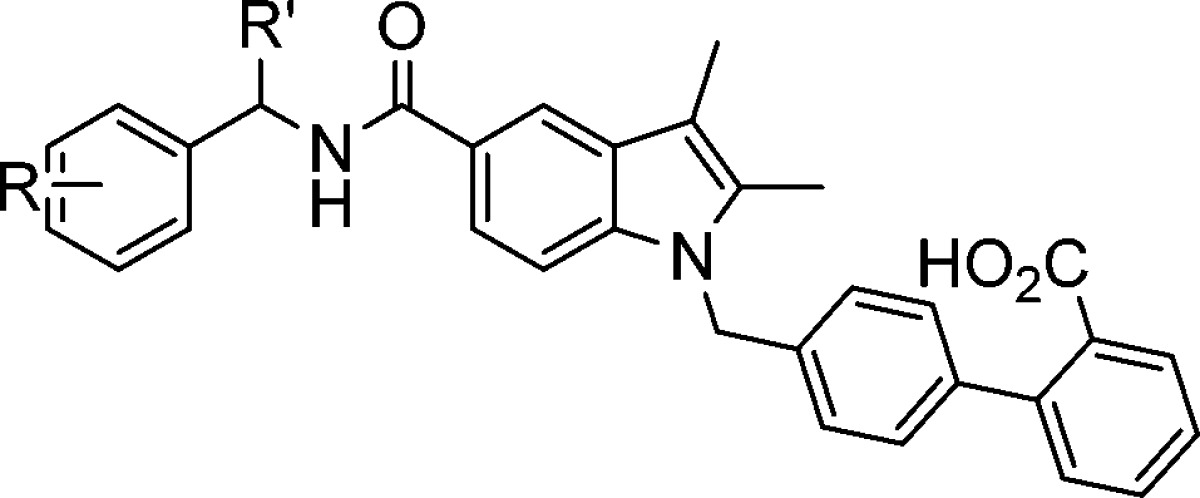
| Cmpd | R | R′ | stereochemistry | Lantha (IC50, nM)a | GAL-4 (EC50, nM)b |
|---|---|---|---|---|---|
| 9034 | H | Et | ± | 0.37 | 13 (40%) |
| 30 | H | Et | S | 3 | 53 (20%) |
| 31 | H | Et | R | 0.13 | 3 (30%) |
| 32 | 4-F | Me | S | 21 | 638 (40%) |
| 33 | 4-F | Me | R | 0.79 | 47 (50%) |
| 34 | 4-OMe | Me | S | 0.90 | 2076 (30%) |
| 35 | 4-OMe | Me | R | 0.54 | 53 (60%) |
| 1664 | 4-NO2 | Me | S | 80 | 4288 (10%) |
| 36 | 4-NO2 | Me | R | 2 | 503 (75%) |
| 37 | 4-Br | Me | S | 28 | 516 (20%) |
| 38 | 4-Br | Me | R | 0.73 | 129 (90%) |
Values are means of at least two experiments. All standard deviations are <20%.
Transactivation at 10 μM.
Table 3. SAR for Phenyl Ring Substitution.


Values are means of at least two experiments. All standard deviations are <20%.
Transactivation at 10 μM.
The next set of analogues incorporated the (S)-alpha-methyl substituent and looked to examine the effect of phenyl substitution in more detail (Table 3). We introduced substitution at the 2-, 3-, and 4-positions of the phenyl ring ranging from small to medium to large with respect to steric bulk. At the 2-position, substitution was tolerated, but analogues were all partial agonists of PPARγ. If the substitution was too large (42), affinity for the receptor is compromised. The 3-position also tolerated substitution, and as the size of the substituent increased, we were able to almost completely remove transactivation (45, 47). The same trend was observed at the 4-position. Small substitution led to potent partial agonists, while analogues containing larger substitution led to antagonists (51, 52).
To put into context the minimal transactivation observed at 10 μM in the GAL-4 assay, the dose response curves for 46 and 51 are included in Figure 3. As can be seen when compared to rosiglitazone, there is essentially no activation of PPARγ despite very good affinity for the receptor. Compound 46 was further profiled for transactivation using full length PPARγ and again showed no activity (Figure 3).
Figure 3.

Transactivation curves for PPARγ antagonists in (A) GAL4-PPARγ and (B) full length PPARγ.
With this information, we fixed the left-hand side of the molecule with 3-isopropylphenyl as in 45 and explored the SAR of the stereogenic center (Table 4). Not surprising, when R = H (53), the compound is a potent partial agonist, in agreement with data from the compounds in Table 1. Trifluoromethyl (54) and ethyl (55) were slightly more potent than α-methyl analogue 45, with a slight increase in transactivation. Most of the substitutions here were tolerated with regards to binding affinity, with the exception of R = iBu (59); however, as the size of the substitution increased beyond ethyl, there seemed to be a noticeable increase in transactivation of PPARγ. These results suggested that the methyl group in the S-configuration was optimal for PPARγ antagonists in this series. Attempts to manipulate the methyl groups at the 2,3-position of the indole core did not offer any advantages with regards to antagonism of PPARγ (data not shown). Removal of one methyl group led to a slight loss in affinity and removal of the second methyl group led to a further drop with no effect on transactivation. Loss in affinity and potential exposure of the indole ring to metabolism led us to maintain the 2,3-dimethylindole substitution in all future analogues.
We also looked at the effect of modifications to the indole core itself on PPARγ potency and transactivation (Table S2 in the Supporting Information). Installation of a benzooxazolone instead of indole as a central core gave 61, which essentially wiped out all potency on PPARγ. A benzoimidazolone core was slightly better tolerated (62) but still led to a 20-fold loss in affinity. A benzimidazole core was investigated and also led to potent compounds with minimal PPARγ transactivation; however, we avoided benzimidazoles of this type in general given their known activity at the angiotensin II receptor.30 Of the several heterocyclic cores examined, none provided any advantages over the indole with regards to potency or activation of PPARγ and were therefore not pursued.
We next investigated the effect of modifying the acid moiety in 45 (Table S3 in the Supporting Information). Replacement with a tetrazole (65) or an oxadiazolone (63, 64) resulted in a slight decrease in PPARγ activity with minimal PPARγ transactivation. The hydroxamic acid (66) and amides (67, 68) were also tolerated. Interestingly, nitrile 69 had no activity.
While these acid bioisosteres maintained affinity for PPARγ with minimal transactivation, several of them proved to be suboptimal with regards to plasma exposure in rodents and were hence abandoned as useful substitutions (Table 5). With several promising PPARγ antagonists to choose from, we investigated oral dosing (p.o.) in mice in an effort to identify potential candidates for in vivo efficacy studies. When compounds were dosed orally at 20 mg/kg, we measured plasma exposure at t = 2 h and t = 6 h. The three carboxylic acids 45, 46, and 51 had better oral exposure than the corresponding acid bioisosteres (Table 5). Based on these results, we chose to evaluate the antidiabetic efficacy of 46 in DIO mice, a model of type-2 diabetes and obesity characterized by hyperglycemia and impaired glucose tolerance. This model more closely mimics the human condition brought about by high-fat Western diets than the genetic models using db/db or ob/ob mice.31
Table 5. In Vivo Exposures of Selected Biphenyl Analogues.
| Cmpda | 45 | 46 | 50 | 51 | 63 | 64 | 65 | 66 | 67 |
|---|---|---|---|---|---|---|---|---|---|
| [plasma] μM, 2 h | 9 | 19 | 0.6 | 5.6 | 0 | 0.2 | 0 | 0.4 | 2.5 |
| [plasma] μM, 6 h | 3 | 2.4 | 0.1 | 1.9 | 0 | 0.01 | 0 | 0.03 | 0.1 |
20 mg/kg p.o.
Male C57BL/6J JAX mice were from Charles River and were maintained on a high fat diet (Research Diets D12451). Mice were dosed orally once a day (q.d.) for 4 days (20 or 40 mg/kg) and fasted on Day 4. On the morning of Day 5, animals were dosed again, and then 180 min later dosed with a glucose challenge (2 g/kg p.o.).
Baseline blood samples were taken immediately from the tail vein before dosing (B1; −180 min) and immediately before the glucose challenge (B2; 0 min). Further blood samples were taken from the tail vein from all groups at 10, 20, 30, 60, and 120 min. Samples were analyzed for plasma glucose and insulin content, and a log transformation was used for insulin.
Analysis was by robust regression and included treatment and assay day as factors and Day 1 body weight, bleeding order, and Day −4 (baseline) plasma glucose or insulin concentration as covariates. Data are shown as adjusted means (n = 8), and standard errors of the mean (SEM) are calculated from the residuals of the statistical models. Comparisons against vehicle p.o. q.d. were by Williams’ test for 46. Significances are denoted by *p < 0.05, **p < 0.01, and ***p < 0.001.
After once daily oral administration of 46 for 5 days, a dose-dependent reduction in fasting glucose was observed (Figure 4). After oral glucose challenge (oGTT), 46 produced a statistically significant improvement in glucose disposal as compared to matched vehicle control mice. Rosiglitazone dosed orally once a day for 5 days (5 mg/kg) was equally efficacious as 46 (40 mg/kg) in the oral glucose tolerance test.
Figure 4.

In vivo efficacy studies with 46 in DIO mice.
In summary, a new class of potent PPARγ antagonists has been developed with oral in vivo efficacy similar to rosiglitazone, a PPARγ full agonist. This represents an alternative and perhaps safer way to target PPARγ for the therapeutic intervention of insulin resistance and type-2 diabetes. A large number of analogues were prepared using convergent synthetic strategies to investigate the SAR of different parts of the molecule. The indole biphenyl carboxylic acid provided an anchor for binding affinity, whereas the stereochemistry and substitution pattern in the 5-carboxamide played a key role in reducing and or eliminating ligand-induced transactivation of the receptor. As such, these compounds bind PPARγ with high affinity but do not activate transcription. This is in contrast to full agonists such as rosiglitazone, which hyperactivate the receptor. We have already reported that antagonists such as SR1664 appear to be devoid of many of the side effects associated with full agonism of the receptor, such as weight gain, edema, and bone mineral loss. It is likely that 46 has a similar profile. We are currently pursuing X-ray crystallography and HDX (hydrogen–deuterium exchange) mass spec techniques to further understand the protein dynamics upon ligand binding. We are also continuing to optimize this scaffold to further improve oral exposure to support chronic dosing studies in obese rodents.
Glossary
ABBREVIATIONS
- AE
adverse events
- DCM
dichloromethane
- DMAC
dimethylacetamide
- DMF
dimethylformamide
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxo-hexafluorophosphate
- HCl
hydrochloric acid
- PPAR
peroxisome proliferator-activated receptors
- PPTS
pyridinium p-toluenesulfonate
- SAR
structure–activity relationship
- T2DM
treatment of type-2 diabetes
- TFA
tifluoroacetic acid
- THF
tetrahydrofuran
- TZD
thiazolidinedione
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00218.
Experimental details for key compounds; lanthascreen and transactivation assays protocols (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
National Institutes of Health grant MH084512 (PI: Rosen), the Abrams Charitable Trust, and the Klorfine Family Fellowship.
The authors declare no competing financial interest.
Supplementary Material
References
- Choi J. H.; Banks A. S.; Kamenecka T. M.; Busby S. A.; Chalmers M. J.; Kumar N.; Kuruvilla D. S.; Shin Y.; He Y.; Bruning J. B.; Marciano D. P.; Cameron M. D.; Laznik D.; Jurczak M. J.; Schurer S. C.; Vidovic D.; Shulman G. I.; Spiegelman B. M.; Griffin P. R. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature 2011, 477 (7365), 477–481. 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinke P. T.; Wood H. B.; Szewczyk J. W. Nuclear Hormone Receptor Modulators for the Treatment of Diabetes and Dyslipidemia. Annu. Rep. Med. Chem. 2006, 41, 99–126. 10.1016/S0065-7743(06)41006-X. [DOI] [Google Scholar]
- Pirat C.; Farce A.; Lebegue N.; Renault N.; Furman C.; Millet R.; Yous S.; Speca S.; Berthelot P.; Desreumaux P.; Chavatte P. Targeting peroxisome proliferator-activated receptors (PPARs): development of modulators. J. Med. Chem. 2012, 55 (9), 4027–4061. 10.1021/jm101360s. [DOI] [PubMed] [Google Scholar]
- Wright M. B.; Bortolini M.; Tadayyon M.; Bopst M. Minireview: Challenges and opportunities in development of PPAR agonists. Mol. Endocrinol. 2014, 28 (11), 1756–1768. 10.1210/me.2013-1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross B. In PPAR agonists: multimodal drugs for the treatment of type-2 diabetes. Best Pract Res. Clin Endocrinol Metab 2007, 2007, 687–710. 10.1016/j.beem.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Wagstaff A. J. Rosiglitazone, Rosiglitazone-A review of its use in the management of type 2 diabetes mellitus. Drugs 2002, 62 (12), 1805–1837. 10.2165/00003495-200262120-00007. [DOI] [PubMed] [Google Scholar]
- Rubenstrunk A.; Hanf R.; Hum D. W.; Fruchart J. C.; Staels B. Safety issues and prospects for future generations of PPAR modulators. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2007, 1771 (8), 1065–1081. 10.1016/j.bbalip.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Tang W. H.; Maroo A. PPARgamma agonists: safety issues in heart failure. Diabetes, Obes. Metab. 2007, 9 (4), 447–454. 10.1111/j.1463-1326.2006.00616.x. [DOI] [PubMed] [Google Scholar]
- Kung J.; Henry R. R. Thiazolidinedione safety. Expert Opin. Drug Saf. 2012, 11 (4), 565–579. 10.1517/14740338.2012.691963. [DOI] [PubMed] [Google Scholar]
- Berger J. P.; Petro A. E.; Macnaul K. L.; Kelly L. J.; Zhang B. B.; Richards K.; Elbrecht A.; Johnson B. A.; Zhou G.; Doebber T. W.; Biswas C.; Parikh M.; Sharma N.; Tanen M. R.; Thompson G. M.; Ventre J.; Adams A. D.; Mosley R.; Surwit R. S.; Moller D. E. Distinct properties and advantages of a novel peroxisome proliferator-activated protein [gamma] selective modulator. Mol. Endocrinol. 2003, 17 (4), 662–76. 10.1210/me.2002-0217. [DOI] [PubMed] [Google Scholar]
- Minoura H.; Takeshita S.; Ita M.; Hirosumi J.; Mabuchi M.; Kawamura I.; Nakajima S.; Nakayama O.; Kayakiri H.; Oku T.; Ohkubo-Suzuki A.; Fukagawa M.; Kojo H.; Hanioka K.; Yamasaki N.; Imoto T.; Kobayashi Y.; Mutoh S. Pharmacological characteristics of a novel nonthiazolidinedione insulin sensitizer, FK614. Eur. J. Pharmacol. 2004, 494 (2–3), 273–281. 10.1016/j.ejphar.2004.04.038. [DOI] [PubMed] [Google Scholar]
- Acton J. J. 3rd; Black R. M.; Jones A. B.; Moller D. E.; Colwell L.; Doebber T. W.; Macnaul K. L.; Berger J.; Wood H. B. Benzoyl 2-methyl indoles as selective PPARgamma modulators. Bioorg. Med. Chem. Lett. 2005, 15 (2), 357–362. 10.1016/j.bmcl.2004.10.068. [DOI] [PubMed] [Google Scholar]
- Liu K.; Black R. M.; Acton J. J. 3rd; Mosley R.; Debenham S.; Abola R.; Yang M.; Tschirret-Guth R.; Colwell L.; Liu C.; Wu M.; Wang C. F.; MacNaul K. L.; McCann M. E.; Moller D. E.; Berger J. P.; Meinke P. T.; Jones A. B.; Wood H. B. Selective PPARgamma modulators with improved pharmacological profiles. Bioorg. Med. Chem. Lett. 2005, 15 (10), 2437–2440. 10.1016/j.bmcl.2005.03.092. [DOI] [PubMed] [Google Scholar]
- Ohashi M.; Oyama T.; Putranto E. W.; Waku T.; Nobusada H.; Kataoka K.; Matsuno K.; Yashiro M.; Morikawa K.; Huh N. H.; Miyachi H. Design and synthesis of a series of alpha-benzyl phenylpropanoic acid-type peroxisome proliferator-activated receptor (PPAR) gamma partial agonists with improved aqueous solubility. Bioorg. Med. Chem. 2013, 21 (8), 2319–2332. 10.1016/j.bmc.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Amato A. A.; Rajagopalan S.; Lin J. Z.; Carvalho B. M.; Figueira A. C.; Lu J.; Ayers S. D.; Mottin M.; Silveira R. L.; Souza P. C.; Mourao R. H.; Saad M. J.; Togashi M.; Simeoni L. A.; Abdalla D. S.; Skaf M. S.; Polikparpov I.; Lima M. C.; Galdino S. L.; Brennan R. G.; Baxter J. D.; Pitta I. R.; Webb P.; Phillips K. J.; Neves F. A. GQ-16, a novel peroxisome proliferator-activated receptor gamma (PPARgamma) ligand, promotes insulin sensitization without weight gain. J. Biol. Chem. 2012, 287 (33), 28169–28179. 10.1074/jbc.M111.332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. H.; Banks A. S.; Estall J. L.; Kajimura S.; Bostrom P.; Laznik D.; Ruas J. L.; Chalmers M. J.; Kamenecka T. M.; Bluher M.; Griffin P. R.; Spiegelman B. M. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 2010, 466 (7305), 451–456. 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggrell S. Do peroxisome proliferation receptor-gamma antagonists have clinical potential as combined antiobesity and antidiabetic drugs?. Expert Opin. Invest. Drugs 2003, 12 (4), 713–716. 10.1517/13543784.12.4.713. [DOI] [PubMed] [Google Scholar]
- Wright H. M.; Clish C. B.; Mikami T.; Hauser S.; Yanagi K.; Hiramatsu R.; Serhan C. N.; Spiegelman B. M. A synthetic antagonist for the peroxisome proliferator-activated receptor gamma inhibits adipocyte differentiation. J. Biol. Chem. 2000, 275 (3), 1873–1877. 10.1074/jbc.275.3.1873. [DOI] [PubMed] [Google Scholar]
- Seargent J. M.; Yates E. A.; Gill J. H. GW9662, a potent antagonist of PPARgamma, inhibits growth of breast tumour cells and promotes the anticancer effects of the PPARgamma agonist rosiglitazone, independently of PPARgamma activation. Br. J. Pharmacol. 2004, 143 (8), 933–937. 10.1038/sj.bjp.0705973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel M.; Wolber G.; Markt P.; Staels B.; Unger T.; Kintscher U.; Gust R. Characterization of new PPARgamma agonists: benzimidazole derivatives-importance of positions 5 and 6, and computational studies on the binding mode. Bioorg. Med. Chem. 2010, 18 (16), 5885–5895. 10.1016/j.bmc.2010.06.102. [DOI] [PubMed] [Google Scholar]
- Herbst L.; Goebel M.; Bandholtz S.; Gust R.; Kintscher U. Characterization of telmisartan-derived PPARgamma agonists: importance of moiety shift from position 6 to 5 on potency, efficacy and cofactor recruitment. ChemMedChem 2012, 7 (11), 1935–1942. 10.1002/cmdc.201200337. [DOI] [PubMed] [Google Scholar]
- Lamotte Y.; Martres P.; Faucher N.; Laroze A.; Grillot D.; Ancellin N.; Saintillan Y.; Beneton V.; Gampe R. T. Jr. Synthesis and biological activities of novel indole derivatives as potent and selective PPARgamma modulators. Bioorg. Med. Chem. Lett. 2010, 20 (4), 1399–1404. 10.1016/j.bmcl.2009.12.107. [DOI] [PubMed] [Google Scholar]
- Taygerly J. P.; McGee L. R.; Rubenstein S. M.; Houze J. B.; Cushing T. D.; Li Y.; Motani A.; Chen J. L.; Frankmoelle W.; Ye G.; Learned M. R.; Jaen J.; Miao S.; Timmermans P. B.; Thoolen M.; Kearney P.; Flygare J.; Beckmann H.; Weiszmann J.; Lindstrom M.; Walker N.; Liu J.; Biermann D.; Wang Z.; Hagiwara A.; Iida T.; Aramaki H.; Kitao Y.; Shinkai H.; Furukawa N.; Nishiu J.; Nakamura M. Discovery of INT131: a selective PPARgamma modulator that enhances insulin sensitivity. Bioorg. Med. Chem. 2013, 21 (4), 979–992. 10.1016/j.bmc.2012.11.058. [DOI] [PubMed] [Google Scholar]
- Minoura H.; Takeshita S.; Yamamoto T.; Mabuchi M.; Hirosumi J.; Takakura S.; Kawamura I.; Seki J.; Manda T.; Ita M.; Mutoh S. Ameliorating effect of FK614, a novel nonthiazolidinedione peroxisome proliferator-activated receptor gamma agonist, on insulin resistance in Zucker fatty rat. Eur. J. Pharmacol. 2005, 519 (1–2), 182–190. 10.1016/j.ejphar.2005.05.042. [DOI] [PubMed] [Google Scholar]
- Chang F.; Jaber L. A.; Berlie H. D.; O’Connell M. B. Evolution of peroxisome proliferator-activated receptor agonists. Ann. Pharmacother. 2007, 41 (6), 973–983. 10.1345/aph.1K013. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Lavan B. E.; Gregoire F. M. Selective Modulators of PPAR-gamma Activity: Molecular Aspects Related to Obesity and Side-Effects. PPAR research 2007, 2007, 32696. 10.1155/2007/32696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acton J. J. 3rd; Akiyama T. E.; Chang C. H.; Colwell L.; Debenham S.; Doebber T.; Einstein M.; Liu K.; McCann M. E.; Moller D. E.; Muise E. S.; Tan Y.; Thompson J. R.; Wong K. K.; Wu M.; Xu L.; Meinke P. T.; Berger J. P.; Wood H. B. Discovery of (2R)-2-(3-{3-[(4-Methoxyphenyl)carbonyl]-2-methyl-6-(trifluoromethoxy)-1H-indol-1 -yl}phenoxy)butanoic acid (MK-0533): a novel selective peroxisome proliferator-activated receptor gamma modulator for the treatment of type 2 diabetes mellitus with a reduced potential to increase plasma and extracellular fluid volume. J. Med. Chem. 2009, 52 (13), 3846–3854. 10.1021/jm900097m. [DOI] [PubMed] [Google Scholar]
- Liu G.; Cogan D. A.; Ellman J. A. Catalytic asymmetric synthesis of tert-butanesulfinamide. Application to the asymmetric synthesis of amines. J. Am. Chem. Soc. 1997, 119 (41), 9913–9914. 10.1021/ja972012z. [DOI] [Google Scholar]
- Liu G.; Cogan D. A.; Owens T. D.; Tang T. P.; Ellman J. A. Synthesis of enantiomerically pure N-tert-butanesulfinyl imines (tert-butanesulfinimines) by the direct condensation of tert-butanesulfinamide with aldehydes and ketones. J. Org. Chem. 1999, 64 (6), 1278–1284. 10.1021/jo982059i. [DOI] [Google Scholar]
- Lamotte Y.; Faucher N.; Sancon J.; Pineau O.; Sautet S.; Fouchet M. H.; Beneton V.; Tousaint J. J.; Saintillan Y.; Ancellin N.; Nicodeme E.; Grillot D.; Martres P. Discovery of novel indazole derivatives as dual angiotensin II antagonists and partial PPARgamma agonists. Bioorg. Med. Chem. Lett. 2014, 24 (4), 1098–1103. 10.1016/j.bmcl.2014.01.004. [DOI] [PubMed] [Google Scholar]
- Nilsson C.; Raun K.; Yan F.; Larsen M. O.; Tang-Christensen M. Laboratory animals as surrogate models of human obesity. Acta Pharmacol. Sin. 2012, 33, 173–181. 10.1038/aps.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.