

Abstract
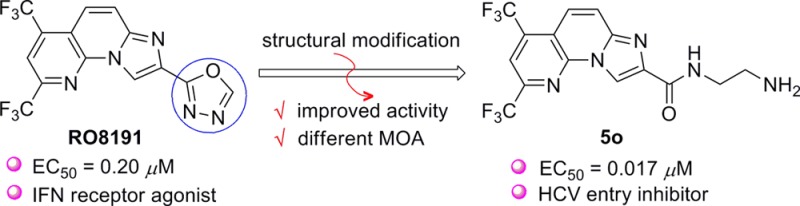
RO8191 represents a newly discovered small-molecule IFN-like agent that displays potent anti-HCV activity. With it as lead, a series of compounds bearing an imidazo[1,2-α][1,8]naphthyridine core and an amide bond-linked side chain were designed and synthesized. These compounds were evaluated on HCV cell culture system (HCVcc-hRluc-JFH1), and some of them exhibited remarkable anti-HCV activity (EC50 = 0.017–0.159 μM) and low toxicity (CC50 > 25 μM). Moreover, it was revealed that these newly identified anti-HCV agents exert their antiviral effect through a distinct mechanism of action from that of RO8191 by targeting the viral entry process. Thus, our study provides a starting point for the development of potential HCV entry inhibitor.
Keywords: Hepatitis C virus, anti-HCV agent, RO8191, IFN-like compound, entry inhibitor
Hepatitis C virus (HCV) is a positive single-stranded RNA virus belonging to the Flaviviridae family.1 Over 170 million people have been infected with HCV and thus are at risk of developing serious liver diseases, such as chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma.2,3 Historically, the standard care of HCV infection was the combinations of pegylated interferon-α (Peg-IFN) and ribavirin (RBV).4 However, its efficiency is largely compromised by several issues, including the moderate sustained virological response (SVR), lack of compliance, and severe side effects.5 Over the past decades, great efforts have been devoted to the development of more effective treatment and prevention of HCV infection,6−8 which culminated in the recent approval of several direct acting antivirals (DAA),9,10 the protease inhibitors11 boceprevir, telaprevir, and simeprevir and the polymerase inhibitor12 sofosbuvir. These new DAAs, in combination of Peg-IFN-α and ribavirin, could dramatically improve the SVR up to 90% across several different HCV genotypes. Albeit such breakthrough, however, there remains an unmet need for the development of new anti-HCV treatments that could have improved resistance profiles and low side effects.
Recently, a small molecule named RO8191 (1) was disclosed by Konishi and co-workers from Chugai Pharmaceutical Co. Ltd., which displays remarkable anti-HCV activity (EC50 = 0.2 μM).13 More importantly, RO8191 exerts its antiviral activity by directly interacting with the type I IFN receptor to drive IFN-stimulated genes (ISG) expression, which then induces the antiviral response of innate immune system. In this regard, RO8191 could be potentially utilized as a small-molecule IFN-substitute in the traditional IFN-α-based antiviral regimens.14 The appealing nature of RO8191 attracts considerable interest from pharmaceutical industry. In 2013, GlaxoSmithKline (GSK) disclosed a systematic structure–activity relationship (SAR) study on RO8191.15 Although over 100 analogues were synthesized by varying the structural variants on the A, B, C and D rings (I, Figure 1), none of the analogues displayed improved anti-HCV activity, indicating that RO8191 had a relative narrow window of SAR. In parallel with this seminal work, we also synthesized a number of RO8191 analogues that bear modified A, B, C, or D ring.16,17 Unfortunately, such efforts also met with limited success.
Figure 1.
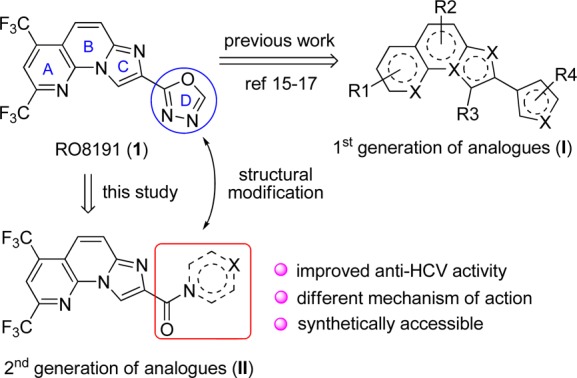
Structure of RO8191 and its analogues.
The above outcomes impelled us to develop the second generation of RO8191 analogues, as represented by structure II (Figure 1). The primary idea is to install an amide bond linker between the imidazo[1,2-α][1,8]naphthyridine scaffold and the 1,3,4-oxadiazole motif. We envisioned that such structural modification could afford several advantages over the lead structure of RO8191. First, it could provide an additional hydrogen bond acceptor, which may facilitate the interaction between the molecule and its biological target. Second, it will increase the molecular flexibility and hydrophilicity, thus improving its pharmacokinetics (e.g., water solubility and oral bioavailability). Last but not least, the second generation analogues are more synthetically accessible and prone to derivatization. Based on this design, we completed the synthesis of a series of compounds that featured an imidazo[1,2-α][1,8]naphthyridine scaffold coupled with an amide bond-derived cyclic or acyclic chain. Fortunately, it turned out that some of these compounds exhibited remarkable anti-HCV activity (EC50 = 0.02–0.05 μM), which were 5–10-fold more potent than RO8191. More interestingly, the preliminary mechanism of action (MOA) investigations revealed that these newly identified anti-HCV compounds most likely functioned as HCV entry inhibitor instead of the expected IFN-like agent.
The general synthetic route toward the designed compounds is depicted in Scheme 1. Thus, 2-amino-5,7-dis(trifluoromethyl)-1,8-naphthyridine 2 was prepared from the commercially available 2,6-diaminopyridine 1, 1,1,1,5,5,5-hexafluoropentane-2,4-dione via an acid-promoted cyclization.18 Subsequently, 2 reacted with methyl bromopyruvate in refluxing acetone to yield the 2,4-bis(trifluoromethyl)imidazo[1,2-a][1,8]naphthyridine-8-carboxylate 3.19 Hydrolysis of 3 in the presence of LiOH/THF/H2O provided the acid 4, which was then coupled with different amines using HATU in DMF to provide the corresponding amides 5.20
Scheme 1. Synthesis of Compounds 5a–5w.

Reagents and conditions: (a) 1,1,1,5,5,5-hexafluoropentane-2,4-dione, acetic acid, and sulfuric acid, reflux, 12 h, 63%; (b) methyl bromopyruvate, acetone, reflux, 12 h, 38%; (c) LiOH, THF/H2O (v/v: 1:1), 50 °C, 6 h, 90%; (d) amine component, HATU, DIPEA, DMF, r.t., 1–6 h, 15–68%.
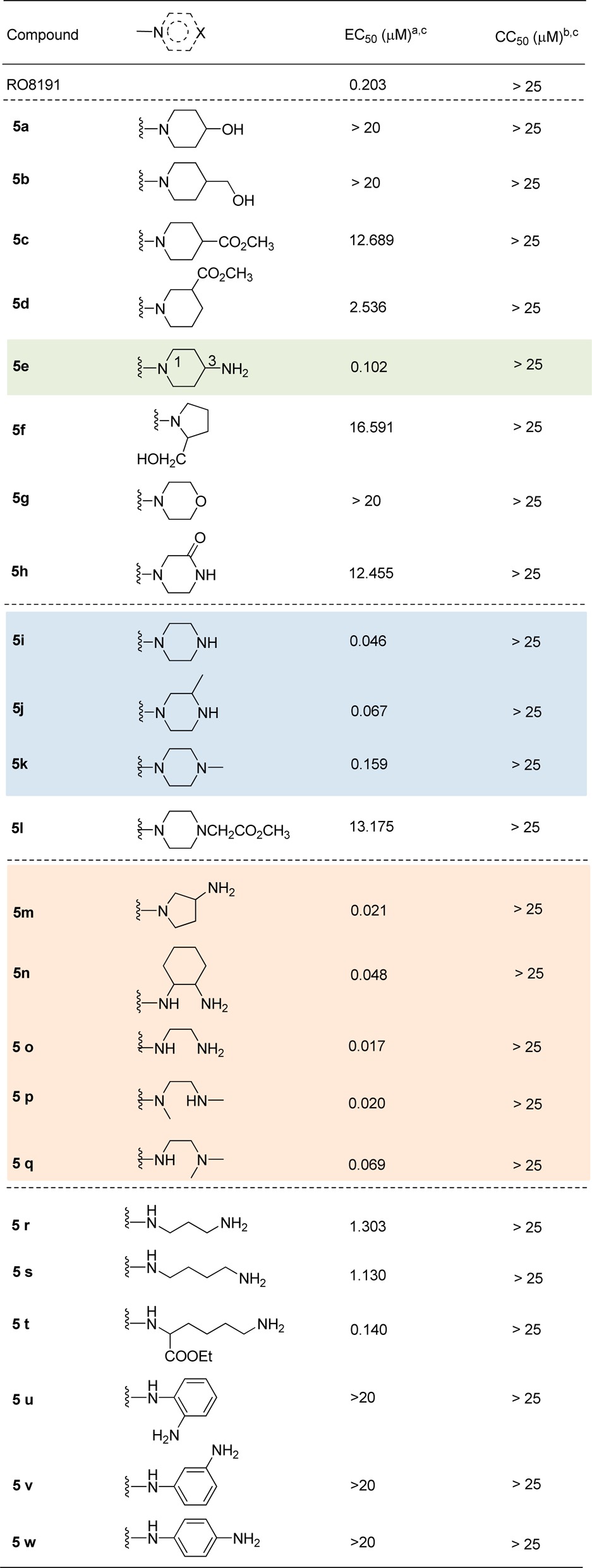
The anti-HCV activities of the synthesized compounds were first evaluated on the HCV cell culture system (HCVcc-hRluc-JFH1) with HCV genotype 2a JFH-1 virus containing a humanized Rellina luciferase reporter gene (Table 1). RO8191 (EC50 = 0.203 μM) were employed as positive control. First, an array of compounds bearing different heterocycles (e.g., piperidine, pyrrolidine, morpholine, or piperazin-2-one) (5a–5h) were examined. To our delight, while most of them failed to give promising results, 5e displayed remarkable anti-HCV activity (EC50 = 0.102 μM), which was 2-fold more potent than RO8191 (EC50 = 0.203 μM). It seemed that the amine moiety on the C-3 position of the piperidine ring played a crucial role for its activity since the other analogues attached with a −OH, −CO2Me, or −CH2OH functionality displayed no or weak activity. This hypothesis was further validated by another group of piperazine-derived compounds (5i–5l). As shown, 5i and 5j, which bear a secondary amine group, displayed even better anti-HCV activity than 5e. Comparably, for 5k and 5l, which carry a tertiary amine, inferior activity was obtained. Following this clue, various other analogues bearing cyclic or acyclic amine (5m–5w) were synthesized and evaluated, from which we could briefly summarize the key elements of the SAR. First, the analogues with a primary or secondary amine functionality showed better activity than those with a tertiary one (e.g., 5m–5p vs 5q). Second, the length of the tether between the amine and amide moieties plays a significant role. As shown, while most of the compounds with a two-carbon tether (5m–5o) displayed highly potent activity, those with a longer tether (three to five carbons, e.g., 5r–5t) afforded decreased potency. Third, it seems that the flexibility of the tether also has influence on the activity. Indeed, different from the aliphatic amine-derived analogues (5m–5t), several aniline-derived ones (5u–5w) proved to be inactive at the concentration of 20 μM. Of note, all of the above compounds displayed no obvious cytotoxicity (CC50 > 25 μM).
Table 1. Anti-HCV Activity of Compounds 5a–5w.
The inhibition of virus luciferase activity.
The reduction of viable cell number.
Calculated by Graphpad Prism 5 software.
Besides the HCV cell culture system, those active compounds with EC50 < 0.2 μM (5e, 5i–5k, and 5m–5q) were further evaluated with HCV replicon cells, which possess a luciferase reporter gene and express the HCV genotype 1b subgenomic replicon. To our surprise, all of the examined compounds display no inhibition on HCV replicons. In sharp contrast, RO8191 was found to be active in both systems. This result indicated that the newly synthesized analogues may exert their anti-HCV activity through a distinct mechanism of action different from that of RO8191.
To get deep insight into the plausible mechanism of action, further investigations were carried out with the most active compound 5o. First, we explored whether it could function as RO8191 to drive the IFN-stimulated genes (ISG) expression. For this end, we transiently transfected IFN-stimulated response element (ISRE) reporter gene into different cells (Vero, Huh7, Huh7.5.1, and HepG2), which serves as an enhancer to promote transcriptional induction by interferon. It was shown that while both RO8191 and IFN-α could induced the ISRE activation, compound 5o showed no effects (Figure S1, SI).
The different behavior of our compounds in the HCV cell culture and replicon system implied that they likely exerted their function in the entry stage of HCV infection. To validate this hypothesis, we conducted the time-of-addition assays to characterize the kinetics of compound activity. Bafilomycin A1, an inhibitor of endosomal acidification, which blocks the final fusion step between the virus envelope and the endosomal membrane, was tested in a parallel assay to define the end point of the entry process. As reported, the inhibition of bafilomycin A1 lasted about 3 h, indicating that HCVcc entry and fusion were completed within this time frame (Figure 2a).21,22 In analogy to bafilomycin, the kinetics of 5o activity revealed that the compound inhibition was exerted within 3 h of infection, indicating that it is an HCV entry inhibitor.
Figure 2.
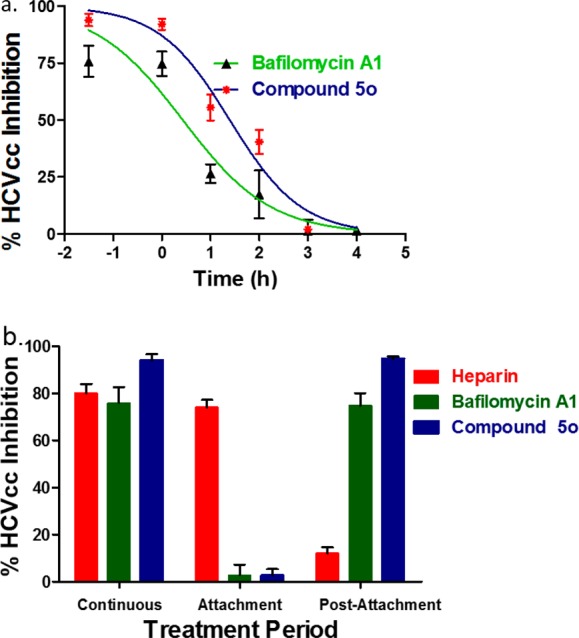
Kinetics of inhibition by 5o as an HCV entry inhibitor. (a) Time-course of inhibition. (b) Inhibitory effect on attachment and postattachment stages.
Furthermore, we turned to explore whether 5o blocks the initial attachment step to heparan sulfate proteoglycans (HSPGs) or a downstream event in the HCV entry process. The hog intestine heparin and bafilomycin A1 were employed as control inhibitors. As previously reported, heparin only had effects on the 4 °C attachment step, while bafilomycin was only effective during the postattachment stage.23,24 It was shown that compound 5o showed little effect on HCVcc attached to HSPGs step, but exerted over 90% inhibition on postattachment infection phase (Figure 2b). Taken together, 5o most likely blocks an event in HCV entry that lies downstream of the attachment to HSPGs, either prior to or during virus-cell fusion stage.
Besides HCV virus, compound 5o was also evaluated in a panel of other viruses including HBV, EV71, HIV, and MERS (Table 2). However, no obvious inhibition was observed at the concentration of 20 μM, showing the antiviral specificity of our compounds as the HCV inhibitor.
Table 2. Activity of 5o in Other Virus Infection Assaysa.
| virus | cell line | EC50 (μM)a | CC50 (μM)a |
|---|---|---|---|
| HBV | HepAD38 | >20 | >20 |
| EV71 | RD | >20 | >20 |
| HIV | Ghost | >20 | >20 |
| MERS | Huh7 | >20 | >20 |
Experiments were done in triplicate with each data point having four parallel test wells.
Finally, the pharmacokinetic profile of 5o was evaluated in rats. The time-course of 5o in plasma following a single oral dose (100 mg/kg) to rats is shown in Figure S2 (Supporting Information), and the key pharmacokinetic parameters were summarized in Table 3. Overall, it was shown that 5o was orally available and had promising pharmacokinetic properties (Tmax = 2.17 ± 0.36 h, T1/2 = 2.93 ± 0.60 h, Cmax = 2.05 ± 0.26 μM, AUC0–24h = 18.98 ± 2.49 μM·h).25
Table 3. Pharmacokinetic Parameters of 5oa.
| pharmacokinetic parameters | compound 5o |
|---|---|
| Tmax (h) | 2.17 ± 0.36 |
| Cmax (μM) | 2.05 ± 0.26 |
| T1/2 (h) | 2.93 ± 0.60 |
| AUC0–24 h (μM·h) | 18.98 ± 2.49 |
| AUC0-∞ (μM·h) | 19.12 ± 2.64 |
The data represent mean ± SD (n = 5). Tmax, time to reach maximal plasma concentration; Cmax, maximal plasma concentration; T1/2, elimination half-life; AUC0–24, area under the curve of plasma concentration from time 0 to 24 h; AUC0-∞, area under the curve of plasma concentration from time 0 to infinity.
In conclusion, with the known small molecule IFN-like agent RO8191 as lead, we have designed and synthesized a series of imidazo[1,2-α][1,8]naphthyridine derivatives, some of which exhibited significant anti-HCV activity (EC50 = 0.02–0.1 μM). Further investigations revealed that these compounds exerted their anti-HCV effect in the viral entry stage, which is distinct from that of RO8191. While the majority of the recently approved DAAs are either protease or polymerase inhibitors, the development of HCV entry inhibitor represents an emerging research field.26 In this context, our work provides a promising entry point for the development of HCV entry inhibitor. Efforts on identifying the underlying biological target of these anti-HCV agents are underway in our laboratory.
Glossary
ABBREVIATIONS
- HCV
hepatitis C virus
- Peg-IFN
pegylated interferon-α
- SVR
sustained virological response
- DAA
direct acting antivirals
- ISG
IFN-stimulated genes
- SAR
structure–activity relationship
- ISRE
IFN-stimulated response element
- HSPGs
heparan sulfate proteoglycans
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00159.
Experimental procedures, characterization data, and NMR-mass spectrum for all new compounds (PDF)
Author Contributions
∥ These authors (H.W. and S.W.) contributed equally to this work. All authors have given approval to the final version of the manuscript.
We gratefully acknowledge the financial supports from the NSFC (21272133, 81470839) and Beijing Natural Science Foundation (2132037).
The authors declare no competing financial interest.
Supplementary Material
References
- Choo Q. L.; Kuo G.; Weiner A. J.; Overby L. R.; Bradley D. W.; Houghton M. Isolation of a cDNA clone derived from a blood borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- Negro F.; Alberti A. The global health burden of hepatitis C virus infection. Liver Int. 2011, 31, 1–3. 10.1111/j.1478-3231.2011.02537.x. [DOI] [PubMed] [Google Scholar]
- Di Bisceglie A. M. Natural history of hepatitis C: its impact on clinical management. Hepatology 2000, 31, 1014–1018. 10.1053/he.2000.5762. [DOI] [PubMed] [Google Scholar]
- Zeuzem S. Interferon-based therapy for chronic hepatitis C: current and future perspectives. Nat. Clin. Pract. Gastroenterol. Hepatol. 2008, 5, 610–622. 10.1038/ncpgasthep1274. [DOI] [PubMed] [Google Scholar]
- Manns M. P.; Wedemeyer H.; Cornberg M. Treating viral hepatitis C: efficacy, side effects, and complications. Gut 2006, 55, 1350–1359. 10.1136/gut.2005.076646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong A. D. The HCV revolution did not happen overnight. ACS Med. Chem. Lett. 2014, 5, 214–220. 10.1021/ml500070q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghany M. G.; Strader D. B.; Thomas D. L.; Seeff L. B. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 2009, 49, 1335–1374. 10.1002/hep.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman W.; Mousa S.; Shiha S.; Mousa S. A. Current status and future directions in the management of chronic hepatitis C. Virol. J. 2012, 9, 57–67. 10.1186/1743-422X-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselah T.; Marcellin P. New direct-acting antivirals’ combination for the treatment of chronic hepatitis C. Liver Int. 2011, 31, 68–77. 10.1111/j.1478-3231.2010.02411.x. [DOI] [PubMed] [Google Scholar]
- Welsch C.; Jesudian A.; Zeuzem S.; Jacobson I. New direct-acting antiviral agents for the treatment of hepatitis C virus infection and perspectives. Gut 2012, 61, 36–46. 10.1136/gutjnl-2012-302144. [DOI] [PubMed] [Google Scholar]
- Wilby K. J.; Partovi N.; Ford J. A.; Greanya E.; Yoshida E. M. Review of boceprevir and telaprevir for the treatment of chronic hepatitis C. Can. J. Gastroenterol. 2012, 26, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofia M. J.; Chang W.; Furman P. A.; Mosley R. T.; Ross B. S. Nucleoside, nuceotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA polymerase. J. Med. Chem. 2012, 55, 2481–2531. 10.1021/jm201384j. [DOI] [PubMed] [Google Scholar]
- Konishi H.; Okamoto K.; Ohmori Y.; Yoshino H.; Ohmori H.; Ashihara M.; Hirata Y.; Ohta A.; Sakamoto H.; Hada N.; Katsume A.; Kohara M.; Morikawa K.; Tsukuda T.; Shimma N.; Foster G. R.; Alazawi W.; Aoki Y.; Arisawa M.; Sudoh M. An orally available, small-molecule interferon inhibits viral replication. Sci. Rep. 2012, 2, 259–267. 10.1038/srep00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedman C. A. M. Current prospects for interferon-free treatment of hepatitis C in 2012. J. Gastroenterol. Hepatol. 2013, 28, 38–45. 10.1111/jgh.12028. [DOI] [PubMed] [Google Scholar]
- Banka A. L.; Botyanszki J.; Burroughs E. G.; Catalano J. G.; Chern W. H.; Dickson H. D.; Gartland M. J.; Hamatake R.; Hofland H.; Keicher J. D.; Moore C. B.; Shotwell J. B.; Tallant M. D.; Therrien J. P.. Compounds and methods for enhancing innate immune responses. WO2013059559A2, 2013.
- Huang S. D.; Qing J.; Wang S.; Wang H.; Zhang L. Q.; Tang Y. F. Design and synthesis of imidazo[1,2-α][1,8]-naphthyridine derivatives as anti-HCV agents via direct C–H arylation. Org. Biomol. Chem. 2014, 12, 2344–2348. 10.1039/c3ob42525h. [DOI] [PubMed] [Google Scholar]
- Unpublished results. For details, see Table S-1 (SI).
- Váradi L.; Gray M.; Groundwater P. W.; Hall A. J.; James A. L.; Orenga S.; Perry J. D.; Anderson R. J. Synthesis and evaluation of fluorogenic 2-amino-1,8-naphthyridine derivatives for the detection of bacteria. Org. Biomol. Chem. 2010, 10, 2578–2589. 10.1039/c2ob06986e. [DOI] [PubMed] [Google Scholar]
- Aginagalde M.; Vara Y.; Arrieta A.; Zangi R.; Cebolla V. L.; Delgado-Camón A.; Cossío F. P. Tandem [8 + 2] cycloaddition-[2 + 6+2] dehydrogenation reactions involving imidazo[1,2-α]pyridines and imidazo[1,2-α]pyrimidines. J. Org. Chem. 2010, 75, 2776–2784. 10.1021/jo9022815. [DOI] [PubMed] [Google Scholar]
- Lu W. F.; Geng D. L.; Sun Z. J.; Yang Z. H.; Ma H. K.; Zheng J. Y.; Zhang X. H. Scaffold hopping approach to a new series of smoothened antagonists. Bioorg. Med. Chem. Lett. 2014, 24, 2300–2304. 10.1016/j.bmcl.2014.03.079. [DOI] [PubMed] [Google Scholar]
- Barth H.; Schafer C.; Adah M. I.; Zhang F.; Linhardt R. J.; Toyoda H.; Kinoshita-Toyoda A.; Toida T.; Van Kuppevelt T. H.; Depla E.; Von Weizsacker F.; Blum H. E.; Baumert T. F. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003, 278, 41003–41012. 10.1074/jbc.M302267200. [DOI] [PubMed] [Google Scholar]
- Basu A.; Kanda T.; Beyene A.; Saito K.; Meyer K.; Ray R. Sulfated homologues of heparin inhibit hepatitis C virus entry into mammalian cells. J. Virol. 2007, 81, 3933–3941. 10.1128/JVI.02622-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldick C. J.; Wichroski M. J.; Pendri A.; Walsh A. W.; Fang J.; Mazzucco C. E.; Pokornowski K. A.; Rose R. E.; Eggers B. J.; Hsu M.; Zhai W.; Zhai G.; Gerritz S. W.; Poss M. A.; Meanwell N. A.; Cockett M. I.; Tenney D. J. A novel small molecule inhibitor of hepatitis C virus entry. PLoS Pathog. 2010, 6, e1001086. 10.1371/journal.ppat.1001086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsoudakis G.; Kaul A.; Steinmann E.; Kallis S.; Lohmann V.; Pietschmann T.; Bartenschlager R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006, 80, 5308–5320. 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kanter C. T. M. M.; Drenth J. P. H.; Arends J. E.; Reesink H. W.; van der Valk M.; de Knegt R. J.; Burger D. M. Viral hepatitis C therapy: pharmacokinetic and pharmacodynamic considerations. Clin. Pharmacokinet. 2014, 53, 409–427. 10.1007/s40262-014-0142-5. [DOI] [PubMed] [Google Scholar]
- When this manuscript was under review, an important reference on this topic appeared elsewhere, see:He S. S.; Lin B.; Chu V.; Hu Z. Y.; Hu X.; Xiao J. B.; Wang A. Q.; Schweitzer C. J.; Li Q. S.; Imamura M.; Hiraga N.; Southall N.; Ferrer M.; Zheng W.; Chayama K.; Marugan J. J.; Liang T. J. Repurposing of the antihistamine chlorcyclizine and related compounds for treatment of hepatitis C virus infection. Sci. Transl. Med. 2015, 7, 282ra49. 10.1126/scitranslmed.3010286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.