

Abstract

A focused high throughput screening for GPR139 was completed for a select 100K compounds, and new agonist leads were identified. Subsequent analysis and structure–activity relationship studies identified (S)-3-chloro-N-(2-oxo-2-((1-phenylethyl)amino)ethyl)benzamide 7c as a potent and selective agonist of hGPR139 with an EC50 = 16 nM. The compound was found to cross the blood–brain barrier and have good drug-like properties amenable for oral dosing in rat.
Keywords: Orphan receptors, GPR139, agonists, glycine amides
G-protein coupled receptors (GPCRs) make for compelling drug targets within discovery research. Indeed, nearly one-third of all approved drugs on the market today target this type of receptor. The sequencing of the human genome has revealed thousands of genes, many of which encode G-protein coupled receptors, offering many new opportunities for novel therapies for unmet medical needs. However, these new GPCRs were identified based solely on their sequence homology to known GPCRs and their corresponding ligands, and thus the biological relevance of these new signaling pathways remains to be determined. Without the ligands for these orphan receptors, it is difficult to understand their physiological role in mammalian systems.
Figure 1.

Structure of GPR139 agonists.
GPR139 is an orphan G-protein coupled receptor that is predominantly expressed in the brain.1−3 Human expression of GPR139 can be found in the habenula, lateral caudate nucleus, and parts of the hypothalamus. The receptor is coupled with Gq signaling and appears to be constitutively active when recombinantly expressed in mammalian cells.3 The closest homologue to GPR139 is GPR142, which is highly expressed in the pancreas and a potential target for the treatment of type II diabetes. Yu et al.4 have reported on a series of phenylalanine base agonists for GPR142 and specified that l-Trp has been identified as the corresponding endogenous ligand. There have also been reports of l-Trp and l-Phe being able to activate GPR139 thus suggesting that putative physiological ligands for GPR139 may have been identified.5,6 GPR139s’ predominant expression in the brain and high degree of sequence homology across many different species suggests that it could play an important role in vertebrate physiology.
In searching for exogenous ligands to explore the function of GPR139, Shi et al.7 have reported on a series of benzohydrazides as potent and selective agonists for the receptor. They were able to elegantly demonstrate that removal of one of the hydrogen bond donors found in 1, thereby reducing the overall TPSA and lowering the MDCK ratio, resulted in an improved brain to plasma ratio for 2 (b/p 0.03 vs 2.8). In addition, Hu et al.8 have also described surrogate ligands, both agonists and antagonists, for GPR139. Isberg et al.6 utilized a pharmacophore model of 1 to identify and screen a set of aromatic dipeptides as potential ligands for GPR139. More recently, Wang et al.9 have reportedly identified several new GPR139 antagonists after screening a 16K set of diverse small molecules.
Our own interest to better understand the pharmacology and physiology of GPR139 led to a focused screening campaign of our 100K diversity set to identify potential small molecule ligands for the receptor. The agonist hit that was selected to investigate is shown in Scheme 1. The hit displayed some very similar functional group characteristics to the known agonist 1. Two carbonyls were present in a 1,4 relationship, separated by two atoms, accompanied by a large flanking hydrophobic surface. In analyzing the structure of 3 the sulfur atom was considered to be a liability. Not only is the sulfur easily oxidized, but the carbonyl of the thioester is reactive toward nucleophilic attack and therefore replaced with the more stable corresponding amide seen in 4 and 7h. Both enantiomers of alpha methyl benzyl amine are readily available, and consequently, it was straightforward to establish if there was a stereochemical preference in the observed activity. Making these simple changes and incorporating the known key binding element of a 3-methoxy into the benzoyl moiety provided 7h, representing a suitable launching point for chemistry to explore these glycine amides. In the present report we wish to describe the synthesis, structure–activity relationship (SAR), and characterization of a new series of glycine benzamides as potent agonists for the GPR139 receptor.
Scheme 1. High Throughput Screening Lead and Initial Optimization.

Compounds 7–9 were prepared as depicted in Scheme 2. Commercially available (S)-alpha methyl benzyl amine was coupled to Boc glycine, using EDCI and Hunig’s base with added HOBt. The protecting group on the amino acid nitrogen was then removed with 4 M HCl in dioxane to provide key intermediate 6.10 Treatment of 6 with the corresponding acid chlorides in DCM with triethylamine as the base thus provided compounds 7a–q.
Scheme 2. Synthesis of Compounds 7–9.

Reagents and conditions: (a) Boc-Gly-OH, EDCI, HOBt, iPr2NEt, DCM; (b) 4 M HCl, dioxane; (c) R1ArCOCl, Et3N, DCM; (d) Boc-NHCH2CHO, NaBH(OAc)3, DCM; (e) 4 M HCl, dioxane; (f) 3-chlorobenzoyl chloride, Et3N, DCM; (g) 3-chlorobenzaldehyde, NaBH(OAc)3, DCM.
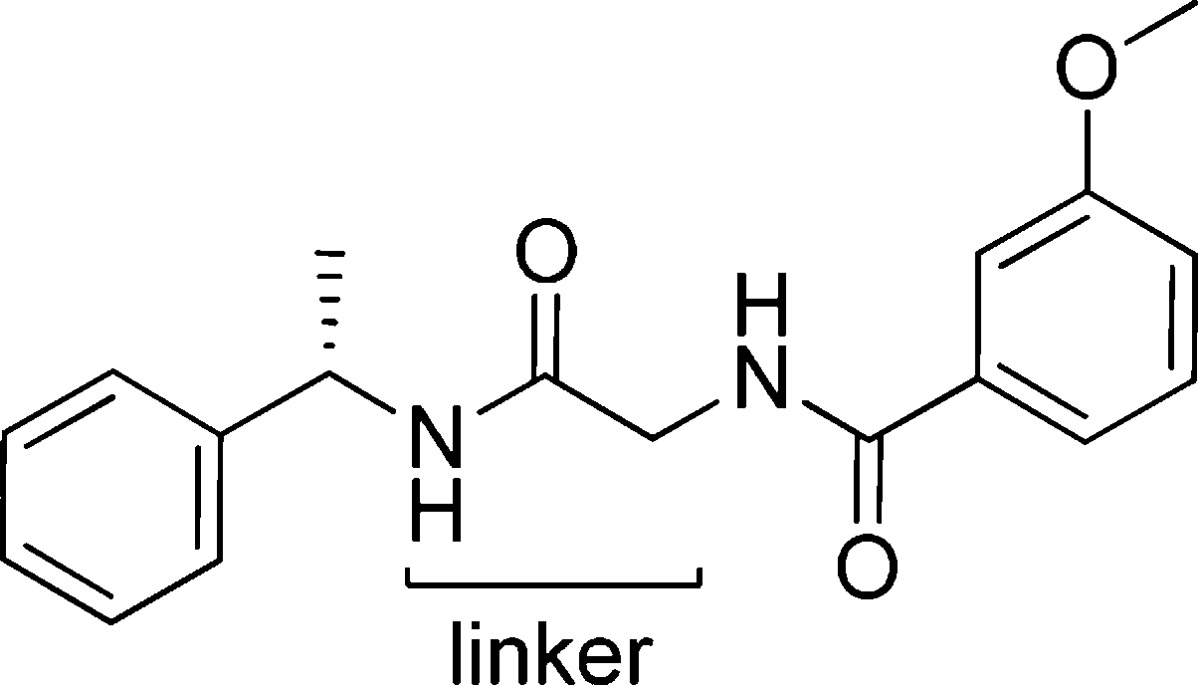
In addition, intermediate 6 was treated with 3-chlorobenzaldehyde and sodium triacetoxyborohydride via reductive amination11 to give compound 9 with the benzamide carbonyl removed. (S)-Alpha methyl benzyl amine was also treated with N-Boc-2-aminoacetaldehyde and sodium triacetoxyborohydride to give the two carbon extension with the distal nitrogen protected. The Boc group was then removed with 4 M HCl in dioxane and the resulting amine coupled with 3-chlorobenzoyl chloride in DCM with triethylamine to obtain compound 8 with the glycine carbonyl removed. Compounds 10–13 found in Table 1 were prepared in a similar fashion to compounds 7a–q shown in Scheme 2. These modifications to the linker portion of the template were carried out by starting with the corresponding Boc-protected amino acids alanine, serine, and phenyl glycine, respectively (only glycine shown).
Table 1. Effect of Linker on Human GPR139 Potency.
| Cmpd | linker | EC50 (nM)a | Emax (%)b |
|---|---|---|---|
| 7h | Gly | 33 ± 7 | 146 |
| 10 | l-Ala | 66 ± 17 | 143 |
| 11 | d-Ala | 440 ± 34 | 88 |
| 12 | l-Ser | 283 ± 46 | 107 |
| 13 | l-Phe | 10 000 | ND |
Data are reported as mean ± SEM from at least three experiments.
The efficacy of l-Trp was set to 100%.
The central linker was explored with a small select set of amino acids: Gly, Ala, Ser, and Phe. Table 1 displays the potency at the human GPR139 receptor of these modifications while holding the 3-methoxy benzamide portion of the molecule constant. The glycine linker was found to provide excellent potency at hGPR139 with an EC50 = 33 nM. Following with that of l-alanine suggested small groups may be allowed in this region of the template. d-Alanine was found to be 4-fold less potent then l-alanine and a preference for the natural configuration was observed. l-Serine was about as potent as d-Ala suggesting that neither linker could achieve the preferred conformation required for efficient binding. The use of l-Phe in the central linker, as found in select GPR142 agonists, was not tolerated. Larger groups in this area gave a pronounced reduction in potency at GPR139.
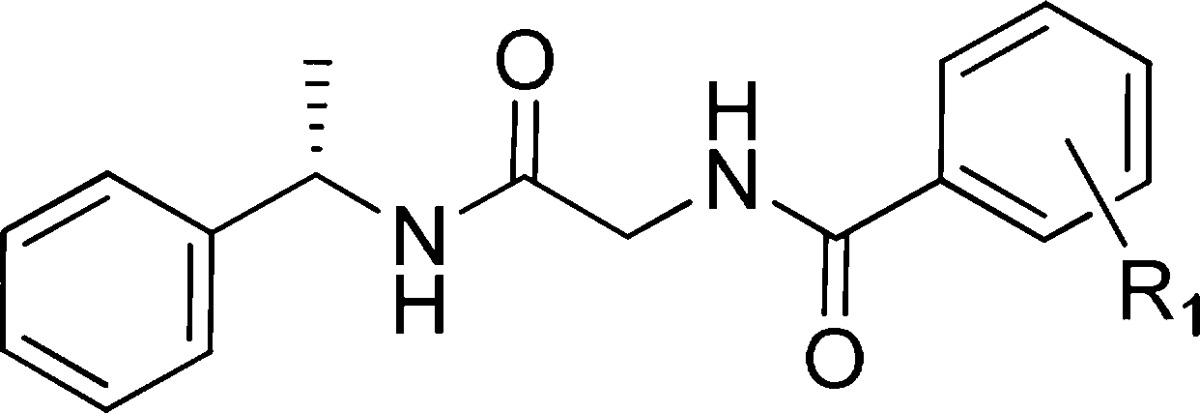
Table 2 contains SARs obtained by exploring substituents around the aryl of the benzamide while holding glycine and (S) alpha methyl benzyl amine constant. A scan at the ortho, meta, and para positions of the aryl benzamide revealed that the addition of a chloro atom anywhere on the ring (7b–d) resulted in an increase or comparable activity to that of the parent phenyl 7a. A clear preference was observed for a chloro positioned meta (7c) to the carbonyl, which gave excellent potency with a hGPR139 EC50 = 16 nM. The meta position was then explored further with both electron withdrawing and electron donating groups. All of the groups examined were found to be tolerated, and there was a clear preference for lipophilic groups. The methyl 7e was found to be nearly as potent as the chloro analogue 7c (EC50 16 nM vs 24 nM) and the trifluoromethoxy 7j slightly less active at hGPR139. The results of this screen led to the discovery of (S)-3-chloro-N-(2-oxo-2-((1-phenylethyl)amino)ethyl)benzamide (7c), which to our knowledge is one of the most potent compounds assayed for the GPR139 receptor to date.
Table 2. Effect of R1 on Human GPR139 Potency.
| Cmpd | R1 | EC50 (nM)a | Emax (%)b |
|---|---|---|---|
| 7a | H | 162 ± 30 | 130 |
| 7b | 2-Cl | 150 ± 23 | 120 |
| 7c | 3-Cl | 16 ± 2 | 138 |
| 7d | 4-Cl | 118 ± 49 | 117 |
| 7e | 3-Me | 24 ± 5 | 137 |
| 7f | 3-CN | 59 ± 13 | 129 |
| 7g | 3-F | 54 ± 11 | 118 |
| 7h | 3-MeO | 33 ± 7 | 146 |
| 7i | 3-CF3 | 52 ± 17 | 134 |
| 7j | 3-OCF3 | 46 ± 22 | 129 |
| 7k | 2,3-diCl | 52 ± 21 | 113 |
| 7l | 3,4-diCl | 193 ± 76 | 106 |
| 7m | 2,5-diCl | 40 ± 14 | 139 |
| 7n | 3,5-diCl | 46 ± 4 | 119 |
| 7o | 3-Cl,5-F | 24 ± 22 | 124 |
| 7p | 3-Br, 5-Cl | 78 ± 8 | 113 |
| 7q | 3,5-diMeO | 32 ± 7 | 149 |
| 8 | >10 000 | ND | |
| 9 | >10 000 | ND | |
| 1 | 43 | 125 |
Data are reported as mean ± SEM from at least three experiments.
The efficacy of l-Trp was set to 100%.
The impact of removing either carbonyl group was also examined. The effects on GPR139 binding suggest that each carbonyl engages in hydrogen bond interactions with the receptor protein. Indeed, deletions of either carbonyl completely abolished GPR139 potency (8 and 9). The corresponding enantiomer of 7c was also prepared and found to be significantly less active at human GPR139 (R enantiomer EC50 = 2.1 μM; structure not shown).
Compound 7c was then profiled in more detail. A comparison of the in vitro ADME data from the initial lead (7h) and compound 7c is shown in Table 3. Both compounds were found to have good extraction ratios in human microsomes. Extraction ratios are a measure of first pass metabolism representing the fraction of compound that is eliminated from blood as it travels through the liver. Compound 7h was found to have a much higher extraction ratio in rat microsomes therefore suggesting that it is metabolically less stable in this species. Both compounds displayed good permeability in the MDCK assay, but compound 7h was found to have a potential efflux issue as determined by the higher basal to apical ratio (5.5 vs 1.7) as compared to 7c. Compounds 7h and 7c were found to be highly soluble at both pH 2 and pH 7 in aqueous media (>35 μM) with no CYP450 inhibition, suggesting no potential drug–drug interaction (DDI) issues. Low protein binding provided for very large drug free fractions in both the plasma and brain for each compound. The initial lead displayed fairly good physical properties representing an excellent starting point for a medicinal chemistry program. Subsequently, it was discovered that by replacing the 3-OMe found in compound 7h with 3-Cl not only improved the rat extraction ratio (rER 0.8 vs rER 0.4) but also lowered the efflux potential.
Table 3. In Vitro ADME Assessment of Compounds 7h and 7c.
| Cmpd | hERa | rERb | MDCKcPapp (1 × 10–6 cm/s) | MDCK ratio (BA/AB) | hPPBd (%free) | rPPBe (%free) | rBBf (%free) |
|---|---|---|---|---|---|---|---|
| 7h | 0.3 | 0.8 | 16 | 5.5 | 14 | 36 | 16 |
| 7c | 0.3 | 0.4 | 32 | 1.7 | 10 | 25 | 9 |
Human extraction ratio.
Rat extraction ratio. Extraction ratios are calculated as in vitro CL/Q where Q is species specific hepatic blood flow.
MDR1-MDCK from Madin Darby canine kidney cells.
Human plasma protein binding.
Rat plasma protein binding.
Rat brain tissue binding.
Compound 7c was found to be clean of any cross reactivity as judged by an external selectivity panel (Eurofins Scientific) of 50 known GPCRs, ion channels, and transporters as well as our own internal whole cell lead generation biology selectivity panel. In addition, compound 7c does not activate rGPR142 to any extent providing for a selective GPR139 agonist. We then examined the pharmacokinetics of compound 7c in the rat to determine if observed in vitro properties translated in vivo. The parameters from the study (1 mg/kg iv; 5 mg/kg po) are shown in Table 4. The IV clearance (53 mL/min/kg) was found to be higher than was predicted in vitro, which was attributed to the lack of amidases in the microsomal assay. Very good systemic exposure was achieved with an oral dose giving an observed Cmax of 317 ng/mL (∼1 μM). More importantly, the compound was able to cross the blood–brain barrier (BBB) with a brain to plasma ratio (b/p) of 1.2, thus providing excellent coverage into the brain compartment.
Table 4. Pharmacokinetics of Compound 7c in the Rat.
| Cmpd | %F | Cmax (ng/mL) | Cla (mL/min/kg) | t1/2b (h) | Vssc (L/kg) | AUC (h·ng/mL) | b/pd (rat) |
|---|---|---|---|---|---|---|---|
| 7c | 49 | 317 | 53 | 2.5 | 2.1 | 683 | 1.2 |
Clearance.
The iv half-life.
Volume of distribution at steady state.
Ratio of brain to plasma conc.
Figure 2.

Total and non-specific binding of [3H]-7c.
Compound 7p was then utilized to prepare [3H]-7c (24.7 Ci/mmol) via reduction of the bromide with tritium through a contract with Moravek Biochemicals (Brea, CA). The radiochemical purity of [3H]-7c was determined to be 99.1% by HPLC analysis with radioactive flow detection. Total binding of [3H]-7c (@10 nM) was carried out in CHO-TRex cells stably expressing the human GPR139 receptor. Nonspecific binding was obtained by blocking with 10 μM 1. The compound was found to have good specific binding in CHO-TRex cells providing an additional tool for exploring the GPR139 receptor.
We have identified a new series glycine benzamides as agonists for GPR139. The SAR studies that were carried out led to the discovery of compound 7c, JNJ-63533054, with an EC50 = 16 nM (138% of max). Furthermore, the compound was found to have good drug-like properties. Compound 7c exhibits good stability in both human and rat microsomes and high solubility in aqueous media, and no DDI potential was found. Additionally, MDCK assay results indicated good cellular permeability with no efflux potential. This is especially desirable since the receptor is located in the CNS. These physical chemical properties then provided for very good pharmacokinetics in the rat. Oral dosing in rat (49%F) provided excellent exposure both systemically and within the CNS. We also prepared [3H]-7c to enable the development of binding and occupancy assays providing very useful tools for exploring GPR139. Further studies to deepen our understanding of the function and pharmacology of GPR139 are ongoing.
Acknowledgments
The authors would like to thank Kevin Coe and Discovery Sciences for ADME support and Mark Seierstad for CADD and informatics support. C.A.D. would also like to thank Christine Gelin for help in the editing of this manuscript.
Supporting Information Available
Experimental details for the synthesis of all compounds, compound characterization, description of the GPR139 assay, and ADME/PK protocols. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00247.
The authors declare no competing financial interest.
Supplementary Material
References
- Vanti W. B.; Nguyen T.; Cheng R.; Lynch K. R.; George S. R.; O’Dowd B. F. Novel human G-protein-coupled receptors. Biochem. Biophys. Res. Commun. 2003, 305 (1), 67–71. 10.1016/S0006-291X(03)00709-5. [DOI] [PubMed] [Google Scholar]
- Gloriam D. E. I.; Schioeth H. B.; Fredriksson R. Nine new human Rhodopsin family G-protein coupled receptors: identification, sequence characterisation and evolutionary relationship. Biochim. Biophys. Acta, Gen. Subj. 2005, 1722 (3), 235–246. 10.1016/j.bbagen.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Matsuo A.; Matsumoto S.-i.; Nagano M.; Masumoto K.-h.; Takasaki J.; Matsumoto M.; Kobori M.; Katoh M.; Shigeyoshi Y. Molecular cloning and characterization of a novel Gq-coupled orphan receptor GPRg1 exclusively expressed in the central nervous system. Biochem. Biophys. Res. Commun. 2005, 331 (1), 363–369. 10.1016/j.bbrc.2005.03.174. [DOI] [PubMed] [Google Scholar]
- Yu M.; Lizarzaburu M.; Motani A.; Fu Z.; Du X.; Liu J.; Jiao X.; Lai S.; Fan P.; Fu A.; Liu Q.; Murakoshi M.; Nara F.; Oda K.; Okuyama R.; Reagan J. D.; Watanabe N.; Yamazaki M.; Xiong Y.; Zhang Y.; Zhuang R.; Lin D. C. H.; Houze J. B.; Medina J. C.; Li L. Aminopyrazole-Phenylalanine Based GPR142 Agonists: Discovery of Tool Compound and in Vivo Efficacy Studies. ACS Med. Chem. Lett. 2013, 4 (9), 829–834. 10.1021/ml4000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak C. A.; Liu C.; Kuei C.. Physiological ligands for GPR139 such as l-tryptophan, l-phenylalanine, or derivatives thereof and their therapeutic use. WO2014152917A2, 2014.
- Isberg V.; Andersen K. B.; Bisig C.; Dietz G. P. H.; Brauner-Osborne H.; Gloriam D. E. Computer-Aided Discovery of Aromatic L-α-Amino Acids as Agonists of the Orphan G Protein-Coupled Receptor GPR139. J. Chem. Inf. Model. 2014, 54 (6), 1553–1557. 10.1021/ci500197a. [DOI] [PubMed] [Google Scholar]
- Shi F.; Shen J. K.; Chen D.; Fog K.; Thirstrup K.; Hentzer M.; Karlsson J.-J.; Menon V.; Jones K. A.; Smith K. E.; Smith G. Discovery and SAR of a Series of Agonists at Orphan G Protein-Coupled Receptor 139. ACS Med. Chem. Lett. 2011, 2 (4), 303–306. 10.1021/ml100293q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L. A.; Tang P. M.; Eslahi N. K.; Zhou T.; Barbosa J.; Liu Q. Identification of surrogate agonists and antagonists for orphan G-protein-coupled receptor GPR139. J. Biomol. Screening 2009, 14 (7), 789–797. 10.1177/1087057109335744. [DOI] [PubMed] [Google Scholar]
- Wang J.; Zhu L.-Y.; Liu Q.; Wang M.-W.; Hentzer M.; Smith G. P. High-throughput screening of antagonists for the orphan G-protein coupled receptor GPR139. Acta Pharmacol. Sin. 2015, 36 (7), 874–878. 10.1038/aps.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroto E. E.; Filippone S.; Suarez M.; Martinez-Alvarez R.; de Cozar A.; Cossio F. P.; Martin N. Stereodivergent Synthesis of Chiral Fullerenes by [3 + 2] Cycloadditions to C60. J. Am. Chem. Soc. 2014, 136 (2), 705–712. 10.1021/ja410408c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Magid A. F.; Carson K. G.; Harris B. D.; Maryanoff C. A.; Shah R. D. Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures. J. Org. Chem. 1996, 61 (11), 3849–3862. 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.