

Abstract
A new series of indole analogues based on our earlier lead compound, 2-(1H-indol-5-yl)-4-(3,4,5-trimethoxyphenyl)-1H-imidazo[4,5-c]pyridine (42), was prepared as tubulin inhibitors in an effort to find a molecule with improved cytotoxic potency and metabolic stability. A series of indolyl-imidazopyridines (IIP) were synthesized and exhibited potent tubulin polymerization inhibitory activity with potent IC50 values ranging from 3 to 175 nM against a panel of human melanoma and prostate cancer cell lines. Among these compounds, the 6-indolyl compound 43 showed improved cytotoxic potency (average IC50 of 9.75 nM vs 55.75 nM) and metabolic stability in human liver microsomes (half-life time was 56.3 min vs. 45.4 min) as compared to previously reported 42. It was also shown to be effective against P-glycoprotein (P-gp) mediated multiple drug resistance (MDR) and taxol resistance.
Keywords: Colchicine-binding site, tubulin polymerization inhibitor, prostate cancer, melanoma, antiproliferative activity, structure−activity relationship (SAR), multiple drug resistance, liver microsomal stability
Microtubules are tubular polymers of tubulin that have been crucial chemotherapeutic targets in a variety of cancers.1 Interfering with tubulin dynamics in tumor cells is a validated approach in developing antimitotic drugs to treat cancers.2 Using potent cytotoxic drugs that inhibit microtubules has been useful in inducing cancer cells to undergo apoptosis through various mechanisms.3 Most of the small molecule antitubulin agents bind to one of the three best characterized binding sites on α,β-tubulin subunits. For example, the taxane, vinca alkaloids, and colchicine binding sites bind to paclitaxel, vinblastine, or colchicine, respectively. Antitubulin agents are also classified according to whether they prevent microtubule degradation (e.g., taxanes, epothilones) or induce microtubule-destabilization (e.g., vinca alkaloids, colchicine).4
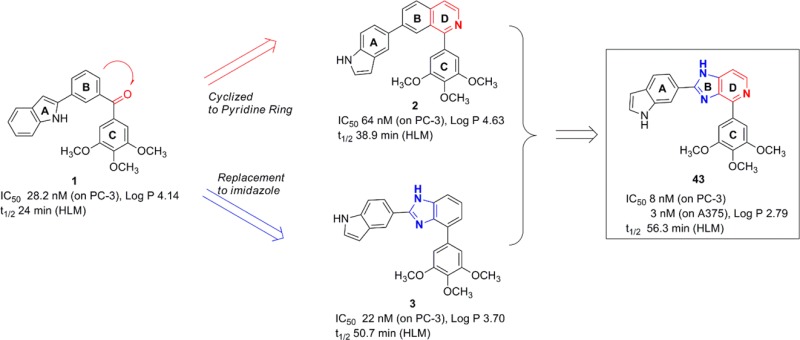
While a number of tubulin inhibitors binding to the taxane or vinca alkaloid sites exhibit high potency and have been approved by the FDA for the treatment of various cancers, the emergence of resistant phenotypes still leads to diminished efficacy in tumors expressing drug efflux transporters.1,5 Compared to the taxane or vinca alkaloid binding sites, targeting the colchicine binding site may provide a better opportunity for structural optimization to overcome such ABC-transporter mediated drug resistance while complying with Lipinski’s Rule of Five.6 Recently, colchicine scaffolds in in vitro studies showed strong cancer inhibition and also overcame resistant phenotypes of carcinoma.7,8 A number of potent tubulin inhibitors targeting the colchicine binding site have been reported, including combretastatin A-4 (CA-4),9 BRP0L075,10 phenstatin,11 ARAP,12 and SD40013 as shown in Figure S1 of the Supporting Information. Following our discovery of (3-(1H-indol-2-yl)phenyl)(1H-indol-2-yl)methanone14 as an antitubulin agent, our group showed several diaryl-ketone chemotypes as tubulin inhibitors that bind to the colchicine domain, including phenyl ring as linker (I-387),15 phenylaminothiazoles (PAT),4 arylbenzoylimidazoles (ABI),16 reverse ABIs (RABI),17 and 4-substituted methoxybenozyl aryl thiazoles (SMART).18 Unsubstituted examples of these chemotypes are shown in Figure S1. Among them, the SMART, ABI, and PAT analogues have shown very strong in vivo efficacy in human melanoma and prostate cancer xenograft models.18 However, the ketone represented a metabolically labile site in human liver microsomes (HLM) studies and thus new approaches were sought to improve antimitotic effects and reduce untoward effects.19 In an effort to improve potency and metabolic stability, we explored general structure 4. As shown in Figure 1, 4 was designed as a fusion of metabolically stable nonketone templates 2(15) and 3.19 The building-blocks for analogues of 4 are indoles and pyrido-imidazoles connected with (3,4,5-trimethoxyphenyl)-(TMP-) template. The imidazole ring is a widely used template in many drugs.20 Also the indole group has been investigated in many antitubulin agents,21 such as 3-formyl-2-phenylindoles, heterocombretastatins, diarylindoles, 2-aroylindoles, D-24851, 2-aryl-3-aroylindoles, 3-aroyl- and 1-aroylindoles, and arylthioindoles.21 We herein report the evaluation of two classes of nonketone antitubulin agents that contain indole and imidazole moieties. Isoquinolines (class I) and imidazopyridines (class II), as shown in Schemes 1 and 2, are bioisosteric chemotypes explored herein as new classes of tubulin inhibitors binding to the colchicine site.
Figure 1.

Hybrid indolyl-imidazopyridine general structure 4 (IIP) derived from a combination of isoquinoline (2) and imidazo-benzene (3) templates.
Scheme 1. Synthesis of Class I (1,7-Disubstituted Isoquinolines) Antitubulin Agents.

Reagents and conditions: (a) 1 equiv of each boric acid 6–8, Pd(PPh3)4, K2CO3, DMF, H2O, reflux; (b) 2 equiv of boric acid 7 or 8, Pd(PPh3)4, K2CO3, DMF, H2O, reflux.
Scheme 2. Synthesis of Class II (2,4-Disubstituted 1H-Imidazo[4,5-c]pyridines) Antitubulin Agents.

Reagents and conditions: (a) n-BuLi, DMF, −78 °C; (b) PhSO2Cl, KOH, TBAHS, DCM, 16 h, room temperature; (c) p-toluenesulfonic acid, 1,4-dioxane, reflux; (d) 8, Pd(PPh3)4, aqueous Na2CO3, THF/MeOH/H2O, 100 °C, MW; (e) aqueous NaOH, EtOH, reflux.
The structure–activity relationship (SAR) was optimized with regard to site of indole attachment to the core motif in an effort to develop new therapeutic candidates for future in vivo studies. 6-Indolyl (43) demonstrated high potency and metabolic stability in vitro. In class I of Scheme 1, five isoquinolines analogues (2, 10–13) with TMP-, 5-indolyl-, or 4-fluorophenyl-groups in A- and/or C-rings were prepared and tested for their cytotoxicity against prostate cancer and human melanoma cell lines,22 and their metabolic stability is discussed. The majority of the isoquinolines derivatives and intermediates were prepared by Suzuki-cross coupling of commercially available aryl halides (5, 9) with boric acids (6–8) using Pd(PPh3)4 as a catalyst. Compounds 7 and 8 were synthesized using 2 equiv of boric acid (7 and 8, respectively).
Scheme 2 shows the synthesis of class II agents, indole-based 2,4-disubstituted 1H-imidazo[4,5-c]pyridines 39–44. Each of the six substitutable positions of the indolyl moiety was synthesized using different indolylaldehydes 14–19 as shown in Scheme 2. The protected aldehydes (20–25) were prepared by known procedures.23 Condensation of the N-protected indolylaldehydes (2023 and 21–25(24)) with pyridodiamine 26 was accomplished using TsOH in 1,4-dioxane, or catalytic amount of c-H2SO4 in toluene/DMF solution using a Dean–Stark trap, to give the desired imidazopyridines 27–32. The Suzuki cross-coupling reaction was performed between this series of protected bromides 27–32 and 3,4,5-trimethoxyphenylboric acid (8) to obtain compounds 33–38.
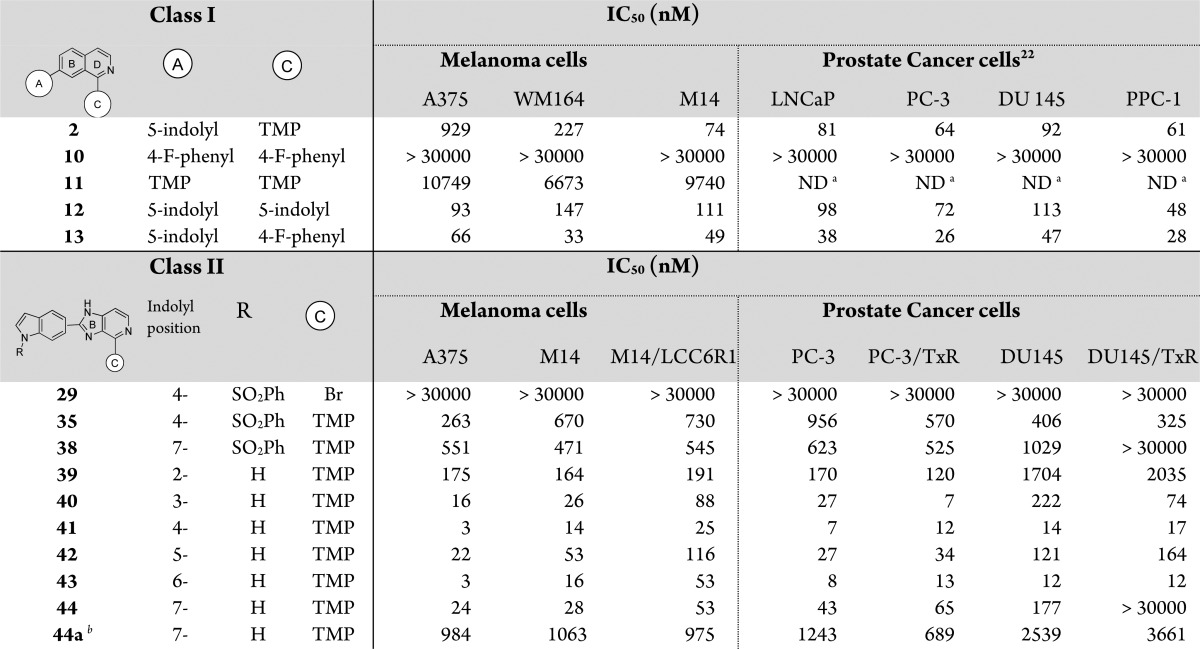
The deprotection of the phenysulfonyl group (SO2Ph) was performed using aqueous NaOH to generate the desired IIP products 39–44. Interestingly, we also found and purified 2-(1H-indol-7-yl)-4-(3,4,5-trimethoxyphenyl)-2,3-dihydro-1H-imidazo[4,5-c]pyridine (44a) when modifying the conditions by heating ethanol to reflux in conditions c (Scheme 2), rather than using 1,4-dioxane. Compound 44a could only be separated at the final step and was tested in our assay system in order to compare it to compound 44. The in vitro cytotoxic/antiproliferative activity of the synthesized class I and II compounds against a panel of human melanoma and prostate cancer cell lines was characterized using the MTS assay (Tables 1). Compounds in class II were further tested in P-gp overexpressing multidrug resistant (MDR) and taxol resistant cancer cell lines. The results for class I showed that compounds with 5-indolyl A-rings (2, 12, and 13, 26–929 nM) had higher growth inhibition potency than compounds with TMP or 4-fluorophenyl A-rings (10 and 11, 10 to >30 μM ranges). Compound 13 was the most potent class I compound bearing a 5-indolyl A-ring with a 4-fluorophenyl C-ring. Compound 13 had potent activity against prostate cell lines in the range of 26 to 47 nM, which was better than that observed with the TMP analogue 2. Class I, with the six-membered B-ring, has increased intramolecular repulsion more than class II. This may be due to the increased steric overlap between the A- and C-rings. Therefore, 4-fluorophenyl of 13 (class I) may have less internal strain as compared to the relatively bulky TMP C-ring of 2. Perhaps rotation of bond connecting the C- and D-rings in class I compounds is not tolerated within the colchicine binding pocket.
Table 1. Antiproliferative Activities of Isoquinoline Analogues (Class I) and 1H-Imidazo[4,5-c]pyridine Analogues (Class II) in Parental and MDR (LLC6R1) or Paclitaxel Resistant (TxR) Cancer Cell Lines.
ND: Not determined.
2-(1H-Indol-7-yl)-4-(3,4,5-trimethoxyphenyl)-2,3-dihydro-1H-imidazo[4,5-c]pyridine, which is dihydro-imidazole on B-ring.
N-Protected class II compounds with TMP as the C-ring (35 or 38) showed moderate activity (average ∼400 nM), but compound 29, which had a bromide substituent instead of the C-ring, was inactive (>30 μM). This indicated again that TMP on the C-ring position was critical for antiproliferative activity. Generally, all the tested compounds with A-ring indole substituents (39–44) showed strong antiproliferative activity against melanoma and prostate cancer cell lines, including taxol resistant PC-3/TxR and DU145/TxR cells, and overcame the P-gp mediated MDR in M14/LCC6R1 cells. In this series, 2-indolyl 39 was less cytotoxic with IC50 values at around a few hundred nanomolar. 3- and 7-Indolyl compounds 40 and 44 presented comparable potency (average IC50 values in the low nanomolar range) to the previously reported 5-indolyl compound 42 (average 55.75 nM). The dihydro-imidazole 44a, which was made as a side product, showed 10–20-fold lower potency in melanoma and prostate cell lines than imidazole analogue 44, indicating that the imidazole ring is an important moiety for inducing apoptosis. This suggests that the planar central ring of 44 is more complementary to the colchicine binding site, which apparently does not tolerate the nonplanar arrangement of the A- and C-rings caused by the dihydro-imidazole of 44a. The most active A-ring indolyl compounds were the 4- and 6-indolyl 41 and 43, which demonstrated IC50 values as low as 3 nM in human melanoma A375 cells. Mechanistic and molecular modeling studies suggested that selected compounds 41, 42, and 43 maintained their mode of action as tubulin polymerization inhibitors as shown in Figure S2 of the Supporting Information. To experimentally determine whether the new analogues maintain their mode of action, we tested the two most potent compounds, 41 and 43, in a microtubule polymerization assay in vitro (at 5 or 10 μM). Vehicle, taxol (5 μM), and colchicine (5 μM) were used as control groups and assayed under the same conditions (Figure 2A). Both 41 and 43 effectively inhibited the tubulin polymerization in a dose-dependent manner. Compound 43 at lower concentration (5 μM) was a more effective inhibitor than 41 at 10 μM. Human melanoma A375 cells and human prostate cancer PC-3 cells were treated with 41 or 43 for 24 h and analyzed through flow cytometry to determine their cell cycle distributions. As shown in the Figure 2B, while the general distributions of A375 cells were not significantly affected by either 41 or 43 at a low concentration of 10 nM, at a higher concentration of 50 nM, 41 and 43 effectively blocked A375 cells at the G2/M phase. The vehicle control group only had 4.0 ± 0.5% of A375 cells distributed in G2/M phase, but 42.0 ± 3.8% or 65.9 ± 2.2% of A375 cells were arrested in G2/M phase for 41 or 43 at 50 nM, respectively. In PC-3 cells, even at the low concentration of 10 nM, 41 or 43 efficiently arrested cells in G2/M phases as shown in Figure S3 of the Supporting Information. This dose-dependent G2/M phase block for 41 and 43 confirms the cytotoxic mechanism of action was conserved. The most potent compounds of each class were selected to be further evaluated for in vitro metabolic stability in human liver microsomes (HLM) and mouse liver microsomes (MLM). As shown in Table 2, Class I B-ring isoquinoline compounds (2 and 13) showed significantly better metabolic stability in HLM (t1/2 = 38.9 and 30.7 min), than was observed in MLM (t1/2 = 2.1 and 2.0 min). The replacement of TMP (for 2) with 4-fluorophenyl group on C-ring (for 13) increased the antiproliferative potency but decreased the metabolic stability slightly. Class II (IIP compounds) 41, 42, and 43 showed favorable metabolic stability in both HLM and MLM. Among them, 43 having an A-ring 6-indolyl substituent and showing the highest anticancer potency, also exhibited the best stability (t1/2 = 56.3 min in HLM and 27.7 min in MLM). This suggests that the adjustment from 5-indolyl 42 to 6-indolyl 43 has improved both antitumor efficacy and metabolic stability.
Figure 2.
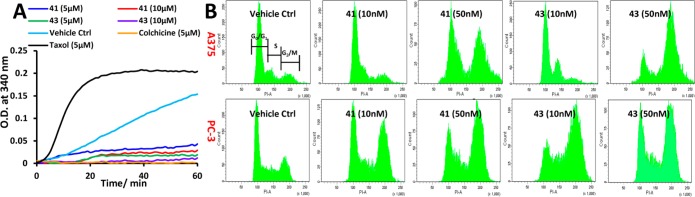
(A) Compounds 41 and 43 effectively inhibited the tubulin polymerization in vitro. The microtubule polymerization was monitored by measuring the absorbance at 340 nm in the absence or presence of drugs (N = 3). A representative experiment is shown. Vehicle control (sky blue); paclitaxel (5 μM) (black); colchicine (5 μM) (brown); 41 (5 μM) (blue), 41 (10 μM) (red); 43 (5 μM) (green), 43 (10 μM) (purple). (B) Compounds 41 and 43 arrested human melanoma A375 and human prostate cancer PC-3 cells in G2/M phase in vitro (N = 3).
Table 2. Half-Lives in Human (HLM) and Mouse (MLM) Liver Microsomes.
|
t1/2 (min) |
|||
|---|---|---|---|
| compd ID | HLM | MLM | |
| class I | 2 | 38.9 ± 3.4 | 2.1 ± 1.1 |
| 13 | 30.7 ± 2.6 | 2.0 ± 0.8 | |
| class II | 41 | 33.1 ± 4.8 | 10.5 ± 1.8 |
| 42 | 45.4 ± 3.0 | 16.1 ± 4.4 | |
| 43 | 56.3 ± 2.3 | 27.7 ± 6.1 | |
In summary, our previous discovery of the 5-indolyl 42 demonstrated that the indolyl-imidazo[4,5-c]pyridine class II chemotype with TMP B-ring could potently bind the colchicine site and additionally provide a metabolic stability benefit. Herein we performed an “indole-walk” study to optimize the best configuration for biological activity on the 4-(3,4,5-trimethoxyphenyl)-1H-imidazo[4,5-c]pyridin-2-yl (IIP template). All six new indolyl derivatives were investigated for their in vitro cytotoxicities in cancer cell lines and stability to metabolism in liver microsomes. We also tested isoquinoline motifs (class I) to compare the structure–activity relationships of cytotoxicity and metabolic stability across nonketone classes I and II. The results showed the pyridine d-ring motif of IIP provided some benefits toward metabolic stability in HLM. Additionally, cytoxicity studies demonstrated compounds of general structure 4 (i.e., class II or IIP compounds) were very potent against the tested tumor cell lines. Among them, 6-indolyl derivative (6-IIP, 43) showed the strongest inhibition activities (IC50 at 3 nM on A375 and 8 nM on PC-3) and best metabolic stability (56.3 min in HLM). We will evaluate the in vivo efficacy of the IIP series, which will be further characterized as potent novel anticancer agents. The IIP series can serve not only as a valuable tool for preclinical research of the modulation of tubulin polymerization dynamics but also as a promising candidate series for clinical development.
Acknowledgments
We thank Dr. Michael L. Mohler at GTx, Inc., for his proofreading and editorial assistance.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00208.
Synthetic procedures, the structures of known colchicine inhibitors (Figure S1), molecular modeling (Figure S2), quantification data for cell cycle analysis (Figure S3), and the analytical spectra for 41, 42, and 43 and their derivatives (Figure S4) (PDF)
Author Contributions
† These authors contributed equally to this work. All authors have given approval to the final version of the manuscript.
This research was supported by the Van Vleet Endowed Professorship (to D.D.M.), NIH grants R01CA148706, 1S10OD010678-01, and RR-026377-01 (to W.L.), and UTHSC Fellowship, Alma & Hal Reagan (to J.W.).
This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interest.
Supplementary Material
References
- Jordan M. A.; Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253. 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Stratton M. R.; Campbell P. J.; Futreal P. A. The cancer genome. Nature 2009, 458, 719. 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracci L.; Schiavoni G.; Sistigu A.; Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15. 10.1038/cdd.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. M.; Chen J.; Lu Y.; Narayanan R.; Parke D. N.; Li W.; Ahn S.; Miller D. D.; Dalton J. T. Pharmacokinetic optimization of 4-substituted methoxybenzoyl-aryl-thiazole and 2-aryl-4-benzoyl-imidazole for improving oral bioavailability. Drug Metab. Dispos. 2011, 39, 1833. 10.1124/dmd.110.036616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipalensuu J.; Tornblom H.; Lindberg G.; Einarsson C.; Sjoqvist F.; Melhus H.; Garberg P.; Sjostrom B.; Lundgren B.; Artursson P. Correlation of gene expression of ten drug efflux proteins of the ATP-binding cassette transporter family in normal human jejunum and in human intestinal epithelial Caco-2 cell monolayers. J. Pharmacol. Exp. Ther. 2001, 299, 2001. [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Cosentino L.; Redondo-Horcajo M.; Zhao Y.; Santos A. R.; Chowdury K. F.; Vinader V.; Abdallah Q. M.; Abdel-Rahman H.; Fournier-Dit-Chabert J.; Shnyder S. D.; Loadman P. M.; Fang W. S.; Diaz J. F.; Barasoain I.; Burns P. A.; Pors K. Synthesis and biological evaluation of colchicine B-ring analogues tethered with halogenated benzyl moieties. J. Med. Chem. 2012, 55, 11062. 10.1021/jm301151t. [DOI] [PubMed] [Google Scholar]
- Fournier-Dit-Chabert J.; Vinader V.; Santos A. R.; Redondo-Horcajo M.; Dreneau A.; Basak R.; Cosentino L.; Marston G.; Abdel-Rahman H.; Loadman P. M.; Shnyder S. D.; Diaz J. F.; Barasoain I.; Falconer R. A.; Pors K. Synthesis and biological evaluation of colchicine C-ring analogues tethered with aliphatic linkers suitable for prodrug derivatisation. Bioorg. Med. Chem. Lett. 2012, 22, 7693. 10.1016/j.bmcl.2012.09.104. [DOI] [PubMed] [Google Scholar]
- Nam N. H. Combretastatin A-4 analogues as antimitotic antitumor agents. Curr. Med. Chem. 2003, 10, 1697. 10.2174/0929867033457151. [DOI] [PubMed] [Google Scholar]
- Kuo C. C.; Hsieh H. P.; Pan W. Y.; Chen C. P.; Liou J. P.; Lee S. J.; Chang Y. L.; Chen L. T.; Chen C. T.; Chang J. Y. BPR0L075, a novel synthetic indole compound with antimitotic activity in human cancer cells, exerts effective antitumoral activity in vivo. Cancer Res. 2004, 64, 2004. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Toki B.; Herald D. L.; Verdier-Pinard P.; Boyd M. R.; Hamel E.; Pettit R. K. Antineoplastic agents. 379. Synthesis of phenstatin phosphate. J. Med. Chem. 1998, 41, 1688. 10.1021/jm970644q. [DOI] [PubMed] [Google Scholar]
- La Regina G.; Bai R.; Coluccia A.; Famiglini V.; Pelliccia S.; Passacantilli S.; Mazzoccoli C.; Ruggieri V.; Sisinni L.; Bolognesi A.; Rensen W. M.; Miele A.; Nalli M.; Alfonsi R.; Di Marcotullio L.; Gulino A.; Brancale A.; Novellino E.; Dondio G.; Vultaggio S.; Varasi M.; Mercurio C.; Hamel E.; Lavia P.; Silvestri R. New pyrrole derivatives with potent tubulin polymerization inhibiting activity as anticancer agents including hedgehog-dependent cancer. J. Med. Chem. 2014, 57, 6531. 10.1021/jm500561a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Go M. L. Antiproliferative properties of piperidinylchalcones. Bioorg. Med. Chem. 2006, 14, 153. 10.1016/j.bmc.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Ahn S.; Hwang D. J.; Barrett C. M.; Yang J.; Duke C. B. 3rd; Miller D. D.; Dalton J. T. A novel bis-indole destabilizes microtubules and displays potent in vitro and in vivo antitumor activity in prostate cancer. Cancer Chemother. Pharmacol. 2011, 67, 293. 10.1007/s00280-010-1319-8. [DOI] [PubMed] [Google Scholar]
- Ahn S.; Duke C. B. 3rd; Barrett C. M.; Hwang D. J.; Li C. M.; Miller D. D.; Dalton J. T. I-387, a novel antimitotic indole, displays a potent in vitro and in vivo antitumor activity with less neurotoxicity. Mol. Cancer Ther. 2010, 9, 2859. 10.1158/1535-7163.MCT-10-0399. [DOI] [PubMed] [Google Scholar]
- Chen J.; Wang Z.; Li C. M.; Lu Y.; Vaddady P. K.; Meibohm B.; Dalton J. T.; Miller D. D.; Li W. Discovery of novel 2-aryl-4-benzoyl-imidazoles targeting the colchicines binding site in tubulin as potential anticancer agents. J. Med. Chem. 2010, 53, 741. 10.1021/jm100884b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M.; Ahn S.; Wang J.; Chen J.; Miller D. D.; Dalton J. T.; Li W. Discovery of 4-Aryl-2-benzoyl-imidazoles as tubulin polymerization inhibitor with potent antiproliferative properties. J. Med. Chem. 2013, 56, 3318. 10.1021/jm4001117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Li C. M.; Wang Z.; Chen J.; Mohler M. L.; Li W.; Dalton J. T.; Miller D. D. Design, synthesis, and SAR studies of 4-substituted methoxylbenzoyl-aryl-thiazoles analogues as potent and orally bioavailable anticancer agents. J. Med. Chem. 2011, 54, 4678. 10.1021/jm2003427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Chen J.; Wang J.; Li C. M.; Ahn S.; Barrett C. M.; Dalton J. T.; Li W.; Miller D. D. Design, synthesis, and biological evaluation of stable colchicine binding site tubulin inhibitors as potential anticancer agents. J. Med. Chem. 2014, 57, 7355. 10.1021/jm500764v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn B. L.; Gill G. S.; Grobelny D. W.; Chaplin J. H.; Paul D.; Leske A. F.; Lavranos T. C.; Chalmers D. K.; Charman S. A.; Kostewicz E.; Shackleford D. M.; Morizzi J.; Hamel E.; Jung M. K.; Kremmidiotis G. Discovery of 7-hydroxy-6-methoxy-2-methyl-3-(3,4,5-trimethoxybenzoyl)benzo[b]furan (BNC105), a tubulin polymerization inhibitor with potent antiproliferative and tumor vascular disrupting properties. J. Med. Chem. 2011, 54, 6014. 10.1021/jm200454y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancale A.; Silvestri R. Indole, a core nucleus for potent inhibitors of tubulin polymerization. Med. Res. Rev. 2007, 27, 209. 10.1002/med.20080. [DOI] [PubMed] [Google Scholar]
- Li W.; Xiao M.; Dalton J. T.; Ahn S.; Miller D. D.; Chen J.; Wang J.. Azoles and related compounds for treatment of cancer and their preparation. UTRF and GTx, 2013. [Google Scholar]
- Mahboobi S.; Uecker A.; Sellmer A.; Cenac C.; Hocher H.; Pongratz H.; Eichhorn E.; Hufsky H.; Trumpler A.; Sicker M.; Heidel F.; Fischer T.; Stocking C.; Elz S.; Bohmer F. D.; Dove S. Novel bis(1H-indol-2-yl)methanones as potent inhibitors of FLT3 and platelet-derived growth factor receptor tyrosine kinase. J. Med. Chem. 2006, 49, 3101. 10.1021/jm058033i. [DOI] [PubMed] [Google Scholar]
- Lai M. J.; Huang H. L.; Pan S. L.; Liu Y. M.; Peng C. Y.; Lee H. Y.; Yeh T. K.; Huang P. H.; Teng C. M.; Chen C. S.; Chuang H. Y.; Liou J. P. Synthesis and biological evaluation of 1-arylsulfonyl-5-(N-hydroxyacrylamide)indoles as potent histone deacetylase inhibitors with antitumor activity in vivo. J. Med. Chem. 2012, 55, 3777. 10.1021/jm300197a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.