

Abstract
Thermobifida fusca is a moderately thermophilic and cellulolytic actinobacterium. It is of particular interest due to its ability to not only produce a variety of biotechnologically relevant enzymes but also serve as an alternative host for metabolic engineering for the production of valuable chemicals from lignocellulosic agricultural wastes. No bacteriophage that infects T. fusca has been reported, despite its potential impacts on the utilization of T. fusca. In this study, an extremely thermostable bacteriophage P1312 that infects T. fusca was isolated from manure compost. Electron microscopy showed that P1312 has an icosahedral head and a long flexible non-contractile tail, a characteristic of the family Siphoviridae. P1312 has a double-stranded DNA genome of 60,284 bp with 93 potential ORFs. Thirty-one ORFs encode proteins having putative biological functions. The genes involved in phage particle formation cluster together in a region of approximately 16 kb, followed by a segment containing genes presumably for DNA degradation/modification and cell wall disruption. The genes required for DNA replication and transcriptional control are dispersed within the rest of the genome. Phylogenetic analysis of large terminase subunit suggests that P1312 is a headful packaging phage containing a chromosome with circularly permuted direct terminal repeats.
Keywords: bacteriophage, Thermobifida fusca, lignocellulosic agricultural wastes, large terminase subunit, amidase, phage purification, endolysins, actinobacteriophage
Introduction
Thermobifida fusca, a moderately thermophilic soil actinobacterium, is known for its ability to produce a battery of cellulolytic enzymes (Maki et al., 2009; Adav et al., 2010; Gomez del Pulgar and Saadeddin, 2014). Sequence analyses of the genome of T. fusca YX strain suggests that it produces nine cellulases (including endocellulase, exocellulase, and cellobiosidase), at least five hemicellulose hydrolysis-related enzymes and many other glycoside hydrolases (Lykidis et al., 2007). Recent studies demonstrated that T. fusca may also secret lignin degradation-promoting enzymes. For example, a copper-containing polyphenol oxidase exhibits an activity for oxidation of phenolic lignin related compounds, and this activity boosts the digestion function of cellulase/xylanase toward sugarcane bagasse (Chen et al., 2013). Besides cellulolytic enzymes, T. fusca also produces a variety of oxidoreductases, such as heme-containing peroxidase (van Blooise et al., 2010) and catalase (Lončar and Fraaije, 2015), which are potentially useful in industries for detoxification and decolorization.
Cellulosic biomass is a low cost, abundant and renewable source for biofuels. To utilize it, great efforts have been devoted to engineer Saccharomyces cerevisiae so that the genetically modified yeasts can ferment xylose, cellobiose, and cello-oligosaccharides for bioethanol production (Katahira et al., 2006; Ha et al., 2011). Nonetheless, directly using the recalcitrant lignocellulose for biofuel production is still a challenging task. T. fusca has potential to be an alternative host for metabolic engineering to transform the sugars embedded in lignocellulose into biofuels and green chemicals. For example, an engineered T. fusca strain was able to convert untreated plant biomass to 1-propanol after an exogenous gene of bifunctional butyraldehyde/alcohol dehydrogenase was inserted into its genome (Deng and Fong, 2011). Despite this encouraging success, more molecular biology tools such as expression vectors and efficient transformation methods need to be developed before T. fusca can be fully modified for the purpose of producing valuable commodities from various cellulosic agricultural wastes.
Bacteriophages have contributed to the development of a variety of molecular tools for biotechnology. For example, the origin of replication from f1 and λ phages was used in the construction of plasmids for single-stranded DNA production (Reece, 2004) and routine recombinant DNA operation, respectively (Boyd and Sherratt, 1995). In addition, recombinases from bacteriophages such as phage λ, P1, and PY54 have been widely used to modify prokaryotic species and to create transgenic animals and plants (Sauer and Henderson, 1988; Nafissi and Slavcev, 2014). On the other hand, bacteriophages have potentials to foul industries that use bacteria to produce fermented products or bioactive molecules. No detailed reports of bacteriophages that infect T. fusca are present in the literature. Thus, we set out to isolate T. fusca-infecting phages with long-term goals to understand the bacterium-phage interactions and look for useful genetic elements for the development of molecular tools specific for T. fusca. A thermostable tailed bacteriophage of the Siphoviridae family was then isolated in this study.
Tailed bacteriophages constitute the Caudovirales order. Phages with contractile tails are further subdivided into the Myoviridae, while those with short and long non-contractile tails are into Podoviridae and Siphoviridae, respectively. Tails are critical to the infection because they contain proteins required for the specific recognition of the hosts and trigger DNA release from the heads. The genomes of tailed bacteriophages are composed of several functional modules. For Siphoviridae, the modular arrangement as follows: packaging, head morphogenesis, tail morphogenesis, lysis, recombination, lytic/lysogenic control, excision, and DNA replication modules is seen repeatedly in numerous temperate phages of Gram-positive low GC-content bacteria (Stevens et al., 2011), whereas lambdoid phages that infect Gram-negative bacteria have a genome consisting of modules in order of packaging, head morphogenesis, tail morphogenesis, recombination, lytic/lysogenic control, DNA replication, and lysis (Campbell, 1994). Characteristics and genome analysis of this newly discovered T. fusca-infecting phage are addressed herein.
Materials and methods
Bacterial strain
T. fusca NTU22 strain (Chen et al., 2013) was routinely cultivated by transferring 107 spores into 50 ml CYC medium (30 g sucrose, 6 g Casamino acids, 3 g NaNO3, 2 g yeast extract, 1 g K2HPO4, 0.5 g KCl, 0.5 g MgSO4·7H2O, 0.01 g FeSO4·7H2O, 1 l distilled water, pH 8.0) and incubated aerobically at 50°C with 200 rpm shaking.
Phage isolation
Compost collected from eight different sites in the suburbs of Taichung city, Taiwan, was the source of phage screening. One gram of the compost was added into 10 ml Luria-Bertani medium (LB) and incubated aerobically at 50°C overnight. One ml aliquot of the sample was irradiated (120 mJ/cm2, 30 s) using a CL-1000 UV crosslinker (UVP, California, USA) before being mixed with 0.1 ml T. fusca spore (108 cfu/ml) and 15 ml CYC media with 0.7% agar. The warm mixture was immediately poured onto a solidified CYC agar plate and incubated at 50°C for 2 days. One of the plaques formed in the bacterial lawn was transferred into 50 ml 1-day-old culture of T. fusca and further incubated at 50°C for 1 day. The bacterial lysate was centrifuged, and the supernatant was sterilized using a 0.45-μm filter and stored at 4°C. The isolated bacteriophage capable of infecting T. fusca was named P1312 hereafter.
Phage titer
Phage titer was estimated by plaque assay method. Briefly, 1 ml phage sample at the 107–109 dilution was mixed with 107 spore of T. fusca in 15 ml pre-warmed (50°C) CYC soft agar medium, and the mixture was poured evenly onto a CYC solid agar medium. The solidified plates were then incubated at 50°C for 2 days, and the number of plaques formed in the lawn of T. fusca was counted to determine the phage titer.
Phage stability, adsorption, and burst size
To determine the structural stability of P1312, the purified phage sample was diluted in water and incubated at temperatures ranging from 60–95°C for the indicated periods of time (10–45 min), and the residual infectivity was determined by the plaque assay. The burst size of P1312 was determined by one-step growth method (Lin et al., 2012) with slight modifications. Briefly, P1312 was added into an overnight culture of T. fusca in 20 mM phosphate buffer (pH 8.0) to give a multiplicity of infection (MOI) of 1.0 according to the initial spore number of T. fusca. The mixture was incubated at room temperature for 10 min to allow the phage particles to attach to the host. The supernatant, after centrifugation at 12,000 rpm for 5 min, was collected to determine the number of unattached phage particles. The percentage of adsorption was calculated using the formula [(initial titer-titer in supernatant)/initial titer × 100%]. The pelleted cells were washed two more times with CYC medium and resuspended in 50 ml of pre-warmed CYC and incubated at 50°C. Aliquots of 1 ml culture were taken at intervals and the phage titers in the clarified supernatant were then determined as aforementioned. The burst size (Bs) of P1312 was calculated as Bs = Pt/Po where Pt is the phage titer at the plateau phase and Po is the initial infective titer, which was estimated on the basis of the plaque-forming units arising from the initially washed cells.
Phage purification
Hundred milliliter broth of T. fusca, which had been cultivated for 1 day, was inoculated with 1 ml of P1312 stock (109 pfu/ml) and the cultivation was continued for another 2 days at 50°C, 200 rpm. The clarified broth, after centrifugation at 12,000 rpm, 4°C, for 10 min, was adjusted to contain 1.5 M NaCl and 10% (w/v) polyethylene glycol (PEG) 8000. The mixture was placed on ice for at least 1 h and subjected to centrifugation at 12,000 rpm, 4°C, for 15 min to precipitate the phage particles. The pellet was resuspended in TBS buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl) that additionally contained 0.25 U/ml DNase I and 10 μg/ml RNase A (Takara Bio). After incubation at 30°C for 1 h, the supernatant was passed through a filter with 0.45 μm pore size and loaded onto a Tricorn 10/600 column packed with Sephacryl S-500HR (Amersham Biosciences). The chromatography was performed with TBS buffer at a flow rate of 0.7 ml/min.
Transmission electron microscopy (TEM)
The purified phage particles on a carbon-coated copper grid were negatively stained with 2% uranyl acetate (pH 4.0) for 10 min and observed at an accelerating voltage of 120 kV under a transmission electron microscope (Jeol LEM-1400).
Genome analysis of phage P1312
Genomic DNA was obtained from the purified phage P1312 after phenol/chloroform extraction and ethanol precipitation. Whole genome sequencing of P1312 was performed using Illumina Miseq (Tri-I Biotech, Inc). The count of reads was 115,622 with the average length of 175 bases per read. The sequence data could be assembled into a circular single contig of 60,284 base pairs using the de novo assembly algorithm of CLC Genomics Workbench (Qiagen). The nucleotide sequence of the genome of P1312 is deposited at GenBank under accession number KT021004. Identification of potential open reading frames (ORFs) within the phage genome was performed by using Bacterial Annotation System (van Domselaar et al., 2005) as well as Glimmer/RBSfinder program (Delcher et al., 1999), and the functions of proteins encode by the ORFs were predicted based on BLASTp program and conserved domain search (http://www.ncbi.nlm.nih.gov/). The probable replication origin of P1312 was predicted by the GenSkew program (http://genskew.csb.univie.ac.at/).
Phylogenetic analysis
Protein sequences of terminase large subunit from a variety of bacteriophages were retrieved from the biological database, National Center for Biotechnology Information (NCBI). Multiple sequence alignments of those terminase sequences were performed by using the ClustalW program with default parameters in MEGA 6.0 version (Tamura et al., 2013). Phylogenetic tree was built by the neighbor-joining method and phylogenies were determined by bootstrap analysis of 10,000 replicates in MEGA 6.0 version.
Identification of proteins associated with P1312 virions
Proteins of P1312, purified via PEG precipitation and gel filtration chromatography, were identified by tandem mass spectrometry using an Applied Biosystems QStar LC-MS/MS spectrometer (Life Technologies Corp., Carlsbad, USA). The obtained spectrometry information was analyzed with Mascot software (Matrix Science Ltd., London, UK) using the NCBI non-redundant database and the specific database created in this study based on the predicted ORFs of phage P1312 (Table 1). The important parameter settings for Mascot analysis were as follows: mass values, monoisotopic; protein mass, unrestricted; peptide mass tolerance, ±0.5 Dalton; fragment mass tolerance, ±0.5 Dalton; and maximal missed cleavages, 2.
Table 1.
Predicted open reading frames (ORFs) of P1312 and predicted database matches.
| ORF | Start | End | Protein kDa (Start)a | Predicted functions | BLASTPb(best match) | Ident (%)c | LC-M/Md |
|---|---|---|---|---|---|---|---|
| 001 | 243 | 980 | 27.8 (V) | Phage head morphogenesis protein | WP_017602198/ | 132/249 | 38 |
| Nocardiopsis lucentensis | (53) | ||||||
| 002 | 987 | 1475 | 18.3 (V) | Hypothetical protein | WP_030728966 | 80/131 | |
| Streptomyces | (61) | ||||||
| 003 | 1462 | 2805 | 50.6 (M) | Phage terminase large subunit | WP_027740749 | 289/435 | 38 |
| Streptomyces | (66) | ||||||
| 004 | 5101 | 2795 | 83.4 (V) | Phage portal protein | WP_040692131 | 299/405 | 14 |
| Nocardiopsis lucentensis | (74) | ||||||
| gp23 | YP_003714730 | 36/148 | |||||
| phiSASD1 phage | (24) | ||||||
| 005 | 4301 | 5080 | 29.0 (L) | Hypothetical protein | WP_040692134 | 45/61 | |
| Nocardiopsis lucentensis | (74) | ||||||
| 006 | 5774 | 5085 | 25.2 (V) | No match | 44 | ||
| 007 | 5121 | 6005 | 31.3 (V) | Capsid | WP_017602203 | 225/283 | 18 |
| Nocardiopsis lucentensis | (80) | ||||||
| gp27 | YP_003714734 | 28/91 | |||||
| phiSASD1 phage | (31) | ||||||
| 008 | 6036 | 6461 | 15.5 (V) | Phage protein | WP_017602204 | 84/138 | 63 |
| Nocardiopsis lucentensis | (61) | ||||||
| gp27 | YP_003714736 | 21/47 | |||||
| phiSASD1 phage | (45) | ||||||
| 009 | 6644 | 6363 | 10.1 (V) | No match | |||
| 010 | 6954 | 6376 | 21.7 (V) | No match | 63 | ||
| 011 | 6487 | 6963 | 18.2 (M) | Hypothetical protein | WP_040692137 | 34/68 | |
| Nocardiopsis lucentensis | (50) | ||||||
| 012 | 6978 | 7340 | 13.3 (M) | Phage head-tail adaptor | WP_017602206 | 80/120 | 66 |
| Nocardiopsis lucentensis | (67) | ||||||
| gp30 | YP_003714737 | 38/94 | |||||
| phiSASD1 phage | (40) | ||||||
| 013 | 7340 | 7732 | 14.8 (V) | Phage tail protein | WP_040692140 | 49/70 | |
| Nocardiopsis lucentensis | (70) | ||||||
| gp32 | YP_003714739 | 43/105 | |||||
| phiSASD1 phage | (41) | ||||||
| 014 | 8508 | 7699 | 29.7 (V) | No match | 36 | ||
| 015 | 7720 | 8157 | 16.2 (V) | Phage tail protein | WP_026128262 | 84/130 | |
| Nocardiopsis lucentensis | (65) | ||||||
| gp33 | YP_003714740 | 44/124 | |||||
| phiSASD1 phage | (35) | ||||||
| 016 | 8512 | 8703 | 7.1 (V) | Phage tail protein | WP_017602209 | 45/62 | |
| Nocardiopsis lucentensis | (73) | ||||||
| gp34 | YP_003714741 | 19/53 | |||||
| phiSASD1 phage | (36) | ||||||
| 017 | 8703 | 9098 | 14.3 (M) | Hypothetical protein | WP_040692145 | 67/116 | |
| Nocardiopsis lucentensis | (58) | ||||||
| gp35 | YP_003714743 | 40/116 | |||||
| phiSASD1 phage | (34) | ||||||
| 018 | 9119 | 9406 | 11.2 (V) | Phage tail protein | WP_017602211 | 48/99 | |
| Nocardiopsis lucentensis | (48) | ||||||
| 019 | 9411 | 9587 | 5.9 (V) | No match | |||
| 020 | 9418 | 14391 | 177.1 (V) | Tail tape measure protein | WP_040703000 | 156/362 | 27 |
| Nocardiopsis salina | (43) | ||||||
| gp37 | YP_003714744 | 19/56 | |||||
| phiSASD1 phage | (34) | ||||||
| 021 | 14388 | 16361 | 71.7 (V) | Phage tail protein | WP_017602213 | 356/672 | |
| Nocardiopsis lucentensis | (53) | ||||||
| gp38 | YP_003714745 | 47/88 | |||||
| phiSASD1 phage | (53) | ||||||
| 022 | 16358 | 19648 | 120.6 (M) | Phosphodiesterase | WP_026128263 | 520/933 | |
| Nocardiopsis lucentensis | (56) | ||||||
| gp38 | YP_003714745 | 126/465 | |||||
| phiSASD1 phage | (27) | ||||||
| 023 | 19670 | 20212 | 19.3 (M) | Hypothetical protein | WP_040692409 | 71/180 | 23 |
| Nocardiopsis lucentensis | (39) | ||||||
| 024 | 20237 | 20497 | 9.6 (M) | No match | |||
| 025 | 20494 | 21270 | 28.4 (M) | O-methyltransferase | YP_024820 | 63/175 | 18 |
| Actinoplanes phage phiAsp2 | (36) | ||||||
| 026 | 21359 | 21267 | 3.2 (M) | No match | |||
| 027 | 21428 | 22423 | 37.2 (V) | Glycosyl/glycerophosphate transferases | WP_017602346 | 147/213 | |
| Nocardiopsis lucentensis | (69) | ||||||
| 028 | 22494 | 23063 | 20.1 (L) | Hypothetical protein | WP_045936534 | 35/120 | |
| Streptomyces sp. NRRL S-104 | (29) | ||||||
| 029 | 23346 | 23068 | 10.5 (M) | Hypothetical protein | WP_017602347 | 43/86 | |
| Nocardiopsis lucentensis | (50) | ||||||
| 030 | 23167 | 23517 | 11.9 (V) | No match | |||
| 031 | 23530 | 23408 | 4.9 (M) | No match | |||
| 032 | 23850 | 23530 | 12.0 (M) | No match | |||
| 033 | 23822 | 24076 | 8.5 (V) | No match | |||
| 034 | 24019 | 23870 | 5.9 (M) | No match | |||
| 035 | 24099 | 24554 | 13.5 (L) | No match | |||
| 036 | 24608 | 24126 | 17.6 (L) | Hypothetical protein | WP_031096358 | 58/114 | |
| Streptomyces | (51) | ||||||
| 037 | 24451 | 24597 | 5.4 (V) | No match | |||
| 038 | 24655 | 24882 | 8.6 (M) | Transcriptional regulator | WP_017946204 | 53/68 | |
| Streptomyces sp. CNS615 | (78) | ||||||
| 039 | 25042 | 25404 | 13.5 (V) | Protein involved in exopolysaccharide biosynthesis | WP_042170120 | 41/124 | |
| Streptomyces sp. NBRC 110035 | (33) | ||||||
| 040 | 25530 | 25973 | 16.3 (L) | Sensory protein kinase CreC | WP_043471037 | 47/109 | |
| Kitasatospora sp. MBT66 | (43) | ||||||
| 041 | 25970 | 26845 | 30.8 (M) | N-acetylmuramoyl-L-alanine amidase/Lysin | WP_017607061, Nocardiopsis | 173/266 | 34 |
| xinjiangensis | (65) | ||||||
| 042 | 27063 | 27344 | 9.9 (L) | Hypothetical protein (Similar to Tfu_2915) | WP_011293338 | 45/73 | |
| Thermobifida fusca | (62) | ||||||
| 043 | 27313 | 28419 | 38.9 (L) | No match | |||
| 044 | 28350 | 27490 | 32.0 (V) | Transcriptional regulator (Helix-turn-helix family protein) | WP_018223844 | 54/131 | |
| Salinispora pacifica | (41) | ||||||
| 045 | 28819 | 28406 | 14.5 (M) | No match | |||
| 046 | 29336 | 28905 | 16.5 (M) | No match | |||
| 047 | 30751 | 29333 | 50.3 (V) | Hypothetical protein | ADJ28745 | 88/289 | |
| Nitrosococcus watsonii C-113 | (30) | ||||||
| 048 | 29466 | 29365 | 3.7 (L) | No match | |||
| 049 | 29438 | 30544 | 42.5 (M) | Transposase, IS605 orfB | WP_016188297 | 364/368 | 29 |
| Thermobifida fusca | (99) | ||||||
| 050 | 30724 | 31998 | 44.1 (L) | No match | 27 | ||
| 051 | 31767 | 30778 | 34.9 (M) | Hypothetical protein | WP_037974524 | 40/107 | 14 |
| Synergistes jonesii | (37) | ||||||
| 052 | 33166 | 31952 | 45.3 (M) | Transcriptional regulator | WP_017615773 | 223/399 | |
| (Helix-turn-helix XRE-family like proteins) | Nocardiopsis salina | (56) | |||||
| 053 | 34868 | 33456 | 50.7 (V) | Replicative DNA helicase | WP_035111511 | 174/451 | 31 |
| Corynebacterium freiburgense | (39) | ||||||
| 054 | 34798 | 35892 | 37.3 (V) | No match | |||
| 055 | 36066 | 34912 | 42.3 (V) | Hypothetical protein | WP_040793245 | 49/130 | |
| Nocardia paucivorans | (38) | ||||||
| 056 | 37130 | 36525 | 22.0 (M) | Hypothetical protein | WP_040271854 | 37/87 | |
| Streptomonospora alba | (43) | ||||||
| 057 | 36735 | 37016 | 9.8 (V) | No match | |||
| 058 | 37102 | 38487 | 45.7 (V) | No match | |||
| 059 | 37779 | 37183 | 22.1 (M) | No match | |||
| 060 | 38460 | 37840 | 23.7 (M) | Hypothetical protein | WP_017541564 | 59/168 | |
| Nocardiopsis halophila | (35) | ||||||
| 061 | 38523 | 40232 | 64.3 (L) | No match | |||
| 062 | 40349 | 38715 | 55.7 (V) | Hypothetical protein | WP_013475335 | 89/160 | |
| Micromonospora sp. L5 | (56) | ||||||
| 063 | 42526 | 40484 | 72.8 (V) | DNA segregation ATPase FtsK/SpoIII | WP_017602285 | 253/481 | |
| Nocardiopsis lucentensis | (53) | ||||||
| 064 | 44174 | 42486 | 58.0 (V) | Hypothetical protein | WP_017602419 | 96/293 | |
| Nocardiopsis lucentensis | (33) | ||||||
| 065 | 43517 | 43747 | 8.7 (M) | No match | |||
| 066 | 44513 | 44346 | 6.2 (V) | No match | |||
| 067 | 45035 | 44577 | 16.5 (V) | ABC-ATPase | WP_037918697 | 51/119 | |
| Streptomyces yeochonensis | (43) | ||||||
| 068 | 45020 | 46795 | 67.3 (L) | Intergrase/Recombinase | YP_005087274 | 155/501 | |
| Rhodococcus phage REQ1 | (31) | ||||||
| 069 | 47265 | 46846 | 15.3 (M) | No match | |||
| 070 | 47870 | 47286 | 22.4 (V) | Hypothetical protein | WP_036322164 | 44/113 | 51 |
| Microbispora sp. ATCC PTA-5024 | (39) | ||||||
| 071 | 47971 | 48198 | 8.6 (M) | Transcriptional regulator | WP_012032242 | 33/63 | |
| (Helix-turn-helix XRE-family like proteins) | Pelotomaculum thermopropionicum | (52) | |||||
| 072 | 48520 | 48948 | 16.5 (M) | No match | |||
| 073 | 49402 | 48866 | 18.2 (L) | No match | |||
| 074 | 49094 | 49405 | 11.2 (V) | No match | |||
| 075 | 49402 | 50229 | 30.7 (M) | No match | 29 | ||
| 076 | 49935 | 49747 | 6.6 (M) | No match | 79 | ||
| 077 | 50446 | 51360 | 33.9 (V) | Exodeoxyribonuclease VIII | WP_017972488 | 156/282 | |
| Actinopolyspora halophila | (55) | ||||||
| 078 | 51357 | 52118 | 28.2 (V) | Hypothetical protein | WP_045740910 | 117/172 | |
| Actinoplanes rectilineatus | (68) | ||||||
| 079 | 52187 | 52939 | 27.5 (V) | DNA polymerase III subunit epsilon | WP_026118477 | 121/234 | |
| (DnaQ-like exonuclease) | Nocardiopsis salina | (52) | |||||
| 080 | 52936 | 53097 | 5.9 (V) | No match | |||
| 081 | 53097 | 53723 | 22.6 (M) | No match | |||
| 082 | 53720 | 54208 | 17.9 (V) | Holliday junction resolvase | WP_033299115 | 102/157 | |
| Nocardiopsis gilva | (65) | ||||||
| 083 | 54205 | 54645 | 16.3 (M) | WhiB family transcriptional regulator | WP_011595908 | 50/107 | |
| Rhodococcus | (47) | ||||||
| 084 | 54648 | 55439 | 30.3 (M) | Hypothetical protein | WP_017602245 | 135/271 | |
| Nocardiopsis lucentensis | (50) | ||||||
| 085 | 55657 | 55343 | 11.0 (M) | No match | 38 | ||
| 086 | 55382 | 56044 | 24.7 (V) | Hypothetical protein | WP_043984960 | 80/215 | |
| Mycobacterium llatzerense | (37) | ||||||
| 087 | 56029 | 57438 | 51.9 (V) | Hypothetical protein | EFE65835 | 135/380 | |
| Streptomyces ghanaensis ATCC 14672 | (36) | ||||||
| 088 | 56919 | 56311 | 21.1 (L) | No match | 40 | ||
| 089 | 57435 | 58103 | 24.4 (M) | Hypothetical protein | WP_030282501 | 30/83 | 63 |
| Streptomyces sp. NRRL B-5680 | (36) | ||||||
| 090 | 58214 | 58504 | 14.5 (V) | No match | 25 | ||
| 091 | 58573 | 58908 | 12.3 (M) | Hypothetical protein | WP_037865813 | 61/118 | |
| Streptomyces sp. NRRL S-1868 | (52) | ||||||
| 092 | 58872 | 58621 | 9.3 (M) | No match | |||
| 093 | 59867 | 59571 | 10.5 (V) | No match |
The first translated amino acid is shown in parentheses.
The matched homologs in Streptomyces phage phiSASD1 are also included although they are not the best hits.
The percentage identity is calculated based on the number of identical amino acid residues (numerator) over the number of compared residues (denominator) and shown in parentheses.
The sequence coverage (%) determined by mass spectrometry of the protein is indicated.
Results and discussion
Isolation and purification of phage P1312
Compost collected from several suburban farms was tested for the presence of phages that infect T. fusca according to the method described in Materials and Methods. A chicken manure sample was found to contain phages that could grow in T. fusca and resulted in lytic plaques in the lawn of the bacterium (Figure 1). After plaque purification and phage propagation, the virions were purified by PEG precipitation and gel filtration using a Sephacryl S-500HR column as described in Materials and Methods. The phage particles could be obtained from the fractions corresponding to the void volume of the chromatography (Figure 2). According to the TEM photos and the genome organization (described below), we believe that the purified sample contained only one type of phage. If there were more than two bacteriophages, more genes for phage specific proteins such as tape measurement protein and large subunit of terminase would be expected.
Figure 1.

Plaques of P1312 in the lawn of T. fusca NTU22.
Figure 2.

Purification of P1312 by gel filtration chromatography. The PEG-precipitated P1312 was loaded into a 50 ml Sephacryl S-500HR column. The proteins were eluted with TBS buffer and monitored with a UV detector. AU is referred to absorbance units.
Infectivity of P1312
Since T. fusca is a moderately thermophile, it was interesting to know the thermal stability of P1312. The phage particles were incubated at 60, 70, 80, 90, and 95°C. Aliquots were withdrawn periodically during the incubation and the residual infectivity was determined by the plaque assay. The infectivity of P1312 remained intact after the incubations at 90°C for 45 min, indicating that P1312 is extremely thermostable (Figure 3). Nonetheless, 5 min incubation at 95°C was able to inactivate P1312 effectively. For one-step growth experiments, P1312 was added into the culture of T. fusca at an MOI of 1. After 10 min incubation, which resulted in 69% adsorption of the phage particles to the host, the infected host cells were transferred into a fresh CYC medium and the phage particles released into the medium during the following cultivation were determined by the plaque assay. There was a latent period of ~45 min before the rapid increase of the phage titer, and lysis was complete by ~120 min (Figure 4). The growth kinetics of P1312 suggests that the average burst size is 57 pfu per infected cell.
Figure 3.

Thermal stability of T. fusca phage P1312. P1312 diluted in water was incubated at 60, 70, 80, 90, and 95°C for the indicated time, and the residual infectivity was determined by the plaque assay.
Figure 4.

One-step growth curve of P1312 at 50°C. Bacterial cultures were infected with a MOI of 1.0. The phage adsorption and culture conditions were as the description in Materials and Methods. Error bars indicate the standard deviation in triplicate samples.
General features of P1312
The morphology of P1312 was visualized using a transmission electron microscope (Figure 5). P1312 has a head in hexagonal outline, plausibly icosahedral, and a long flexible non-contractile tail, suggesting that P1312 belongs to the family Siphoviridae. The head is approximately 56 nm in diameter, and the tail is approximately 250 nm long and 11 nm wide. The complete nucleotide sequence of the genome of P1312 was determined with Illumina Miseq and assembled into a circular genome of 60,284 base pairs using the assembly algorithm of CLC Genomics Workbench. The GC content of the phage genome is 65.9%, close to 67.5% of the host's chromosome (Lykidis et al., 2007). The possible ORFs in the genome were predicted by Bacterial Annotation System and Glimmer/RBSfinder program. Integrating the computing results indicated the presence of 93 ORFs, arranged on both strands. Among them, 36 ORFs start with methionine, while 44 and 13 ORFs start with valine and leucine, respectively. The biggest ORF encodes a protein containing 1657 amino acid residues; by contrast, the smallest one encodes a peptide of 30 amino acids. The general information of the proteins encoded by the ORFs regarding their lengths, putative functions, and homologs found by BLASTp are listed in Table 1. The phage head morphogenesis protein-encoding ORF was arbitrarily assigned as the first ORF, followed by terminase genes, so that the presented gene order is similar to that of many bacteriophages such as Salmonella bacteriophage ES18 (Casjens et al., 2005) and Enterococcus faecalis bacteriophage ϕEf11 (Stevens et al., 2011). Thirty-one ORFs encode proteins that contain acknowledged functional domains or motifs, while 23 ORFs encode hypothetical proteins that have homologs found in other phages, prophages or bacteria, despite the lack of putative conserved domains. The rest of the predicted ORFs encode proteins sharing no significant identity with proteins in the databases; therefore, some of them may represent false positives.
Figure 5.
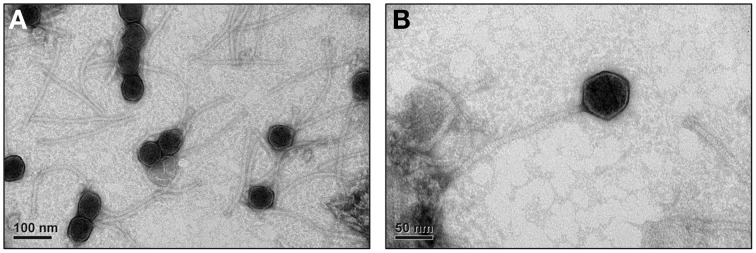
Electron microgram of P1312 virions. The phage particles were prepared, negatively stained and examined by electron microscope as described in Materials and Methods. (A) The broad view of the phage particles. (B) The close-up of a single phage particle.
The purified phage particles were subjected to protein identification by LC-MS/MS. This method identified 24 out of the 93 predicted proteins, including many phage structural proteins and enzymes such as N-acetylmuramoyl-L-alanine amidase and replicative DNA helicase (Table 1). Nine identified proteins (encoded by ORF6, 10, 14, 50, 75, 76, 85, 88, and 90) actually do not have significant homologs in the databases; therefore, they may represent novel proteins. It is noteworthy that either DNase I or RNase A, which were used in the pretreatment prior to the size exclusion chromatography, was not detected. The absence of the nucleases in the phage preparation suggests that the chromatographic method could effectively separate the soluble proteins from the virions.
Provisional functions of P1312 proteins
Genome organization of P1312 is illustrated in Figure 6. ORFs are displayed on both DNA strands with many of them overlapping. The cluster encompassing ORF1–21 is responsible for phage particle formation. ORF1 produces the head morphogenesis protein. The product of ORF3 contains an ATP-binding cassette transporter nucleotide-binding domain and shares similarity to the large subunit of phage terminases. ORF4 is supposed to produce the phage portal protein, the junction between the phage head and tail proteins, through which DNA passes during packaging and injection. ORF7 encodes a phage capsid protein. The product of ORF8 belongs to a family composed of proteins from a variety of bacteriophages. The region from ORF12 to ORF21 probably is involved in tail formation. In details, ORF12 directs the synthesis of the phage head-tail adaptor, while ORF13, 15, 16, 17, 18, and 21 likely encode the phage tail components. ORF20, the largest gene in the genome, encodes the tail tape measure protein. For lambdoid phages, the size of the tape measurement protein corresponds to the tail length by a fairly constant of 0.15 nm per amino acid residues (Katsura, 1990). The average tail length of P1312 is approximately 250 nm, accordant to the size of the phage tail tape measure protein. Many of the proteins encoded by the assumed head and tail gene modules share similarities to the proteins encoded by Streptomyces phage phiSASD1 (Wang et al., 2010) (Table 1). The comparison of the modules between P1312 and phiSASD1 is shown in Figure 7. Despite the resemblance, a couple of differences in the gene order are noticed. First, the directions of portal protein gene are opposite. Second, the N terminus of gp38 of phiSASD1 shares a small similar region with a tail protein of P1312 (product of ORF21), while the C terminus has a region similar to the CBM_4_9 domain present in the phosphodiesterase of P1312 (product of ORF22). Apart from the head and tail gene modules, the other modules share less similarity between P1312 and phiSASD1.
Figure 6.

Map of the genome of P1312. The apparently circular genome is open arbitrarily at the upstream region of the head morphogenesis protein-encoding gene. Rightward arrowed boxes denote the ORFs encoded by the forward strand, while leftward arrowed boxes represent those encoded by the complementary strand. ORFs colored by red, blue, green, and magenta direct the synthesis of structural proteins, enzymes involved in DNA degradation/modification and cell wall modifications, enzymes participating in phage DNA replication, and transcriptional factors, respectively. The orange arrow denotes the presumed origin of DNA replication. Ticks on the scale are at intervals of 500 bp.
Figure 7.

Comparison of the head and tail gene modules between P1312 and phiSASD1. The percentage identity between the compared proteins (see details in Table 1) is shown within the gray shadow.
The segment from ORF22 to ORF49 is notable for producing enzymes for a variety of functions such as DNA degradation/modification and cell wall disruption. ORF22 encodes a protein of 1096 amino acid residues. This protein is predicted to contain an N-terminal signal peptide, a metallophosphatase (MPP) domain and a domain related to carbohydrate-binding module (CBM) family 4_9 at the C-terminus. Proteins in the MPP superfamily are functionally diverse; the members include Mre11/SbcD-like exonucleases, Dbr1-like RNA lariat debranching enzymes, YfcE-like phosphodiesterases, purple acid phosphatases (PAPs), YbbF-like UDP-2,3-diacylglucosamine hydrolases and acid sphingomyelinases according to the conserved domain database, NCBI. By the N-terminal signal peptide and the C-terminal CBM, the product of ORF22 may be secreted through the cell membrane and anchors within the cell wall architecture. The MPP domain is thought to have a DNA degradation activity that may prevent the superinfection of the host by other bacteriophages. The cell wall of T. fusca mainly contains peptidoglycan and polyglycerolphosphate-lipoteichoic acid (Rahman et al., 2009); the latter is covalently linked to the outer leaflet of the cytoplasmic membrane. Therefore, the alternative function of the MPP domain may be involved in the hydrolysis of the cell wall by hydrolyzing the phosphodiester bonds in the teichoic acid polymers. ORF25 directs the synthesis of a putative class I adenosylmethionine-dependent methyltransferase. Presumably, this enzyme has an activity for nucleic acid modification. ORF27 encodes a protein similar in amino acid sequence to glycosyl/glycerophosphate transferase, a protein responsible for the polymerization of the main chain of the cell wall-associated teichoic acid. The product of ORF27, as being a phage protein, is thus assumed to interfere with, rather than promote, the host's function of teichoic acid synthesis.
The protein encoded by ORF41 is a putative N-acetylmuramoyl-L-alanine amidase, presumably able to disintegrate the peptidoglycan of the host cell by cleaving the amide bond between N-acetylmuramoyl and L-alanine. It is noteworthy that this phage protein does not contain a secretory signal sequence, consistent with a long-term observation that the endolysin produced by double stranded DNA bacteriophages requires a small membrane protein, known as a holin, to permeabilize the membrane for its access to the peptidoglycan (Young et al., 2000). Interestingly, we found that this putative amidase was associated with the phage particles according to the results of tandem mass spectrometry. This association may reflect an artifact caused by an unspecific interaction of the protein to the virion. Alternatively, it may implicate an involvement of the amidase in the infection step by assisting the phage to inject its DNA into the host cell. Actually, tail-associated peptidoglycan-degrading enzymes involved in localized cell wall degradation have been evidenced in a number of bacteriophages such as Tuc2009 (Kenny et al., 2004) and ϕ29 (Xiang et al., 2008). ORF042 encodes a 93-amino acid polypeptide with two transmembrane helices predicted by the TMPred program (Hofmann and Stoffel, 1993). It is tempting to assume that this polypeptide serves as the holin for the phage amidase to pass through the membrane. ORF49 encodes a transposase belonging to IS605 family, which is also present in the genome of T. fusca and many other bacteria. Presumably, phage P1312 acquired this gene from its bacterial host.
Genes responsible for DNA replication and transcriptional regulation are situated sporadically in the remaining half of the genome. Presumably, ORF53 directs the synthesis of a DnaB-like replicative helicase that unwinds the DNA duplex at the replication fork. ORF63 encodes a protein whose C-terminus shares similarity to DNA translocase ftsK of Actinoplanes, suggesting the involvement of the protein in DNA segregation. The protein encoded by ORF68 contains a putative serine recombinase domain that is usually found associated with pfam00239 in putative integrases/recombinases of mobile genetic elements of diverse bacteria and phages. Therefore, this protein may catalyze the integration and excision of the phage genome in and from the host chromosome, respectively. It is noteworthy that P1312 behaved as a lytic bacteriophage in this study; therefore, the culture condition may disfavor the integration function of the protein probably through suppressing the protein expression. The protein encoded by ORF77 is related to the PDDEXK superfamily, and appears to be an exonuclease VIII. The product of ORF79 is a DnaQ-like exonuclease, belonging to DEDDh 3′-5′ exonuclease family. Probably, it acts as a proofreading subunit (epsilon) of polymerase III for the DNA replication of P1312. ORF 82 encodes a protein of RusA superfamily that can resolve Holliday junction intermediates by its endonuclease activity. BLASTp analysis also suggests the presence of five transcriptional regulators, encoded by ORF38, 44, 52, 71, and 83. P1312 may use them to control the gene expressions, in terms of transcriptional timing and level, for its successful proliferation. The probable replication origin of P1312 was predicted by the GenSkew program, an application for computing and plotting nucleotide skew data. The resulting GC-skew plot (not shown) suggests that the region around the nucleotide 38440 (close to ORF60) could be the initiation site for replication.
DNA packaging strategy
All known tailed-bacteriophage virions contain a single linear dsDNA chromosome, because the passage of the portal protein is not wide enough to allow two parallel dsDNAs to be threaded simultaneously into the head during packaging and out of the virion during injection. As a member of Siphoviridae, P1312 probably contains a linear genome, rather than a circular one as determined by whole genome sequencing using Illumina Miseq. Six types of termini of the chromosomes in tailed-bacteriophage virions have been studied (Casjens and Gilcrease, 2009); they are (1) single-stranded cohesive ends, (2) circularly permuted direct terminal repeats, (3) short, non-permuted, direct terminal repeats, (4) long, non-permuted, direct terminal repeats, (5) terminal host DNA sequences, and (6) covalently bound terminal proteins. These different types of ends reflect varied DNA replication strategies and depend on terminase actions during DNA packaging. A previous bioinformatic analysis indicated that the different functional classes of phage-encoded terminases can usually be predicted from the amino acid sequence of large terminase subunits (Casjens et al., 2005). According to the determined nucleotide sequence, the termini of type 5 and 6 were precluded from the genome of P1312. To have a clue about the most probable packaging strategy employed by P1312, the large terminase subunit of P1312 was compared, in amino acid sequence, to those of bacteriophages with known chromosomal termini. Based on the resulting phylogenetic tree (Figure 8), P1312 is classified into a clan that uses the P22-like headful strategy for DNA packaging. Accordingly, the genomic termini of P1312 are possibly circularly permuted direct terminal repeats. To support this proposition, the phage genome was digested with a variety of restriction enzymes, and the digested products were heated at 80°C for 15 min, followed by a fast or slow cooling process. The samples were then analyzed by agarose gel electrophoresis (Figure 9). If the genome has single-stranded cohesive ends, the two restriction fragments, which have a single cohesive terminus, will join together and appear as a larger fragment in the slow chilled sample. That the restriction patterns of P1312 did not alter in response to the different cooling processes excludes the possibility of the presence of cohesive ends. For headful packaging phages that contain terminally redundant and circularly permutated chromosomes, the restriction pattern may consist of all the fragments expected from a circular genome plus a submolar pac fragment as in the case of P22 (Casjens and Gilcrease, 2009). However, if the headful terminase makes imprecise series initiation cleavage, as is the case for sf6 and ES18, no visible pac fragment will be detected in the restriction pattern. Instead, the terminal fragments will show as blur backgrounds between bands due to their variable lengths (Casjens and Gilcrease, 2009). The restriction patterns created by the various restriction enzymes in Figure 9 are consistent with the predicted results based on a circular P1312 genome. No pac fragment was observed in the electrophoresis gel in this study; however, blur backgrounds were actually present. This result supports the proposition that P1312 is a P22-like headful packaging phage.
Figure 8.

Neighbor-joining phylogenetic tree of large terminase subunit amino acid sequences. The sequences, except that from P1312, were classified as described previously (Casjens et al., 2005), and bootstrap analysis was performed with 1000 repetitions. The node of phylogenetic tree shows the bootstrap confidence values above 50%.
Figure 9.

Restriction analysis of P1312 DNA. The phage DNA was completely digested with BamHI, BstXI, PvuI, PvuII, SacI or SphI, and the products were analyzed by 0.8% agarose gel electrophoresis. Lane M indicate DNA marker, 1-kb DNA Ladder. f and s indicate that the digests were heated to 80°C for 15 min and then cooled fast or slow to room temperature, respectively.
Concluding remarks
Bacteriophage P1312 is the first well-described phage to infect T. fusca. The tolerance to high temperature indicates that P1312 has adapted to the thermophilic stage of composting as T. fusca. In general, P1312 has a genome organization similar to Siphoviridae of Gram-positive bacteria; however, a notable difference is the direction of the portal protein-encoding gene, which renders the genome of P1312 unique. Nocardiopsis lucentensis, an actinomycete lives in saline soil habitats (Yassin et al., 1993), is the number 1 ranked organism that provides best-matched homologs with the proteins encoded by the putative ORFs of P1312. This fact suggests the presence of prophages in N. lucentensis or implies a possibly evolutionary origin of P1312. P1312 may represent a valuable resource for understanding the molecular interaction between T. fusca and its co-evolutionary bacteriophages.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants MOST 103-2313-B-005-032-MY3 from the Ministry of Science and Technology, Taiwan, ROC. We would also like to express our gratitude to Professor Michael Bagdasarian, Michigan State University, for his help in the preparation of the manuscript.
References
- Adav S. S., Ng C. S., Arulmani M., Sze S. K. (2010). Quantitative iTRAQ secretome analysis of cellulolytic Thermobifida fusca. J. Proteome Res. 9, 3016–3024. 10.1021/pr901174z [DOI] [PubMed] [Google Scholar]
- Boyd A. C., Sherratt D. J. (1995). The pCLIP plasmids: versatile cloning vectors based on the bacteriophage lambda origin of replication. Gene 153, 57–62. 10.1016/0378-1119(94)00788-T [DOI] [PubMed] [Google Scholar]
- Campbell A. (1994). Comparative molecular biology of lambdoid phages. Annu. Rev. Microbiol. 48, 193–222. 10.1146/annurev.mi.48.100194.001205 [DOI] [PubMed] [Google Scholar]
- Casjens S. R., Gilcrease E. B. (2009). Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol. Biol. 502, 91–111. 10.1007/978-1-60327-565-1_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens S. R., Gilcrease E. B., Winn-Stapley D. A., Schicklmaier P., Schmieger H., Pedulla M. L., et al. (2005). The generalized transducing Salmonella bacteriophage ES18: complete genome sequence and DNA packaging strategy. J. Bactriol. 187, 1091–1104. 10.1128/JB.187.3.1091-1104.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. Y., Hsieh Z. S., Cheepudom J., Yang C. H., Meng M. (2013). A 24.7-kDa copper-containing oxidase, secreted by Thermobifida fusca, significantly increasing the xylanase/cellulase-catalyzed hydrolysis of sugarcane bagasse. Appl. Microbiol. Biotechnol. 97, 8977–8986. 10.1007/s00253-013-4727-y [DOI] [PubMed] [Google Scholar]
- Delcher A. L., Harmon D., Kasif S., White O., Salzberg S. L. (1999). Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27, 4636–4641. 10.1093/nar/27.23.4636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y., Fong S. S. (2011). Metabolic engineering of Thermobifida fusca for direct aerobic bioconversion of untreated lignocellulosic biomass to 1-propanol. Metab. Eng. 13, 570–577. 10.1016/j.ymben.2011.06.007 [DOI] [PubMed] [Google Scholar]
- Gomez del Pulgar E. M., Saadeddin A. (2014). The cellulolytic system of Thermobifida fusca. Crit. Rev. Microbiol. 40, 236–247. 10.3109/1040841X.2013.776512 [DOI] [PubMed] [Google Scholar]
- Ha S.-J., Galazka J. M., Kim S. R., Choi J.-H., Yang X., Seo J.-H., et al. (2011). Engineered Saccharomyces cerevisiae capable of simultaneous cellobiose and xylose fermentation. Proc. Natl. Acad. Sci. U.S.A. 108, 504–509. 10.1073/pnas.1010456108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K., Stoffel W. (1993). TMbase - a database of membrane spanning proteins segments. Biol. Chem. Hoppe-Seyler 374, 166. 1400476 [Google Scholar]
- Katahira S., Mizuike A., Fukuda H., Kondo A. (2006). Ethanol fermentation from lignocellulosic hydrolysate by a recombinant xylose- and cellooligosaccharide-assimilating yeast strain. Appl. Microbiol. Biotechnol. 72, 1136–1143. 10.1007/s00253-006-0402-x [DOI] [PubMed] [Google Scholar]
- Katsura I. (1990). Mechanism of length determination in bacteriophage lambda tails. Adv. Biophys. 26, 1–18. 10.1016/0065-227X(90)90004-D [DOI] [PubMed] [Google Scholar]
- Kenny J. G., McGrath S., Fitzgerald G. F., van Sinderen D. (2004). Bacteriophage Tuc2009 encodes a tail-associated cell wall-degrading activity. J. Bacteriol. 186, 3480–3491. 10.1128/JB.186.11.3480-3491.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. R., Chiu C. W., Chang F. Y., Lin C. S. (2012). Characterization of a new phage, termed ΦA318, which is specific for Vibrio alginolyticus. Arch. Virol. 157, 917–926. 10.1007/s00705-012-1244-8 [DOI] [PubMed] [Google Scholar]
- Lončar N., Fraaije M. W. (2015). Not so monofunctional–a case of thermostable Thermobifida fusca catalase with peroxidase activity. Appl. Microbiol. Biotechnol. 99, 2225–2232. 10.1007/s00253-014-6060-5 [DOI] [PubMed] [Google Scholar]
- Lykidis A., Mavromatis K., Ivanova N., Anderson I., Land M., DiBartolo G., et al. (2007). Genome sequence and analysis of the soil cellulolytic actinomycete Thermobifida fusca YX. J. Bacteriol. 189, 2477–2486. 10.1128/JB.01899-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki M., Leung K. T., Qin W. (2009). The prospects of cellulase producing bacteria for the bioconversion of lignocellulosic biomass. Int. J. Biol. Sci. 5, 500–516. 10.7150/ijbs.5.500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nafissi N., Slavcev R. (2014). Bacteriophage recombination systems and biotechnical applications. Appl. Microbiol. Biotechnol. 98, 2841–2851. 10.1007/s00253-014-5512-2 [DOI] [PubMed] [Google Scholar]
- Rahman O., Pfitzenmaier M., Pester O., Morath S., Cummings S. P., Hartung T., et al. (2009). Macroamphiphilic components of thermophilic actinomycetes: identification of lipoteichoic acid in Thermobifida fusca. J. Bacteriol. 191, 152–160. 10.1128/JB.01105-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece R. J. (2004). Analysis of Genes and Genomes. Hoboken, NJ: John Wiley & Sons. [Google Scholar]
- Sauer B., Henderson N. (1988). Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. U.S.A. 85, 5166–5170. 10.1073/pnas.85.14.5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens R. H., Ektefaie M. R., Fouts D. E. (2011). The annotated complete DNA sequence of Enterococcus faecalis bacteriophage φEf11and its comparison with all available phage and predicted prophage genomes. FEMS Microbiol. Lett. 317, 9–26. 10.1111/j.1574-6968.2010.02203.x [DOI] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blooise E., Torres Pazmiño D. E., Winter R. T., Fraaije M. W. (2010). A robust and extracellular heme-containing peroxidase from Thermobifida fusca as prototype of a bacterial peroxidase superfamily. Appl. Microbiol. Biotechnol. 86, 1419–1430. 10.1007/s00253-009-2369-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Domselaar G. H., Stothard P., Shrivastava S., Cruz J. A., Guo A., Dong X., et al. (2005). BASys: a web server for automated bacterial genome annotation. Nucleic Acids Res. 33, W455–W459. 10.1093/nar/gki593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Qiao X., Liu X., Zhang X., Wang C., Zhao X., et al. (2010). Complete genomic sequence analysis of the temperate bacteriophage phiSASD1 of Streptomyces avermitilis. Virology 403, 78–84. 10.1016/j.virol.2010.03.044 [DOI] [PubMed] [Google Scholar]
- Xiang Y., Morais M. C., Cohen D. N., Bowman V. D., Anderson D. L., Rossmann M. G. (2008). Crystal and cryoEM structural studies of a cell wall degrading enzyme in the bacteriophage ϕ29 tail. Proc. Natl. Acad. Sci. U.S.A. 105, 9552–9557. 10.1073/pnas.0803787105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassin A. F., Galinski E. A., Wohlfarth A., Jahnke K.-D., Schaal K. P., Trüper H. G. (1993). A new actinomycete species, Nocardiopsis lucentensis sp. nov. Int. J. Syst. Evol. Microbiol. 43, 266–271. 11526036 [Google Scholar]
- Young R., Wang I.-N., Roof W. D. (2000). Phages will out: strategies of host cell lysis. Trends Microbiol. 8, 120–128. 10.1016/S0966-842X(00)01705-4 [DOI] [PubMed] [Google Scholar]