

Abstract
The contiguity and phase of sequence information are intrinsic to obtain complete understanding of the genome and its relationship to phenotype. We report the fabrication and application of a novel nanochannel design that folds megabase lengths of genomic DNA into a systematic back-and-forth meandering path. Such meandering nanochannels enabled us to visualize the complete 5.7 Mbp (1 mm) stained DNA length of a Schizosaccharomyces pombe chromosome in a single frame of a CCD. We were able to hold the DNA in situ while implementing partial denaturation to obtain a barcode pattern that we could match to a reference map using the Poland-Scheraga model for DNA melting. The facility to compose such long linear lengths of genomic DNA in one field of view enabled us to directly visualize a repeat motif, count the repeat unit number, and chart its location in the genome by reference to unique barcode motifs found at measurable distances from the repeat. Meandering nanochannel dimensions can easily be tailored to human chromosome scales, which would enable the whole genome to be visualized in seconds.
I. INTRODUCTION
Conventional genomics methods, such as those that were used in the sequencing of the human reference genome handle and analyze molecules in bulk. Methods such as cloning or PCR are needed to isolate and produce sufficient quantities of DNA to be detected. However, the lengths of genomic DNA that can be handled by these methods are limited and cannot extend to whole chromosomes.
Analyzing genomic information by imaging long linear single DNA molecules was first introduced by the groups of Bensimon1 (dynamic molecular combing) and Schwartz2 (optical mapping) by stretching the DNA on surfaces. Notably, optical mapping has been employed at the finishing stage of genome sequencing projects.
While these methods stretch DNA on a surface, subsequent fluidic approaches have analyzed DNA in extensional flows.3–6 A recent variant of flow stretching is implemented by attaching a large bead at the end of the DNA.7 As the DNA is pulled into a small channel, the bead blocks further motion of the DNA and ensures that the DNA can be fully stretched and kept in one place for monitoring of dynamic processes. DNA molecules can also be tethered in both or a single end by chemical bonds to create DNA curtains as pioneered by Greene.8 The technique is combined with a lipid bilayer for passivation to enable studies of protein-DNA interactions without non-specific binding of the proteins.
Elongation due to confinement in nanochannels has been used to address questions in polymer physics and is advantageous for single molecule genomics. It can be performed by confining the DNA in one dimension by using a nanoslit9,10 or in two dimensions by using a nanoscale channel.11,12 It does not require external forces to perform the stretching and after a short relaxation time a steady state is reached, at which the DNA molecule remains elongated at equilibrium for an extended period.13 In addition, the extension of the DNA molecule scales linearly with the contour length of the DNA molecule and thereby provides a correlation between spatial positions along the polymer and within the genome. This has led to launch of single molecule mapping in nanochannels commercially, although the current platform available is restricted to analyzing genomic sub-fragments in parallel rather than complete lengths of DNA from whole chromosomes. Similarly, even though modern sequencing methods have begun to use single molecule analysis, the kilobase length scales that are now amenable14,15 are a sub-fraction of the lengths of chromosomes (Mbps). The problem with non-complete lengths is that de novo assembly of sub-sequences up to human chromosome length scales has been to-date computationally intractable and is confounded in diploid genomes by the sub-sequences originating from a mixture of homologous chromosomes, which means that haplotypes cannot be easily resolved.
Even though nanochannel systems exhibit a large potential, they have so far only been used for visualization of relatively short DNA strands when compared to the genome/chromosome sizes of most organisms. An interesting approach to handle long DNA is the dumbbell approach where a long DNA straddles a slit.10 Due to the excluded volume forces of the blobs at opposing sides of the slit, full stretching is achieved over a section of the DNA. However, a major limitation of this and other current designs is that the DNA is stretched in just one direction, which allows only limited segments of native genomic molecules to be observed in a single frame of the camera. These methods do not make efficient use of the real-estate on a CCD chip. In a recent study16 where megabase lengths of genomic DNA were stretched, up to 20 frames of a typical 512 × 512 chip needed to be stitched together to make a panoramic composite image. The vast majority of pixels in each image were devoid of useful information. More pertinent to the high-throughput needs of genomics, the longer the length of the genomic DNA to be visualized the longer the time it takes.
In the work reported here, we have drastically reduced the number of frames required to image large genomes by using serpentine or meandering nano-channels to systematically re-fold the DNA along a meandering path, so that a larger fraction of the pixels of the CCD chip can be used to collect useful information. This design is used to image DNA molecules in the Mbp range within a single CCD image. We show that DNA refolded in the meandering channel design can be used in combination with partial melting to create a high-resolution map 17 along an entire eukaryotic chromosome. We have developed new computational tools to extract single molecule optical maps from the images (supplementary material in Ref. 18).
II. MATERIALS AND METHODS
A. Experimental setup
A fluorescence microscopy system incorporating a Nikon Eclipse TE2000 inverted microscope, a 100 × N.A. 1.40 oil immersion objective, a standard FITC fluorescence filter set and an EMCCD camera (Andor iXon DV-897) and a standard mercury short arc lamp (100 W) was used.
A setup similar to the one described in Ref. 17 was used to mount the chip to the microscope, heating of the DNA sample, and control of the flow within the micro- and nano-channels. The setup included a fused-silica chip containing the channel system (made as explained below), a chip holder (also referred to as a chuck), a heater system, and a homebuilt pressure-control system for flow manipulation.
Each chip has two 1 × 5 μm2 channels for transporting the DNA to the array of nano-channels spanning between the micro-channels. Inlet holes at the ends of the micro-channels creates a connection to the macro world.
The chuck (designed to support the chip) was made of Zeonor 1060R (Sigolis AB, Sweden), due to its high resistance to chemicals, low inherent fluorescence, and high transparency. Reservoirs in the chuck connected to the inlet holes on the chip enable easy loading and manipulation of the sample during experiments. For managing the flow inside the chip, nitrogen gas was used to create an overpressure at the appropriate reservoir. Nitrogen gas was used to keep oxygen concentration at a minimum and thereby minimizing the photo damaging of the molecules.
The heating system consists of a temperature controller (336 Cryogenic Temperature Controller, Lake Shore, USA), an aluminum cap, a cartridge heater, and a thermocouple. Both the cartridge heater and thermocouple were positioned inside the aluminum cap which in turn was placed in contact with the backside of the chip through a hole spanning the center of the chuck. As a result, a reproducible thermal contact between the heater and the chip was achieved and a feedback loop created to ensure a stable temperature. To further increase the heat transfer thermally conductive grease (Omegatherm, Omega, USA) was applied at all interfaces in the heating system.
B. Chip production
The fabrication of the nanofluidic chip comprises the following processing steps, based on photolithography (PL), electron-beam lithography (EBL), reactive ion etching (RIE), sandblasting, fusion bonding, and dicing: (1) Alignment marks for PL and EBL were etched 500 nm deep into a 4-in. fused silica wafer of 500 μm thickness (from HOYA Corporation, USA), using fluorine based (50 sccm Ar, 50 sccm CHF3, 30 m Torr, 150 W RF-power) RIE and a photo resist (S1813, Shipley, USA) etch mask. Subsequently, the wafer was cleaned in piranha acid (H2SO4 2:1 H2O2) at 120 °C for 10 min, rinsed in DI-water and dried under nitrogen stream. (2) Aligned to the marks fabricated during the first precessing step, meandering grooves of 250 nm width were etched 250 nm into the substrate with fluorine based RIE (5 sccm Ar, 50 sccm CF4, 10 sccm CHF3, 8 mTorr, 100 W RF-power, 100 W ICP-power), using an electron-beam resist (ZEP520A, Nippon Zeon Co. Ltd., Japan) as an etch mask. After RIE, the resist was stripped under the same conditions as before. (3) Repeating the processing of the first step, 1 μm deep grooves were fabricated, overlapping with the ends of the meandering structures. After sealing the fluidic system, these grooves will form microfluidic channels that allow reagents to be brought to the nano-channels, which are formed by the sealed, meandering grooves etched during the previous step. (4) To fabricate inlet and outlet holes, Nitto SW-tape (Nitto Denko, Japan) was glued on both sides of the processed wafer. The tape served as a soft mask during sandblasting on the backside and as protection against contamination on the frontside. Small openings of approximately 1 mm diameter were cut into the tape at the desired position of the in/outlets, and through-holes were sandblasted using fused aluminum oxide powder of 50 μm particle size. (5) After removing the protective tape, the processed wafer was cleaned again in piranha etch (see above) and, together with a second unprocessed wafer of 175 μm thickness (from University Wafers, USA), immersed for 10 min first in RCA II (5:1:1 H2O: HCl: H2O2) and then in RCA I solution (5:1:1 H2O: NH2OH: H2O2) at 120 °C, rinsed in DI and dried under nitrogen stream. The two wafers were then brought together and covalently fusion bonded by annealing them at 1100 °C in nitrogen atmosphere for 6 h. (7) Finally, nanofluidic chips of 25 × 25 mm2 were diced from the bonded wafers, using a 200 μm wide resin-bonded blade with 40 μm-diamonds (Dicing Blade Technology, USA).
C. Sample preparation
The experiments were performed with chromosomes from Schizosaccharomyces pombe (S. pombe) (CHEF DNA Size Marker, from Bio-Rad). The DNA was delivered in agarose gel as an electrophoresis size standard comprising chromosomes of lengths 3.5 Mbp, 4.6 Mbp, and 5.7 Mbp. To avoid degradation of the DNA molecules, they were stored within the agarose gel at 8 °C until use. Prior to experiments DNA was stained with YOYO-1 fluorescent dye (LifeTechnologies, USA) at a ratio of 1 dye molecule per 6 base pairs. A simultaneous staining and gel-extraction process was performed in a buffer consisting of 10 mM NaCl in 0.05 × TBE (4.5 mM Tris, 4.5 mM Boric acid, and 0.1 mM EDTA) and YOYO-1. In this process, small pieces of agarose gel, carefully cut with a sharp knife, containing DNA were placed in a large volume staining buffer and left at room temperature (20 °C) over night during which the DNA diffused out from the gel. Just prior to experiments the running buffer was created by adding 2-mercaptoethanol (Sigma-Aldrich, MO, USA) and formamide (Sigma-Aldrich, MO, USA) directly to the staining solution to final concentrations of 3% and 50%, respectively.
D. Experimental procedure
Prior to each new experiment, a buffer consisting of 5 mM NaCl in 0.025x TBE, 3% (v/v) 2-mercaptoethanol and 50% formamide was flushed through the channels for at least 15 min to ensure correct buffer conditions before introducing the DNA. After introduction of DNA, the flow rate was kept as low as possible at all times to minimize shearing of the molecules. Once the DNA molecules reached the inlets to the nano-channels pressure was applied to both the reservoirs connected to the micro-channel containing the DNA. In this way, the buffer was forced to flow through the nano-channels dragging the DNA with it (Figure 1). After introduction into the nano-channels, the molecules were either imaged immediately to collect information about the relaxation behavior or heated for 10 min to create a barcode pattern before imaging.
FIG. 1.

(a) Overview of chip design showing how the micro-channels (blue) connect the reservoirs to the meandering nano-channels (purple). (b) Zoom in of the meander channel array. The pillar array initially placed at the inlets to the nano-channels is also visualized. (c) Zoom in of the meandering part of the nano-channels. (d) Image of one bend in meandering channel. (e) Cross section of an open nano-channel prior to bonding. (f) Cross section of nano-channel after closing of the channel.
Despite the use of thermally conductive grease, the measured temperature varied slightly from the actual temperature in the channels. Also the temperature measured for DNA denaturation to be initiated may potentially vary from day to day due to variations in buffer conditions and quality of formamide. During the melting experiment, the temperature was therefore slowly and step-wise increased from 29 °C up to 45 °C. Several different molecules were studied at each temperature to find the optimal melting conditions in each case. To ensure a stable temperature during data collection, the system was equilibrated for 15 min in darkness at each new temperature before data collection was initiated.
E. Image analysis
Image analysis is required for extracting time-traces from the meandering nano-channels. To this end, our algorithm first performed top-hat filtering using a linear structuring element to correct for uneven background illumination, then rotated the image to have the linear parts vertical. Thereafter, a parameterized meander contour based on the known nanolithography mask was placed on top of the picture with a vertical scale-factor, a horizontal scale-factor, up-down orientation, and mean position as fitting parameters, see supplementary material in Ref. 18 for details. This procedure allowed for extraction of a time-trace for the melting map by walking along the parameterized meander contour. In this procedure, a 7 pixel wide window was used in the direction perpendicular to the meander orientation.
F. Time-trace alignment
Once the meander contour had been extracted at each time frame, a kymograph was generated, with time on the vertical axis and distance along the contour on the horizontal axis. However, due to random diffusive processes, the DNA had undergone thermally induced stretching and contracting at the local and global levels, resulting in a “fuzzy” barcode.17 Thus, before further analysis could be performed, the kymograph had to undergo an alignment procedure. For this purpose, we applied our novel alignment algorithm, WPAlign.19 Our new approach brings down computational costs considerably compared to previous procedures.17 Importantly, for MBp-sized genomes as of interest here, the old approach is estimated to require more than a day to perform its task, whereas our new method aligns a barcode in less than a minute.19 Briefly, the algorithm identifies the most distinctive “feature” by treating the kymograph as an intensity landscape and finding the shortest vertical path through it. This feature is then aligned, and the process is called recursively on the portions of the kymograph to the left and to the right of the newly-aligned feature. The process eventually terminates when the left and right regions are smaller than a threshold width given by the width of a typical feature.
G. Comparison between experiments and theory
The Poland-Scheraga model20 and the Fixman-Freire approximation were used to compute DNA melting maps. During the calculations, stability parameters from Ref. 21 and a loop exponent of c = 1.76 were used with the end base-pairs assumed clamped. An elevated temperature by an amount ΔT = F 62 °C was used17 in the theory in order to account for the F = 50% formamide by volume, i.e., for the simulation we used a temperature 31 °C greater than that in the experiments.
The output from the theory is a base pair resolution probability map pds(s). To handle the different flexibilities for single-stranded and double-stranded regions, we used the approach from Ref. 17 together with a Gaussian convolution with a width equal to the optical point spread function 200 nm to yield smoothed theoretical prediction. When comparing experiments and theory, both experiments and theory are scaled to be on average zero and to have variance one.17,22 Experimental time averages were then compared to theory using subsequence dynamic time warp23 with horizontal and vertical “stretching” costs set to 10 and the diagonal cost set to 1, thus heavily biasing against stretching, yet allowing for, for instance, channel defects causing a varying nanochannel width. The required stretching, determined by this process, was then applied to the full kymographs for visualization (see Fig. 6).
FIG. 6.
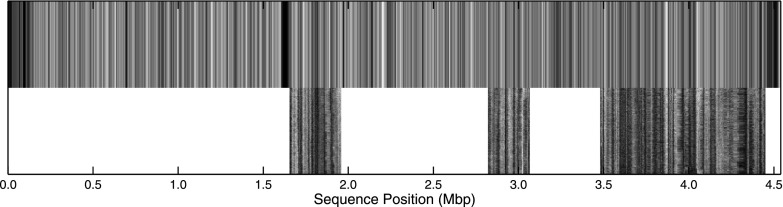
Comparison between convoluted theoretical DNA melting prediction and the experiment shown in Fig. 4. To make this comparison, we split the experiment into three regions: (1) 20 μm–150 μm, (2) 240 μm–300 μm, and (3) 450 μm–680 μm. This was done because we believe cutting events occurred between these regions. Each of these regions was then matched to the theoretical profile (chromosome 2) independently (i.e., without bias against overlapping) using subsequence dynamic time warping. All three regions align to the theoretical profile in the order seen from the experiment (Fig. 4), supporting the hypothesis that cutting events occurred between them.
III. RESULTS AND DISCUSSION
A. Nano-channel design
Direct imaging of DNA molecules in the Mbp range was enabled by folding nano-channels into a long meandering pattern. Each meandering channel could be imaged within a single field of view and was designed to hold up to 10 Mbp of DNA stretched to 30% extension (1 mm) (Figures 1 and 4). To simplify the analysis, the meandering part of the nano-channel was designed with 50 μm long straight parts parallel to each other and connected with soft bends. The bends were in turn designed with a 1 μm long straight part connected to two 90° turns to minimize stress on the DNA molecules in the meander-channels.
FIG. 4.

(Top, left) Time-averaged raw fluorescence intensity image of a melted S. pombe chromosome. (Top, middle) The precise location of the meandering nano-channel was found and highlighted using our meander-finding algorithm. (Top, right) Nano-lithography pattern. (Middle) Raw kymograph extracted by our tool by “walking along” the meander contour. In this procedure, a 7 pixel wide window was used in the direction perpendicular to the meander orientation. (Bottom) The alignment procedure, WPAlign, from Ref. 19 applied to the raw kymograph.
The higher resistance of longer channels together with the high risk of shearing the molecules during entry into the nano-channels at high pressures led to a design where the inlets of several meander-channels were kept in close proximity of each other. This increased the resulting bulk flow rate towards the nano-channels decreasing the time needed for the introduction of DNA ensuring that the bulk flow rate exceeded the diffusional rate of the end of molecule that was to be introduced.
A micro-post array spanning across half the micro-channel was initially placed at the inlets to the nano-channels for pre-stretching of the molecules, and thereby easing their introduction into the nano-channels (Figure 2).24 However, for the long chromosomes used in this study the micro-post array turned out to be more of an unneeded hindrance than an aid. The large DNA molecules followed the flow lines around the micro-post array and thereby ended up further from the inlets than they would have in the absence of the array. When forced to travel trough the array, the DNA molecules became entangled around the pillars which prevented further movement towards the nano-channels. DNA molecules in the Mbp range have a radius of gyration in the order of a micron and therefore a slight elongation due to confinement was apparent in the 1 μm deep micro-channel. Furthermore, the parabolic flow profile inside the channel served to stretch the large DNA molecules since different parts of the molecule experience different flow rates.
FIG. 2.

DNA behavior with and without a micro-post array in the micro-channel. (a) A bright field image showing the micro-post array at the nano-channel inlets. (b) A fluorescence microscopy image of long DNA molecules around the pillars in the micro-post array. Due to their large size, the DNA molecules follow the flow lines around the pillars along the micro-channel rather than entering the array. (c) During the introduction into the nano-channels the DNA molecules become entangled around the pillars within the array which prevents them from entering the nano-channels. (d) The parabolic velocity profile in the micro-channel pre-stretches the long DNA molecules rendering the pillar array unnecessary. The scale bars are 15 μm.
B. Handling long DNA
As a proof of principle, an entire chromosome (5.7 Mbp) from S. pombe was imaged in a 250 × 250 nm2 meander channel (Figures 3(a) and 3(b)). Figure 3(a) shows the DNA molecule directly after introduction into the meander-channel. Due to the shear experienced during introduction into the nano-channels, the molecule was highly extended. When the flow was stopped, the molecule started to relax towards its equilibrium state and after 10 min the molecule had relaxed considerably (3(b)). However, due to the slow relaxation for long molecules25 it still had not reached its equilibrium state and was extended to 50%, compared to the expected 30% extension for a relaxed molecule.
FIG. 3.

Relaxation of long S. pombe DNA stained with YOYO-1 and imaged using a fluorescence microscope. The molecules were stretched in 250 × 250 nm2 meander channels. (a) The image shows a 5.7 Mbp long S. pombe chromosome highly extended due to the shear experienced during loading. (b) The same molecule as in (a) but after 10 min in the meander-channel. It is clear that the molecule has relaxed considerably. (c) Time-trace of a relaxing DNA molecule with a repetitive melting pattern along the molecule. The slow relaxation enables these high resolution images of relaxing molecules. The white vertical line represents a meander bend from which no data has been extracted. The scale bars represent 15 μm.
Requirements for obtaining a sharp pattern structure in an image include a movement of the sample at slower rate than the exposure time during imaging. The long relaxation times for chromosomal length DNA therefore enables imaging of fine structures on molecules that have not yet reached equilibrium and thereby gaining a higher resolution than would be possible for relaxed molecules in equal sized channels (Figure 3(c)).
C. Mapping to theory
Combining melting mapping with meandering nano-channels has led to direct visualization of a barcode pattern along DNA molecules in the Mbp range. As a proof of principle we show a melting mapping pattern along the 4.6 Mbp S. pombe chromosome (Figure 4). Our meander-finding algorithm correctly identified the meander in Figure 4 (top, left) as is visualized in Figure 4 (top, middle). The time-trace in Figure 4 middle was extracted using our tool by “walking along” the meander contour and successfully extracting the barcode pattern along the DNA confined in the nano channel (Figure 4, bottom).
An important problem in melting mapping is the photo cutting of DNA. In this case, it is not fully clear whether a dark area is locally melted DNA or just discontinuous space in between two DNA strands. For small fragments diffusion would reveal a cut, but for long DNA it may be difficult to tell the difference. A simple edge detection algorithm along the aligned DNA identified three different regions (20 μm–150 μm, 240 μm–300 μm, and 450 μm–680 μm) along the barcode shown in Figure 4. These three regions were then independently aligned to a theoretical profile for S. pombe chromosome 2, and they successfully aligned in the order seen in the experiment with no bias against overlapping. This suggests that the extended dark regions in Figure 4 may have resulted from cutting events.
Melting of whole chromosomes can be used to determine the number of repetitive parts as well as their lateral placement, something that can be difficult when only smaller parts are analyzed. The left end of the time-trace in Figure 5 includes a repetitive part (magnified in the bottom image). In order to check for expected repetitive regions in the barcodes of the three S. pombe chromosomes based on sequence data, we calculated theoretical barcodes. Based on a simple repetitive region finding method, see supplementary material in Ref. 18, we found no repetitive regions with of the order 40 repeats in these theory barcodes.
FIG. 5.

(Top) Time-trace of an S. pombe chromosome containing a repetitive part with approximately 40 repeats in the left part of the image. (Bottom) A magnification of the repetitive part of the molecule. The scale bar is 15 μm.
IV. CONCLUSIONS
We have shown that meandering nano-channels substantially ease direct imaging of DNA molecules in the Mbp range, by demonstrating a 5.7 Mbp DNA molecule at 50% extension within a single field of view. In the current design, one of the meandering parts is almost long enough (1 mm) to hold the entire genome from Saccharomyces cerevisiae (12.1 Mbp). With an optimized denser design, we expect to be able to hold at least 20 mm of DNA per field of view (at 50% stretching corresponding to 120 Mbp; this length would be enough to contain the whole of most of the human chromosomes, while the longer chromosomes, could be covered by spooling the molecule through and taking a second image in a dumbbell arrangement of the DNA, which may also be used for achieving an improved degree of stretching of the DNA). This would allow us to image the entire diploid human genome (6.4 Gbp) with a mere 50 frames. Together with our algorithms for extracting and aligning barcodes from our meandering nano-channel, the novel channel design is a first step towards an automated tool for whole chromosome analysis.
We have demonstrated that molecules in the meandering channels can yield useful bioanalytical information by creating melting barcodes along the elongated DNA, which have been validated as a means for determining meaningful genomic information.16 In order to validate the experimental barcodes, we compared experiments to a theoretical reference barcode, obtained using the Poland-Scheraga model with S. pombe DNA sequences as input, and found good agreement. We could identify fragments of the genome corresponding to sequence positions 1.7 Mbp–1.9 Mbp, 2.8 Mbp–3.1 Mbp, and 3.5 Mbp–4.4 Mbp. What is particularly striking is that the matching patterns can easily be discerned by eye (see Figure 6).
Furthermore, within the single field of view, we have detected and localized a region of repetitive DNA in the single S. pombe chromosome, which cannot be seen in published sequences of S. pombe. The long-range view of genomic DNA made possible by our approach allows us to count the number of repeated segments within the region. This finding points to the method's unique potential for detecting large structural variations in a single molecule.
The meandering nanochannel map technique allows for the characterization of large fragments of chromosomal DNA from individual cells for characterization of disease associated rearrangements of the genome. They will also open up for identification of single microorganisms, which will be important for rapid pathogen diagnosis and metagenomic studies without the need for culturing the cells or DNA amplification. Here, a range of different labeling schemes can be used for mapping relevant information along the DNA, including melt mapping as we show here,17 competitive binding,26 nick translation,9,12 fluorocode,27 and labels for epigenetic patterns.28 We have shown the visualization of whole microbial chromosomes, which will allow analysis of whole microbial genomes without the need to assemble smaller fragments. The fabrication of longer meander structures will open up the intriguing prospect of extending the approach to human-scale chromosomes and assembly-free genome analysis.
ACKNOWLEDGMENTS
This work was supported by the Swedish Research Council (VR) Grant Nos. 2007-584 and 2007-4454, the EU's seventh Framework Programme (7RP/2007-2013) under Grant Agreement No. 201418 (READNA), a project grant from VINNOVA (Grant No. P35735-1), NanoLund, and the Crafoord Foundation Grant Nos. 2008-0841 and 2005-1123. K.U.M. acknowledges support from the UK Royal Society Theo Murphy Blue Skies Award. T.A. acknowledges Swedish Research Council, Grant No. 2009-2924 and The Carl Tryggers Foundation, Grant No. 12:13.
References
- 1. Bensimon A., Simon A., Chiffaudel A., Croquette V., Heslot F., and Bensimon D., “ Alignment and sensitive detection of DNA by a moving interface,” Science 265(5181), 2096–2098 (1994). 10.1126/science.7522347 [DOI] [PubMed] [Google Scholar]
- 2. Schwartz D. C., Li X. J., Hernandez L. I., Ramnarain S. P., Huff E. J., and Wang Y. K., “ Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping,” Science 262(5130), 110–114 (1993). 10.1126/science.8211116 [DOI] [PubMed] [Google Scholar]
- 3. Perkins T. T., Smith D. E., Larson R. G., and Chu S., “ Stretching of a single tethered polymer in a uniform-flow,” Science 268(5207), 83–87 (1995). 10.1126/science.7701345 [DOI] [PubMed] [Google Scholar]
- 4. Quake S. R., Babcock H., and Chu S., “ The dynamics of partially extended single molecules of DNA,” Nature 388(6638), 151–154 (1997). 10.1038/40588 [DOI] [PubMed] [Google Scholar]
- 5. Tegenfeldt J. O., Bakajin O., Chou C.-F., Chan S., Austin R. H., Fann W., Liou L., Chan E., Duke T., and Cox E. C., “ Near-field scanner for moving molecules,” Phys. Rev. Lett. 86(7), 1378–1381 (2001). 10.1103/PhysRevLett.86.1378 [DOI] [PubMed] [Google Scholar]
- 6. Chan E. Y., Goncalves N. M., Haeusler R. A., Hatch A. J., Larson J. W., Maletta A. M., Yantz G. R., Carstea E. D., Fuchs M., Wong G. G., Gullans S. R., and Gilmanshin R., “ DNA mapping using microfluidic stretching and single-molecule detection of fluorescent site-specific tags,” Genome Res. 14, 1137–1146 (2004). 10.1101/gr.1635204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sriram K. K., Yeh J.-W., Lin Y.-L., Chang Y.-R., and Chou C.-F., “ Direct optical mapping of transcription factor binding sites on field-stretched lambda-DNA in nanofluidic devices,” Nucleic Acids Res. 42(10), e85 (2014). 10.1093/nar/gku254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qi Z., Redding S., Lee J.-Y., Gibb B., Kwon Y., Niu H., Gaines W.-A., Sung P., and Greene E.-C., “ DNA sequence alignment by microhomology sampling during homologous recombination,” Cell 160(5), 856–869 (2015). 10.1016/j.cell.2015.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jo K., Dhingra D. M., Odijk T., de Pablo J. J., Graham M. D., Runnheim R., Forrest D., and Schwartz D. C., “ A single-molecule barcoding system using nanoslits for DNA analysis,” Proc. Natl. Acad. Sci. U.S.A. 104(8), 2673–2678 (2007). 10.1073/pnas.0611151104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kounovsky-Shafer K. L., Hernandez-Ortiz J. P., Jo K., Odijk T., de Pablo J. J., and Schwartz D. C., “ Presentation of large DNA molecules for analysis as nanoconfined dumbbells,” Macromolecules 46(20), 8356–8368 (2013). 10.1021/ma400926h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tegenfeldt J. O., Prinz C., Cao H., Chou S., Reisner W. W., Riehn R., Wang Y. M., Cox E. C., Sturm J. C., Silberzan P., and Austin R. H., “ The dynamics of genomic-length DNA molecules in 100-nm channels,” Proc. Natl. Acad. Sci. U.S.A. 101(30), 10979–10983 (2004). 10.1073/pnas.0403849101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lam E. T., Hastie A., Lin C., Ehrlich D., Das S. K., Austin M. D., Deshpande P., Cao H., Nagarajan N., Xiao M., and Kwok P.-Y., “ Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly,” Nat. Biotechnol. 30(8), 771–776 (2012). 10.1038/nbt.2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reisner W., Morton K., Riehn R., Wang Y., Yu Z., Rosen M., Sturm J., Chou S., Frey E., and Austin R., “ Statics and dynamics of single DNA molecules confined in nanochannels,” Phys. Rev. Lett. 94(19), 196101 (2005). 10.1103/PhysRevLett.94.196101 [DOI] [PubMed] [Google Scholar]
- 14. Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B., Bibillo A., Bjornson K., Chaudhuri B., Christians F., Cicero R., Clark S., Dalal R., Dewinter A., Dixon J., Foquet M., Gaertner A., Hardenbol P., Heiner C., Hester K., Holden D., Kearns G., Kong X. X., Kuse R., Lacroix Y., Lin S., Lundquist P., Ma C. C., Marks P., Maxham M., Murphy D., Park I., Pham T., Phillips M., Roy J., Sebra R., Shen G., Sorenson J., Tomaney A., Travers K., Trulson M., Vieceli J., Wegener J., Wu D., Yang A., Zaccarin D., Zhao P., Zhong F., Korlach J., and Turner S., “ Real-time DNA sequencing from single polymerase molecules,” Science 323(5910), 133–138 (2009). 10.1126/science.1162986 [DOI] [PubMed] [Google Scholar]
- 15. Huddleston J., Ranade S., Malig M., Antonacci F., Chaisson M., Hon L., Sudmant P. H., Graves T. A., Alkan C., Dennis M. Y., Wilson R. K., Turner S. W., Korlach J., and Eichler E. E., “ Reconstructing complex regions of genomes using long-read sequencing technology,” Genome Res. 24(4), 688–696 (2014). 10.1101/gr.168450.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marie R., Pedersen J. N., Bauer D. L. V., Rasmussen K. H., Yusuf M., Volpi E., Flyvbjerg H., Kristensen A., and Mir K. U., “ Integrated view of genome structure and sequence of a single DNA molecule in a nanofluidic device,” Proc. Natl. Acad. Sci. 110(13), 4893–4898 (2013). 10.1073/pnas.1214570110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reisner W., Larsen N. B., Silahtaroglu A., Kristensen A., Tommerup N., Tegenfeldt J. O., and Flyvbjerg H., “ Single-molecule denaturation mapping of DNA in nanofluidic channels,” Proc. Natl. Acad. Sci. U.S.A. 107(30), 13294–13299 (2010). 10.1073/pnas.1007081107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.See supplementary material at http://dx.doi.org/10.1063/1.4923262E-BIOMGB-9-019503 for details on the image processing.
- 19. Noble C., Nilsson A. N., Freitag C., Beech J. P., Tegenfeldt J. O., and Ambjörnsson T., “ A fast and scalable kymograph alignment algorithm for nanochannel-based optical DNA mappings,” PLoS One 10(4), e0121905 (2015). 10.1371/journal.pone.0121905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poland D. and Scheraga H. A., Theory of Helix-Coil Transitions in Biopolymers: Statistical Mechanical Theory of Order-Disorder Transitions in Biological Macromolecules, Molecular Biology ( Academic Press, 1970). [Google Scholar]
- 21. Krueger A., Protozanova E., and Frank-Kamenetskii M. D., “ Sequence-dependent basepair opening in DNA double helix,” Biophys. J. 90(9), 3091–3099 (2006). 10.1529/biophysj.105.078774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Welch R. L., Sladek R., Dewar K., and Reisner W., “ Denaturation mapping of Saccharomyces cerevisiae,” Lab Chip 12(18), 3314–3321 (2012). 10.1039/c2lc40504k [DOI] [PubMed] [Google Scholar]
- 23. Müller M., Information Retrieval for Music and Motion ( Springer, 2007). [Google Scholar]
- 24. Cao H., Tegenfeldt J. O., Austin R. H., and Chou S. Y., “ Gradient nanostructures for interfacing microfluidics and nanofluidics,” Appl. Phys. Lett. 81(16), 3058 (2002). 10.1063/1.1515115 [DOI] [Google Scholar]
- 25. Mannion J. T., Reccius C. H., Cross J. D., and Craighead H. G., “ Conformational analysis of single DNA molecules undergoing entropically induced motion in nanochannels,” Biophys. J. 90(12), 4538–4545 (2006). 10.1529/biophysj.105.074732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nyberg L. K., Persson F., Berg J., Bergström J., Fransson E., Olsson L., Persson M., Stålnacke A., Wigenius J., Tegenfeldt J. O., and Westerlund F., “ A single-step competitive binding assay for mapping of single DNA molecules,” Biochem. Biophys. Res. Commun. 417(1), 404–408 (2012). 10.1016/j.bbrc.2011.11.128 [DOI] [PubMed] [Google Scholar]
- 27. Neely R. K., Dedecker P., Hotta J. I., Urbanaviciute G., Klimasauskas S., and Hofkens J., “ DNA fluorocode: A single molecule, optical map of DNA with nanometre resolution,” Chem. Sci. 1(4), 453–460 (2010). 10.1039/c0sc00277a [DOI] [Google Scholar]
- 28. Michaeli Y., Shahal T., Torchinsky D., Grunwald A., Hoch R., and Ebenstein Y., “ Optical detection of epigenetic marks: Sensitive quantification and direct imaging of individual hydroxymethylcytosine bases,” Chem. Commun. 49(77), 8599–8601 (2013). 10.1039/c3cc42543f [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- See supplementary material at http://dx.doi.org/10.1063/1.4923262E-BIOMGB-9-019503 for details on the image processing.