

Abstract
We hypothesized that hyperglycemia-induced mitochondrial dysfunction and oxidative stress are closely associated with amyloid-β peptide (Aβ) toxicity in endothelial cells. Brain microvascular endothelial cells from rat (RBMEC) and mice (MBMEC) were isolated from adult Sprague-Dawley rats and homozygous db/db (Leprdb/Leprdb) and heterozygous (Dock7m/Leprdb) mice, and cultured under normo- and hyperglycemic conditions for 7 d followed by 24 h exposure to Aβ1-40. Some experiments were also performed with two mitochondrial superoxide (O2•−) scavengers, MitoTempo and Peg-SOD. Cell viability was measured by the Alamar blue assay and mitochondrial membrane potential (Δ Ψ m) by confocal microscopy. Mitochondrial O2•− and hydrogen peroxide (H2O2) production was assessed by fluorescence microscopy and H2O2 production was confirmed by microplate reader. Hyperglycemia or Aβ1-40 alone did not affect cell viability in RBMEC. However, the simultaneous presence of high glucose and Aβ1-40 reduced cell viability and Δ Ψ m, and enhanced mitochondrial O2•− and H2O2 production. MitoTempo and PEG-SOD prevented Aβ1-40 toxicity. Interestingly, MBMEC presented a similar pattern of alterations with db/db cultures presenting higher susceptibility to Aβ1-40. Overall, our results show that high glucose levels increase the susceptibility of brain microvascular endothelial cells to Aβ toxicity supporting the idea that hyperglycemia is a major risk factor for vascular injury associated with AD.
Keywords: Alzheimer’s disease, amyloid-β peptide, brain endothelial cells, mitochondria, type 2 diabetes
1. INTRODUCTION
Modern improvements in health care, increased life expectancy, and the proportion of the aged population in the developed world [1] has led to an abrupt rise in the prevalence of age-associated diseases such as Alzheimer’s disease (AD). Sporadic AD, which represents the majority of AD cases, is a late-onset disease primarily affecting people over 65 years old. This disease is characterized by a progressive cognitive decline with behavioral changes culminating in a complete loss of control of bodily functions and death [2]. The deposition of amyloid-β (Aβ) peptide as senile plaques and neurofibrillary tangles formed mainly by hyperphosphorylated tau protein are characteristic neuropathologic hallmarks of AD. Although research has focused primarily on Aβ peptide and the mechanisms underlying its production and toxic effects on neurons [3], evidence shows that cerebral vascular endothelium is an early target in AD [4].
Type 2 diabetes (T2D) is one of the major risk factors for AD with chronic hyperglycemia being one of its hallmarks. High levels of glucose activate several deleterious pathways culminating in several metabolic and cellular abnormalities [5]. Indeed, under a hyperglycemic state, high levels of reactive oxygen species (ROS) are produced activating several deleterious pathways, such as the activation of the transcription factor nuclear factor κ B (NFκB) enhancing monocytes adhesion to the vessel walls, which may initiate an atherosclerotic state [6]. Furthermore, an increase in endothelin-1 and a decrease in nitric oxide levels can contribute to increased blood-brain barrier permeability, inflammation, vasoconstriction, and, consequently, neuronal damage [7].
An extensive literature shows that hyperglycemia has deleterious effects on endothelial cells. Indeed, Rogers and coworkers [8] showed that human umbilical vein endothelial cells exposed to high glucose showed the accelerated appearance of markers of senescence jointly with reduced endothelial nitric oxide synthase expression and activity. Furthermore, several studies demonstrated that hyperglycemia enhances ROS production, which plays a major role in the etiology of diabetes complications, such as endothelial dysfunction [9, 10]. Recent findings confirmed that hyperglycemia-derived ROS mediated apoptosis of vascular endothelial cells [11, 12]. It was also reported that high glucose increased the permeability of endothelial cells of umbilical veins [13] or microvessels [14] in humans.
Since T2D is a risk factor for AD and chronic hyperglycemia is a hallmark of diabetes, we postulated that elevated blood glucose could compromise the cerebral vascular endothelium and enhance the toxicity of Aβ. Furthermore, we hypothesized that endothelial mitochondria might be adversely affected by hyperglycemia and initiate, via mechanisms involving enhanced production ROS, the destructive effects of Aβ. Therefore, we investigated whether high glucose potentiates the effects of Aβ1-40 in rat (RBMEC) and mice (MBMEC) brain microvascular endothelial cells derived from adult Sprague Dawley (SD) rats and diabetic db/db mice, respectively.
MATERIAL AND METHODS
The animal protocol was approved by the Institutional Animal Care and Use Committee of Tulane University School of Medicine. All experiments complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. For RBMEC culture, 10 week old, male SD rats (n = 6) were obtained from Harlan laboratory; for MBMEC culture, 11 week old db/db mice, a homozygous mouse model for the diabetes spontaneous mutation (Leprdb), and the heterozygous mice (Dock7m/Leprdb) were obtained from Jackson Laboratory. Rodents were housed in the animal care facility and received standard rat or mice chow and tap water ad libitum.
Brain microvascular endothelial cell isolation
Briefly, animals were decapitated under deep anesthesia, and the brain cortices were freed from meninges, homogenized, and digested. The homogenate was redistributed in 20% bovine serum albumin and was centrifuged at 1,000 g for 20 min to yield cortical microvessels. The microvessels were washed in Dulbecco’s modified Eagle medium (DMEM) further digested, layered on a continuous 33% Percoll gradient, and centrifuged again at 1,000 g for 10 min. The band of cerebral microvascular endothelial cells was aspirated, washed, and was then seeded onto collagen IV and fibronectin-coated glass-bottom culture dishes (MatTek, Ashland, MA, USA) and plates (BD Falcon, Bedford, MA). A 300 μl volume of cells was added to each well and allowed to seed for 24 h. Then, the medium was changed and Puromycin (4 μg/ml) was added for 48 h to avoid the proliferation of P-glycoprotein negative contaminating cells [15]. At day 3, the cell medium was replaced by fresh medium with different glucose concentrations and was then changed every 48 h. The cell culture medium consisted of DMEM supplemented with 20% fetal bovine plasma-derived serum, 2 mM glutamine, 1 ng/ml basic fibroblast growth factor, 50 μg/ml endothelial cell growth supplement, 100 μg/ml heparin, 5 μg/ml vitamin C, and antibiotics. We have previously demonstrated the purity of our cultures consisting of >95% BMECs, verified by positive immunohistochemistry for von Willebrand factor and by negative immunochemistry for glial fibrillary acidic protein (GFAP) and α-smooth muscle actin [16].
Cell culture and treatments
The RBMEC were exposed to 5, 25, and 30 mM of glucose for 7 days at 37°C and MBMEC were maintained in 5 or 30 mM glucose medium for 7 days at 37°C. The glucose concentrations and incubation period were selected based upon previous studies with 5 mM being considered a normoglycemic concentration and 30 mM as hyperglycemia [17, 18]. Five or 10μM of Aβ1-40 was added at day 6. To elucidate the involvement of mitochondrial ROS in Aβ-induced toxicity, RBMECs were co-incubated with Aβ1-40 and antioxidants: 100 μM mitochondrial-targeted antioxidant (MitoTempo) or 100 units/ml of an enzyme involved in O2•− removal (PEG-SOD). Osmotic controls in RBMEC were done with 25 mM mannitol and Aβ40-1 was used as peptide negative control.
Assessment of cell viability
Cell viability was determined using the Alamar Blue (a soluble, stable, and non-toxic redox indicator that is used to evaluate metabolic function and cellular health) assay. One hour before the termination of an experiment, a 10% solution of Alamar blue was added to the culture medium. After 1 h incubation at 37°C, the supernatant was collected and the absorbance was measured at 570 nm and 600 nm using a microplate reader (SpectraMax uQuant microplate Reader, BioTek, Winooski, VT) [19]. Cell viability (% of control) was calculated according to the formula (A570 − A600) of treated cells × 100/(A570 − A600) of control cells.
Measurement of mitochondrial ROS production
Live staining of RBMEC for mitochondrial superoxide (O2•−) production was performed using MitoSOX (Molecular Probes, Eugene, OR), a cell permeable probe that accumulates in mitochondria and fluoresces following oxidation by O2•−, as described by Knorr et al. [20]. MitoSOX was dissolved in DMSO and cells were incubated with a solution of MitoSOX in DMEM phenol-free to a final concentration of 5 μM, at 37°C, for 20 min in a light-protected coverslip chamber. Then, cells were rinsed and fresh DMEM phenol-free was added. Cells were immediately examined by confocal microscopy. Images were obtained with a Leica SP2 AOB laser confocal microscope (Heidelberg, Germany) for a period of no longer than 20 min.
Measurement of H2O2 levels
H2O2 levels were measured using the Amplex™ Red-horseradish peroxidase assay kit, as previously described [21]. This assay utilizes horseradish peroxidase to catalyze the H2O2-dependent oxidation of non-fluorescent Amplex™ Red to fluorescent resorufin red. Briefly, 50 μM Amplex™ Red reagent and 0.1 U/ml peroxidase in DMEM phenol-free were added to cells and incubated for 1 h at 37°C protected from light [22]. Fluorescence was read at 565 nm wavelength at 37°C in an automatic microplate reader (FLUOstar OPTIMA microplate reader, BMG Labtech, Offenburg, Germany) equipped with a thermally controlled compartment and results were expressed as % of control.
Measurement of mitochondrial membrane potential (Δ Ψ m)
Rhodamine 123 (RHD123) (Molecular Probes, Eugene, OR), a fluorescent cationic dye, was used to monitor changes in Δ Ψ m. After treatment, cells were washed with phosphate-buffered saline and incubated with 2.5 μM RHD123 in DMEM phenol-free for 45 min at 37°C. After incubation, the medium was replaced with fresh DMEM phenol-free and fluorescence was read using a FLUOstar OPTIMA microplate reader (BMG Labtech, Offenburg, Germany) and basal fluorescence was monitored at 505 nm excitation and 525 nm emission wavelengths. Immediately after the initial reading, the mitochondrial uncoupler CCCP (5μM) was added to induce complete depolarization. The difference between the initial fluorescence and the final fluorescence was used to evaluate Δ Ψ m. Results were expressed as percentage of control fluorescence. This protocol was also performed for live imaging and cells were examined by confocal microscopy. Images were obtained with a Zeiss 7 Live laser scanning confocal microscope (Jena, Germany) for a period of no longer than 10 min.
Statistical analysis
Results are presented as means ± SEM of the indicated number of experiments. Statistical significance was determined using the One Way ANOVA test for multiple comparisons, followed by the post hoc Tukey test.
RESULTS
Hyperglycemia increases the susceptibility of endothelial cells to Aβ peptide toxicity
Under chronic hyperglycemia, RBMEC did not show any significant alteration in cell viability (Fig. 1A). However, under hyperglycemic conditions, the viability of RBMEC decreased by approximately 40% when exposed to 10 μM Aβ1-40 whereas cells under normoglycemic conditions (5 mM glucose) did not show any significant alterations (Fig. 1A). Interestingly, MBMEC from homozygous but not heterozygous db/db mice presented an increased susceptibility to Aβ1-40 at both 5 and 10 μM (Fig. 1B) showing that endothelial cells isolated from mice under an in vivo hyperglycemic state are more vulnerable to Aβ1-40 toxicity. For RBMEC, MitoTempo and Peg-SOD were equally effective in protecting cell viability with a 1.75 fold increase in cell viability (Fig. 1C). Peg-Catalase and L-NAME were also able to modestly reverse cell death, but with less efficacy (data not shown).
Fig. 1.
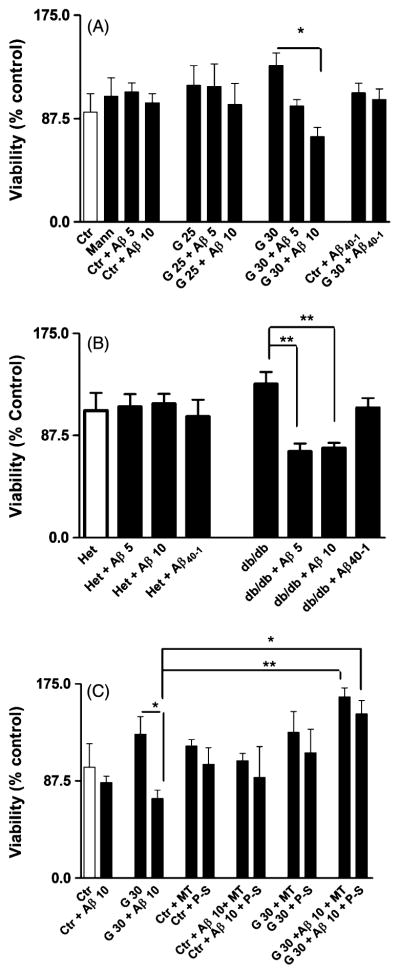
Effects of glucose and Aβ on viability of RBMEC and MBMEC. A) High levels of glucose alone did not affect cell viability, however, high glucose increased susceptibility to Aβ toxicity. B) In MBMEC from db/db mice but not from lean mice, both high and low glucose increased toxicity to Aβ toxicity. Here we only show low glucose data since there were no differences in responses within the obese and lean groups. C) When treated with MitoTempo and PEG-SOD, cell viability was restored to values close to control treated cells. Where Ctrl is Control, Mann is Mannitol, Aβ 5 is Aβ peptide 1–40 (5 μM), Aβ 10 is Aβ peptide 1–40 (10 μM), G 25 is 25 mM Glucose, G 30 is 30 mM Glucose, MT is MitoTempo, P-S is PEG-SOD, Aβ40-1 is Aβpeptide 40-1 (10 μM), Het is heterozygote culture and db/db is diabetic mice culture. Data are combined from 5 independent cultures. Statistical significance: *p < 0.05; **p < 0.01, and ***p < 0.001.
Hyperglycemia-induced ROS production is enhanced by Aβ exposure
The RBMEC under hyperglycemic conditions presented higher levels of mitochondrial O2•− production (Fig. 2G), which were exacerbated in the presence of Aβ1-40 (Fig. 2J). As expected, MitoTempo and Peg-SOD effectively prevented the increase in O2•− levels (Fig. 2H, I, K, and L).
Fig. 2.

Role of ROS in promoting toxicity to Aβ. Representative MitoSOX fluorescence images of RBMEC treated with 5 mM (A–F) or 30 mM (G–L) glucose in the presence (D–F; J–L) or absence (A–C; G–I) of Aβ peptide are shown. Furthermore, MitoTempo (B,E,H,K) and PEG-SOD (C,F,I,L) representative fluorescence images are also shown. Data are combined from 4 independent cultures.
Hyperglycemia alone did not change H2O2 levels (Fig. 3). However, Aβ1-40 promoted an increase in H2O2 levels, in both normo- and hyperglycemic conditions, an effect that was prevented by MitoTempo and Peg-SOD (Fig. 3).
Fig. 3.

Effects of hyperglycemia and Aβ on H2O2. High levels of glucose alone did not increase H2O2 levels however Aβ incubation led to a significant increase in H2O2 levels in both 5 mM and 30 mM glucose. When treated with MitoTempo and PEG-SOD, H2O2 levels reverted to values near control cells values. Data are combined from 5 independent cultures. Statistical significance: *p < 0.05.
Aβ leads to a decrease in mitochondrial membrane potential under chronic hyperglycemia
The plate reader assay showed that RBMEC exposed to high glucose did not present significant alterations in Δ Ψ m. However, those cells co-incubated with Aβ1-40 showed a significant loss of Δ Ψ m (Fig. 4). The antioxidants MitoTempo and Peg-SOD prevented the loss of Δ Ψ m; however, Peg-SOD was more effective in preserving Δ Ψ m (Fig. 4).
Fig. 4.

Effects of hyperglycemia and Aβ on Δ Ψ m. A) High levels of glucose did not affect mitochondrial membrane potential (Δ Ψ m). However, in the presence of Aβ peptide a significant drop in Δ Ψ m was observed. MitoTempo and PEG-SOD restored Δ Ψ m in 30 mM glucose. Data are combined from 5 independent cultures. Statistical significance: **p < 0.01. Also representative images of RHD123 fluorescence images of RBMEC treated with 5 mM (B1, B2) or 30 mM (B3, B4) glucose in the presence (B2, B4) or absence (B1, B3) of Aβ peptide (10 μM) are shown.
DISCUSSION
The major finding of our study is that chronic hyperglycemia enhances brain microvascular endothelial cell susceptibility to Aβ peptide exposure through mitochondrial changes that seem to be initiated by increased mitochondrial O2•− production. Additionally, the results obtained with MitoTempo and Peg-SOD seemed to corroborate this hypothesis, since these antioxidants were proved to be effective in preventing high glucose and Aβ toxicity. Thus, our study provides a mechanistic basis for the linkage between T2D and the enhanced damage to cerebral endothelial cells as a cause for AD.
T2D is one of the major risk factors for the development of neurodegenerative diseases. Over 60% of T2D patients are estimated to suffer from neurological disorders [23–25]. Furthermore, studies report that people with T2D are 1.39 times more likely to develop AD [26]. In fact, previous studies from our laboratory showed that diabetic and AD mice present a similar profile of behavioral, cognitive, vascular, and mitochondrial abnormalities [27, 28]. Interestingly, diabetic mice presented a significant increase in Aβ levels in brain cortex and hippocampus [27, 28]. Accordingly, Kolluru and coworkers [29] reported that vascular dysfunction is a major player in the establishment of diabetes-associated complications. However, studies on the cerebral microvasculature exposed to hyperglycemia are not as advanced as those on peripheral vessels [30].
Hyperglycemia per se did not affect viability or other parameters in RBMEC. However, the combination of high glucose plus Aβ1-40 peptide induced a significant decrease in cell viability in a dose-dependent manner, whereas the viability of RBMEC cells was not significantly changed when exposed to Aβ1-40 alone. The absence of Aβ1-40 toxicity seems, in some extent, contradictory with most published studies describing the toxic effects of the peptide. However, when we analyzed the results, we realized that the control maintenance medium is supplemented with high levels of glucose hiding the possible non-toxic, physiological roles of Aβ1-40 [31, 32]. To prove that chronic hyperglycemia increases the susceptibility of brain endothelial cells to Aβ1-40, we also tested the effects of this peptide in MBMEC isolated from diabetic db/db mice. The db/db mice are polyphagic, polydipsic, and polyuric and demonstrate an uncontrolled rise in blood sugar, severe depletion of the insulin-producing β-cells of the pancreatic islets, and death by 10 months of age. Interestingly, a substantial effect on db/db MBMEC viability occurred regardless of glucose level in culture when exposed to Aβ peptide. Additionally, the experiments on MBMEC demonstrate that these endothelial cells retain a distinct phenotype in culture consistent with chronic exposure to hyperglycemia. Previous studies also reported that Aβ neurotoxicity was exacerbated during hypoglycemia/hyperglycemia (≦2 mM/≧30 mM) [33]. Hyperglycemia alone did not interfere with Δ Ψ m but, in the presence of Aβ1-40, we observed a significant decrease in Δ Ψ m (Fig. 4). These results are in agreement with a study performed by Moreira et al. [34] showing that brain mitochondria isolated from T2D Goto-Kakizaki rats did not show significant alterations in Δ Ψ m, but a significant drop in this parameter occurred when mitochondria were exposed to Aβ1-40 peptide.
Furthermore, we showed that the increased susceptibility of RBMEC under high glucose exposure to Aβ is mediated by mitochondrial O2•−. Indeed, it was previously suggested that mitochondrial overproduction of ROS due to hyperglycemia may increase the susceptibility of endothelial cells to injury [35]. In fact, we observed a significant increase in mitochondrial O2•− when cells were exposed to 30 mM glucose and this increase was exacerbated by Aβ1-40. A significant increase in H2O2 levels was also observed in both normoglycemic and hyperglycemic cells exposed to Aβ peptide but it is unclear whether this ROS was produced directly or via dismutation of O2•−. These results are consistent with previous studies showing that, under physiologic conditions, cells maintain the redox balance through the generation and elimination of ROS, however, when redox homeostasis is disturbed, oxidative stress may lead to aberrant cell death and contribute to disease development including neurodegenerative diseases [36]. To confirm that mitochondrial ROS play a key role in the alterations observed in cells exposed to 30 mM glucose and Aβ we tested several antioxidants: MitoTempo, Peg-SOD, Peg-catalase, L-NAME, and apocynin (an inhibitor of NADPH oxidases). The MitoTempo and Peg-SOD completely prevented the loss of cell viability induced by high glucose plus Aβ1-40, contrasting with the other antioxidants which only exerted a partially protective effect (data not shown). As expected, MitoTempo and Peg-SOD normalized O2•− levels in cells exposed to high glucose or high glucose plus Aβ1-40. These observations suggest that mitochondrial O2•− has a key role in mediating high glucose and Aβ toxicity. Previous studies showed that MitoTempo is a SOD mimetic with a mechanism of action similar to SOD in O2•− detoxification, which is, however, specifically targeted to mitochondria [37]. Furthermore, both MitoTempo and Peg-SOD were able to prevent the drop in Δ Ψ m and the increase in H2O2 levels in cells exposed to high glucose plus Aβ1-40. Interestingly, Peg-SOD was more effective in restoring Δ Ψ m than MitoTempo. This may be due to the fact that Peg-SOD is also able to detoxify the O2•− produced at other locations besides mitochondria. Although mitochondria are major sources of ROS, we cannot exclude the potential contribution of other cytosolic sources [38], which may potentiate mitochondrial dysfunction and ROS production [39]. Concerning H2O2 levels, no alterations were observed in the presence of Peg-SOD in control conditions (Fig. 3). However, Peg-SOD reversed Aβ-induced increase in H2O2 levels under hyperglycemic conditions. The reduction in H2O2 levels seems contradictory since Peg-SOD converts O2•− to H2O2. Liochev and Fridovich [40] suggested that an increase in O2•− dismutation would prevent the formation of H2O2 by other reactions. This is supported by studies performed in cell lines overexpressing CuZnSOD, which show reduced levels of H2O2 [41]. It was also reported that Peg-SOD treatment increased catalase activity in cancer cells [42], an additional mechanism that could justify the decrease in H2O2 levels. Additionally, MitoTempo was able to decrease H2O2 levels (Fig. 4). Dikalova and coworkers [43] showed that in addition to O2•− scavenging, MitoTempo has the capacity to reduce mitochondrial ROS production by normalizing mitochondrial respiration. In agreement with our results, Liang et al. [44] also showed that MitoTempo was able to recover Δ Ψ m through the inhibition of the mitochondrial permeability transition pore.
In summary, our results show that brain endothelial cells under chronic hyperglycemia are more susceptible to Aβ toxicity, an effect that seems to be mediated by mitochondrial ROS. Furthermore, we have shown that our in vitro model of chronic hyperglycemia mimics diabetic conditions. This study supports the idea that diabetes is a risk factor for AD.
Acknowledgments
We thank Dan Liu for technical assistance. We thank Nancy Busija, M.A., CCC-SLP, for editing the manuscript. This work was supported by the grants HL-030260, HL-065380, HL077731, and HL093554.
Footnotes
Authors’ disclosures available online (http://www.j-alz.com/disclosures/view.php?id=1839).
References
- 1.Taqui AM, Itrat A, Qidwai W, Qadri Z. Depression in the elderly: Does family system play a role? A cross-sectional study. BMC Psychiatr. 2007;7:57. doi: 10.1186/1471-244X-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaturvedi RK, Beal MF. Mitochondrial approaches for neuroprotection. Ann N Y Acad Sci. 2008;1147:395–412. doi: 10.1196/annals.1427.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arab L, Sadeghi R, Walker DG, Lue L, Sabbagh MN. Consequences of aberrant insulin regulation in the brain: Can treating diabetes be effective for Alzheimer’s disease. Curr Neuropharmacol. 2011;9:693–705. doi: 10.2174/157015911798376334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merlini M, Meyer EP, Ulmann-Schuler A, Nitsch RM. Vascular β-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAβ mice. Acta Neuropathol. 2011;122:293–311. doi: 10.1007/s00401-011-0834-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Correia S, Carvalho C, Santos MS, Seiça R, Oliveira CR, Moreira PI. Mechanisms of action of metformin in type 2 diabetes and associated complications: An overview. Mini Reviews Med Chem. 2008;8:1343–1354. doi: 10.2174/138955708786369546. [DOI] [PubMed] [Google Scholar]
- 6.Yorek MA. The role of oxidative stress in diabetic vascular and neural disease. Free Radic Res. 2003;37:471–480. doi: 10.1080/1071576031000083161. [DOI] [PubMed] [Google Scholar]
- 7.Carvalho C, Correia SC, Santos RX, Cardoso S, Moreira PI, Clark TA, Zhu X, Smith MA, Perry G. Role of mitochondrial-mediated signaling pathways in Alzheimer disease and hypoxia. J Bioenerg Biomembr. 2009;41:433–440. doi: 10.1007/s10863-009-9247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogers SC, Zhang X, Azhar G, Luo S, Wei JY. Exposure to high or low glucose levels accelerates the appearance of markers of endothelial cell senescence and induces dysregulation of nitric oxide synthase. J Gerontol A Biol Sci Med Sci. 2013 doi: 10.1093/gerona/glt033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–1662. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- 10.Sun J, Pu Y, Wang P, Chen S, Zhao Y, Liu C, Shang Q, Zhu Z, Liu D. TRPV1-mediated UCP2 upregulation ameliorates hyperglycemia-induced endothelial dysfunction. Cardiovasc Diabetol. 2013;12:69. doi: 10.1186/1475-2840-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hulsmans M, Van Dooren E, Holvoet P. Mitochondrial reactive oxygen species and risk of atherosclerosis. Curr Atheroscler Reports. 2012;14:264–276. doi: 10.1007/s11883-012-0237-0. [DOI] [PubMed] [Google Scholar]
- 12.Kapitulnik J, Benaim C, Sasson S. Endothelial cells derived from the blood-brain barrier and islets of langerhans differ in their response to the effects of bilirubin on oxidative stress under hyperglycemic conditions. Front Pharmacol. 2012;3:131. doi: 10.3389/fphar.2012.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dang L, Seale JP, Qu X. High glucose-induced human umbilical vein endothelial cell hyperpermeability is dependent on protein kinase C activation and independent of the Ca2+-nitric oxide signalling pathway. Clin Exp Pharmacol Physiol. 2005;32:771–776. doi: 10.1111/j.1440-1681.2005.04266.x. [DOI] [PubMed] [Google Scholar]
- 14.Scalia R, Gong Y, Berzins B, Zhao LJ, Sharma K. Hyperglycemia is a major determinant of albumin permeability in diabetic microcirculation: The role of mu-calpain. Diabetes. 2007;56:1842–1849. doi: 10.2337/db06-1198. [DOI] [PubMed] [Google Scholar]
- 15.Perriére N, Demeuse P, Garcia E, Regina A, Debray M, Andreux J-P, Couvreur P, Scherrmann J-M, Temsamani J, Couraud P-O, Deli MA, Roux F. Puromycin-based purification of rat brain capillary endothelial cell cultures. Effect on the expression of blood-brain barrier-specific properties. J Neurochem. 2005;93:279–289. doi: 10.1111/j.1471-4159.2004.03020.x. [DOI] [PubMed] [Google Scholar]
- 16.Domoki F, Kis B, Gáspár T, Bari F, Busija DW. Cerebromicrovascular endothelial cells are resistant to L-glutamate. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1099–R1108. doi: 10.1152/ajpregu.90430.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castilho ÁF, Aveleira CA, Leal EC, Simões NF, Fernandes CR, Meirinhos RI, Baptista FI, Ambrósio AF. Heme oxygenase-1 protects retinal endothelial cells against high glucose- and oxidative/nitrosative stress-induced toxicity. PloS one. 2012;7:e42428. doi: 10.1371/journal.pone.0042428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Somjen D, Paller CJ, Gayer B, Kohen F, Knoll E, Stern N. High glucose blocks the effects of estradiol on human vascular cell growth: Differential interaction with estradiol and raloxifene. J Steroid Biochem Mol Biol. 2004;88:101–110. doi: 10.1016/j.jsbmb.2003.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neves SS, Sarmento-Ribeiro AB, Simões SP, Pedroso de Lima MC. Transfection of oral cancer cells mediated by transferrin-associated lipoplexes: Mechanisms of cell death induced by herpes simplex virus thymidine kinase/ganciclovir therapy. Biochim Biophys Acta. 2006;1758:1703–1712. doi: 10.1016/j.bbamem.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 20.Knorr M, Hausding M, Kröller-Schuhmacher S, Steven S, Oelze M, Heeren T, Scholz A, Gori T, Wenzel P, Schulz E, Daiber A, Münzel T. Nitroglycerin-induced endothelial dysfunction and tolerance involve adverse phosphorylation and S-Glutathionylation of endothelial nitric oxide synthase: Beneficial effects of therapy with the AT1 receptor blocker telmisartan. Arterioscler Thromb Vasc Biol. 2011;31:2223–2231. doi: 10.1161/ATVBAHA.111.232058. [DOI] [PubMed] [Google Scholar]
- 21.Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 22.Santiago APSA, Chaves EA, Oliveira MF, Galina A. Reactive oxygen species generation is modulated by mitochondrial kinases: Correlation with mitochondrial antioxidant peroxidases in rat tissues. Biochimie. 2008;90:1566–1577. doi: 10.1016/j.biochi.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 23.Ristow M. Neurodegenerative disorders associated with diabetes mellitus. J Mol Med. 2004;82:510–529. doi: 10.1007/s00109-004-0552-1. [DOI] [PubMed] [Google Scholar]
- 24.Zhu H, Li F, Yu W-J, Wang W-J, Li L, Wan L-D, Le Y, Ding W-L. Effect of hypoxia/reoxygenation on cell viability and expression and secretion of neurotrophic factors (NTFs) in primary cultured schwann cells. Anat Rec (Hoboken) 2010;293:865–870. doi: 10.1002/ar.21105. [DOI] [PubMed] [Google Scholar]
- 25.Yan J, Zhang Z, Shi H. HIF-1 is involved in high glucose-induced paracellular permeability of brain endothelial cells. Cell Mol Life Sci. 2012;69:115–128. doi: 10.1007/s00018-011-0731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu F-P, Lin K-P, Kuo H-K. Diabetes and the risk of multi-system aging phenotypes: A systematic review and meta-analysis. PloS one. 2009;4:e4144. doi: 10.1371/journal.pone.0004144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carvalho C, Cardoso S, Correia SC, Santos RX, Santos MS, Baldeiras I, Oliveira CR, Moreira PI. Metabolic alterations induced by sucrose intake and Alzheimer’s disease promote similar brain mitochondrial abnormalities. Diabetes. 2012;61:1234–1242. doi: 10.2337/db11-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carvalho C, Machado N, Mota PC, Correia SC, Cardoso S, Santos RX, Santos MS, Oliveira CR, Moreira PI. Type 2 diabetic and Alzheimer’s disease mice present similar behavioral, cognitive, and vascular anomalies. J Alzheimers Dis. 2013;35:623–635. doi: 10.3233/JAD-130005. [DOI] [PubMed] [Google Scholar]
- 29.Kolluru GK, Bir SC, Kevil CG. Endothelial dysfunction and diabetes: Effects on angiogenesis, vascular remodeling, and wound healing. Int J Vasc Med. 2012;2012:918267. doi: 10.1155/2012/918267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerbone AM, Macarone-Palmieri N, Saldalamacchia G, Coppola A, Di Minno G, Rivellese AA. Diabetes, vascular complications and antiplatelet therapy: Open problems. Acta Diabetol. 2009;46:253–261. doi: 10.1007/s00592-008-0079-y. [DOI] [PubMed] [Google Scholar]
- 31.Broersen K, Jonckheere W, Rozenski J, Vandersteen A, Pauwels K, Pastore A, Rousseau F, Schymkowitz J. A standardized and biocompatible preparation of aggregate-free amyloid beta peptide for biophysical and biological studies of Alzheimer’s disease. Protein Eng Des Sel. 2011;24:743–750. doi: 10.1093/protein/gzr020. [DOI] [PubMed] [Google Scholar]
- 32.Manzoni C, Colombo L, Bigini P, Diana V, Cagnotto A, Messa M, Lupi M, Bonetto V, Pignataro M, Airoldi C, Sironi E, Williams A, Salmona M. The molecular assembly of amyloid aβ controls its neurotoxicity and binding to cellular proteins. PloS one. 2011;6:e24909. doi: 10.1371/journal.pone.0024909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Song X, Takata T, Miichi Y, Yokono K, Sakurai T. Amyloid-β neurotoxicity restricts glucose window for neuronal survival in rat hippocampal slice cultures. Exp Gerontol. 2010;45:904–908. doi: 10.1016/j.exger.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 34.Moreira PI, Santos MS, Moreno AM, Seiça R, Oliveira CR. Increased vulnerability of brain mitochondria in diabetic (Goto-Kakizaki) rats with aging and amyloid-beta exposure. Diabetes. 2003;52:1449–1456. doi: 10.2337/diabetes.52.6.1449. [DOI] [PubMed] [Google Scholar]
- 35.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trachootham D, Lu W, Ogasawara MA, Nilsa R-DV, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10:1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoehn KL, Salmon AB, Hohnen-Behrens C, Turner N, Hoy AJ, Maghzal GJ, Stocker R, Van Remmen H, Kraegen EW, Cooney GJ, Richardson AR, James DE. Insulin resistance is a cellular antioxidant defense mechanism. Proc Natl Acad Sci U S A. 2009;106:17787–17792. doi: 10.1073/pnas.0902380106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee S-J, Ryter SW, Xu J-F, Nakahira K, Kim HP, Choi AMK, Kim YS. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am J Respir Cell Mol Biol. 2011;45:867–873. doi: 10.1165/rcmb.2010-0352OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park J, Lee J, Choi C. Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PloS one. 2011;6:e23211. doi: 10.1371/journal.pone.0023211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liochev SI, Fridovich I. Effects of overproduction of superoxide dismutase on the toxicity of paraquat toward Escherichia coli. J Biol Chem. 1991;266:8747–8750. [PubMed] [Google Scholar]
- 41.Teixeira HD, Schumacher RI, Meneghini R. Lower intracellular hydrogen peroxide levels in cells overexpressing CuZn-superoxide dismutase. Proc Natl Acad Sci U S A. 1998;95:7872–7875. doi: 10.1073/pnas.95.14.7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sen S, Kawahara B, Chaudhuri G. Maintenance of higher H2O2 levels, and its mechanism of action to induce growth in breast cancer cells: Important roles of bioactive catalase and PP2A. Free Radic Biol Med. 2012;53:1541–1551. doi: 10.1016/j.freeradbiomed.2012.06.030. [DOI] [PubMed] [Google Scholar]
- 43.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang HL, Sedlic F, Bosnjak Z, Nilakantan V. SOD1 and MitoTEMPO partially prevent mitochondrial permeability transition pore opening, necrosis, and mitochondrial apoptosis after ATP depletion recovery. Free Radic Biol Med. 2010;49:1550–1560. doi: 10.1016/j.freeradbiomed.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]