

Abstract
Wnt pathway deregulation is a common characteristic of many cancers. Only colorectal cancer predominantly harbours mutations in APC, whereas other cancer types (hepatocellular carcinoma, solid pseudopapillary tumours of the pancreas) have activating mutations in β-catenin (CTNNB1). We have compared the dynamics and the potency of β-catenin mutations in vivo. Within the murine small intestine (SI), an activating mutation of β-catenin took much longer to achieve Wnt deregulation and acquire a crypt-progenitor cell (CPC) phenotype than Apc or Gsk3 loss. Within the colon, a single activating mutation of β-catenin was unable to drive Wnt deregulation or induce the CPC phenotype. This ability of β-catenin mutation to differentially transform the SI versus the colon correlated with higher expression of E-cadherin and a higher number of E-cadherin:β-catenin complexes at the membrane. Reduction in E-cadherin synergised with an activating mutation of β-catenin resulting in a rapid CPC phenotype within the SI and colon. Thus, there is a threshold of β-catenin that is required to drive transformation, and E-cadherin can act as a buffer to sequester mutated β-catenin.
Keywords: APC, β-catenin, colorectal cancer, E-cadherin
See also: GT Chen & ML Waterman (September 2015)
Introduction
Mutation of the APC (adenomatous polyposis coli) gene is the most common event in colorectal cancer (CRC). A recent study where a large number of CRCs (200) were sequenced showed mutation in over 70% of CRC (TCGA, 2012). It is proposed that the reason for APC mutation in CRC is due to its role as a negative regulator of the Wnt signalling pathway. APC is part of a large multiprotein destruction complex that targets β-catenin (CTNNB1, CATNB1) for degradation. In the absence of a Wnt ligand, APC in complex with AXIN, casein kinase 1 (CK1) and GSK3beta is required for the phosphorylation of β-catenin by GSK3, which targets β-catenin for degradation. Following a Wnt signal or APC mutation, this complex does not form and β-catenin is no longer targeted for degradation and accumulates in the nucleus. Within the nucleus, β-catenin interacts with T-cell factor-1/lymphoid-enhancing factor-1 (TCF/LEF1) transcription factors to drive transcription of TCF/LEF1 target genes (Kinzler & Vogelstein, 1996; Clevers, 2006).
CRC is unusual compared to other cancers in that loss of function mutations in APC are much more frequent than activating mutations in β-catenin (i.e. in exon 3), ∼75% compared to ∼5%, respectively (TCGA, 2012). In other cancers, such as hepatocellular cancer, activating mutations in the β-catenin gene are much more frequent, where the phosphorylation sites that target β-catenin for degradation are mutated (Cui et al, 2003; Edamoto et al, 2003; Ahn et al, 2014). As APC is a large protein, it has been hypothesised that functions other than just Wnt deregulation may be crucial for tumour initiation. These include microtubule binding, mitosis and actin cytoskeleton regulation (Nathke, 2004).
β-catenin is also an essential part of the adherens junction at the membrane where it binds to E-cadherin. Loss of β-catenin from the intestine therefore leads not only to a loss of Wnt signalling (and stem cells) but also to reduced adhesion alongside cell loss from the villus (Ireland et al, 2004). The inter-relationship between β-catenin:E-cadherin and the β-catenin:TCF4 complexes has been a subject of much study and debate. Loss of E-Cadherin in cancer cells is often associated with markers of “EMT” and an upregulation of β-catenin TCF4 signalling, which is thought to happen at late stages of tumour progression.
However, despite this, there is a lack of definitive in vivo evidence that E-cadherin levels are able to limit the transforming capacity of β-catenin accumulation in tumour initiation. Presumably, this is due to the fact that increases in free β-catenin would be degraded by the destruction complex.
Given the centrality of the APC tumour suppressor to CRC, we decided to use a genetic approach in vivo and in vitro to address the differences in Wnt activation by either mutation of β-catenin or mutation of the destruction complex (APC or GSK3). We have previously shown that genetic deletion of both copies of the Apc tumour suppressor (Apcfl/fl) rapidly generates a crypt-progenitor cell-like phenotype (CPC) within the intestinal epithelium, with cells failing to differentiate, retaining proliferative capacity and failing to migrate up the crypt-villus axis (Sansom et al, 2004). This is associated with a relocalisation of β-catenin to the nucleus and the expression of functionally important Wnt target genes such as cMyc (Sansom et al, 2007).
We therefore investigated a number of alternative approaches to deregulation of Wnt signalling within the intestinal epithelium and assessed functional and Wnt signalling outputs. Taken together, our data show that mutation of the destruction complex, either by loss of APC or by loss of GSK3, leads to a rapid Wnt deregulation with accumulation of β-catenin in the small intestine and the colon. Surprisingly, a single activating mutation of β-catenin takes much more time to develop a phenotype in the small intestine and is unable to transform the colon. We show that this is due to high E-cadherin levels in the colon, which act as a sink for the mutated β-catenin. Reduction in E-cadherin or mutation of both copies of β-catenin swamps E-cadherin and then leads to rapid transformation of both the small intestine and colon.
Results
Apc loss, Gsk3 loss and homozygous mutation of β-catenin are each sufficient to induce a crypt-progenitor phenotype
Our previous data showed that Apc loss was sufficient to induce β-catenin activation in the mouse intestine. The resulting Wnt signalling deregulation is characterised by a crypt-progenitor cell (CPC) phenotype with high nuclear β-catenin, increased proliferation and upregulation of Wnt target genes in both the small intestine and colon (Sansom et al, 2004; Hinoi et al, 2007; Fig 1A).
Figure 1. Apc loss, GSK3 loss and homozygous mutation of β-catenin are sufficient to induce a rapid crypt-progenitor phenotype, but not a single β-catenin mutation.

- Wild-type mice have small defined crypts in the small intestine with little nuclear β-catenin at the bottom of the crypt. The small intestine of AhCreER APCfl/fl, AhCreER Catnblox(ex3)/lox(ex3) (day 5) or AhCreER Gsk3alphafl/fl betafl/fl (day 6) show the crypt-progenitor cell (CPC) phenotype with increased crypt size (red bar) and nuclear β-catenin (arrows) along the crypt-villus axis. Scale bar, 100 μm.
- A heterozygous activation of β-catenin (AhCreER Catnblox(ex3)/+) shows no increase in crypt size or nuclear β-catenin at days 5–10. At day 15 post-induction, accumulation of nuclear β-catenin (arrows) becomes evident with a dramatic CPC phenotype at about day 25. Scale bar, 100 μm.
- Culture of small intestinal crypts of WT and VilCreER Apcfl/fl (or AhCreER Apcfl/fl) mice with/without R-spo1 shows the dependence of the Wnt agonist in WT organoids but not in Apc-deficient spheres. Representative photos were taken at day 5 in culture. Black scale bar, 50 μm.
- At day 5 post-induction, only crypts from AhCreER Catnblox(ex3)/lox(ex3) but not AhCreER Catnblox(ex3)/+ survive in culture without the addition of R-spo1. At day 10 post-induction, we observed a mixed phenotype of more organoid-like structures in AhCreER Catnblox(ex3)/+ compared to spheres from Catnblox(ex3)/lox(ex3) crypts in the first week of culture. Black scale bar, 50 μm.
- Quantification of organoids/spheres at day 10 post-induction (N = 2 or 3 mice per genotype, mean of 6 technical replicates per mouse).
To confirm our finding that inactivation of the destruction complex was sufficient to initiate transformation of the small intestine and colon, we deleted GSK3 and assessed if it would drive a similar phenotype to that of Apc loss. There are two isoforms of GSK3, GSK3alpha (GSK3A) and GSK3beta (GSK3B), and data from ES cells suggested that in the absence of one isoform, the other can compensate (Doble et al, 2007).
We generated AhCreER Gsk3alphafl/fl Gsk3betafl/fl mice, which upon induction with β-naphthoflavone and tamoxifen recombine in the crypts of the small intestine and to a lesser extent in the colon (Kemp et al, 2004). Complete genetic ablation of Gsk3 produced a phenotype that recapitulated Apc deficiency within the intestinal epithelium (Figs A and EV1A). Six days after Gsk3 deletion, the intestines adopted the CPC phenotype with nuclear localisation of β-catenin and an induction of Wnt target genes. This similarity to the Apc loss phenotype suggested that the main function of GSK3 within the intestine is to control Wnt signalling. Apc heterozygous mice develop intestinal tumours on the loss of the remaining copy of Apc. Given there are 2 different GSK3 isoforms, 4 mutations would be required to produce a situation where Wnt would be deregulated. We asked, if only 1 allele of GSK3 needed to be lost, whether these mice would undergo intestinal tumourigenesis. To do this, we generated mice lacking 3 alleles of GSK3 (AhCre GSK3alphafl/fl GSK3betafl/+ and AhCre Gsk3alphafl/+ Gsk3betafl/fl mice) and found following Cre deletion a significant fraction of these mice spontaneously developed intestinal tumours, in contrast to the single isoform mice (AhCre Gsk3betafl/fl; Fig EV1B and C).
Figure EV1. Only deletion of both Gsk3alpha and Gsk3beta leads to crypt-progenitor-cell phenotype.
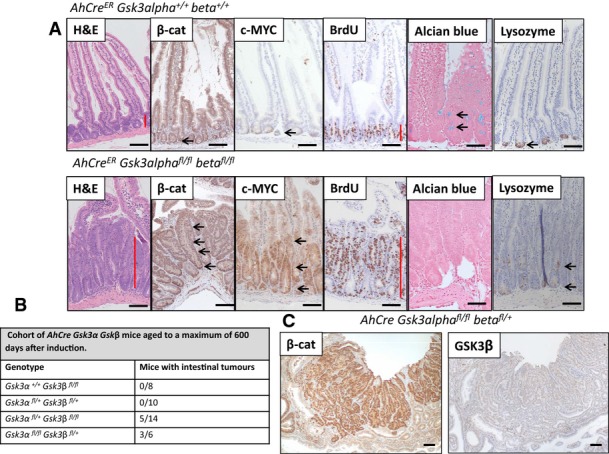
- Small Intestine of mice at day 6 after induction. Loss of Gsk3alpha and Gsk3beta (AhCreER Gsk3alphafl/fl Gsk3betafl/fl) leads to accumulation of nuclear β-catenin and upregulation of cMyc (arrows). The crypt-progenitor cell phenotype (red bar) is characterised by increased proliferation (BrdU) and perturbed differentiation/localisation of goblet and Paneth cells (Alcian Blue, Lysozyme respectively, arrows). Scale bar, 100 μm.
- Table shows cohort of AhCre Gsk3alpha Gsk3beta mice aged until signs of intestinal tumour burden, genotypes as indicated. Note, mice homozygous for Gsk3beta deletion, or with only one copy of GSK3alpha and GSK3beta, did not develop intestinal tumours.
- An adenoma from an aged mouse deficient for 3 alleles of Gsk3alpha and Gsk3beta (AhCre GSK3alphafl/fl betafl/+) showing that it developed after loss of the remaining GKS3beta allele. Scale bar, 100 μm.
Similarly to loss of the destruction complex, activating mutation of both copies of the β-catenin allele (AhCreER Catnblox(ex3)/lox(ex3))showed the CPC phenotype with nuclear localisation of β-catenin and increased proliferation along the crypt-villus axis (Figs A and EV2A). Exon 3, which encodes the GSK3 phosphorylation sites that target β-catenin for degradation, is flanked by loxP sites, so the expression of Cre recombinase within the intestinal epithelium results in deletion of exon 3 and hence, β-catenin can no longer be targeted for degradation (Harada et al, 1999).
Figure EV2. Single copy activation of β-catenin only slowly transforms the intestine.
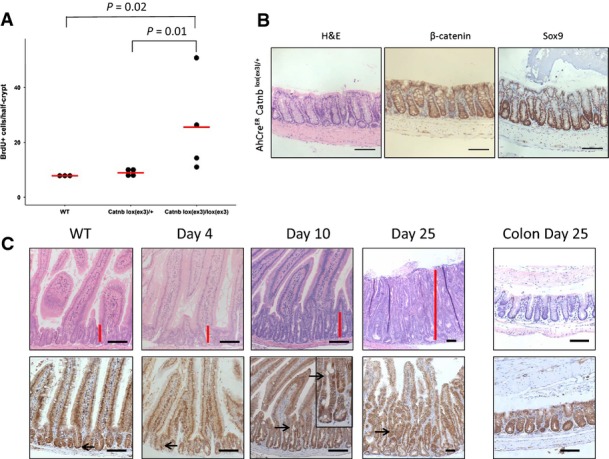
- Only activation of both alleles of β-catenin led to hyperproliferation in the small intestine. Proliferation of wild-type (WT), AhCreER Catnblox(ex3)/+ and AhCreER Catnblox(ex3)/lox(ex3) mice 5 days after induction was scored by counting the number of BrdU-positive cells/half-crypt. N ≥ 3 per group, at least 25 crypts per mouse were scored, P-value of one-sided Mann–Whitney U-test.
- Activation of one of copy β-catenin in an aged AhCreER Catnblox(ex3)/+ at day 25 post-induction with no phenotype in the colon. For comparison to a WT colon, see Appendix Fig S3. Scale bar, 100 μm.
- Activation of one copy of β-catenin in VilCreER Catnblox(ex3)/+ leads to the same crypt-progenitor cell phenotype (red bar) with similar kinetics as observed in AhCreER Catnblox(ex3)/+ mice. Scale bar, 100 μm.
We investigated the impact of a single β-catenin mutation, which is analogous to the situation found in human patients. In contrast to Apc deletion (AhCreER Apcfl/fl), activation of a single copy of β-catenin did not yield a robust CPC phenotype at day 5 or day 10. Immunohistochemical staining of β-catenin showed very little nuclear localisation and no increase in proliferation (Figs B and EV2A).
At day 15, nuclear β-catenin became evident in AhCreER Catnblox(ex3)/+ mice and a number of crypts became enlarged. At time points past day 20, a single copy of Catnblox(ex3)/+ was able to induce a robust CPC phenotype in the small intestine (Fig 1B).
To probe the kinetics of β-catenin activation, we used intestinal “organoid” crypt culture (Sato et al, 2009). Previous studies have shown that once Apc is lost, intestinal cultures no longer require the addition of R-spondin or Wnt ligand produced by the Paneth cells. They also no longer bud into “crypt-like” structures, but form sphere structures instead (Fig 1C). The cultures therefore provide an excellent system to compare β-catenin exon 3 deletion to the loss of Apc. We therefore examined the capacity of AhCreER Catnblox(ex3)/+ and AhCreER Catnblox(ex3)/lox(ex3) crypts (from the small intestine) which were induced in vivo to form organoids/spheres in culture and their R-spondin1 dependence. In contrast to organoids where Apc was deleted, AhCreER Catnblox(ex3)/+ crypts did not show R-spondin1 independence and died in culture when plated 5 days following Cre recombination in vivo (Fig 1D). In contrast, AhCreER Catnblox(ex3)/lox(ex3) formed spheres in an equivalent manner to homozygous Apc deletion, independent of R-spondin1. Crypts sampled at later time points (day 10) from AhCreER Catnblox(ex3)/+ mice were able to survive in culture without R-spondin. At this time point, we observed a more organoid-like structure phenotype (∼85%) than the expected typical round spheres (∼15%) (Fig 1E). This early intermediate phenotype of crypts suggests that absolute levels of Wnt signalling in crypts with one copy of β-catenin were not as high as either mutation of both alleles of β-catenin or Apc loss. In contrast, crypts from AhCreER Catnblox(ex3)/lox(ex3) almost completely formed spheres, and only few organoid-like structures were observed.
Single copy activation of β-catenin does not transform the colon either immediately or at longer time points
We next examined the colons from the AhCreER Catnblox(ex3)/+ mice. In contrast to AhCreER Apcfl/fl mice, which rapidly develop a CPC phenotype with nuclear β-catenin accumulation and Wnt target gene activation, this did not occur in the AhCreER Catnblox(ex3)/+ mice. Colons from AhCreER Catnblox(ex3)/+ mice even 25 days post-induction did not show nuclear β-catenin or Wnt target gene activation despite a clear CPC phenotype in the small intestine (Fig EV2B).
To examine this phenomenon more carefully, we used the VilCreER transgene to drive recombination as this delivers much higher recombination in the colorectal epithelium than the AhCreER transgene. Four days post-recombination, a CPC phenotype was observed in the colons (and the small intestine) of VilCreER Catnblox(ex3)/lox(ex3) and VilCreER Apcfl/fl mice with increased levels of nuclear β-catenin and SOX9. However, no phenotype or accumulation of β-catenin or Sox9 was observed in the colons (or small intestines) of VilCreER Catnblox(ex3)/+ mice at this time point (Fig EV3). The dramatic CPC phenotype in the SI and colon meant both the VilCreER Catnblox(ex3)/lox(ex3) and VilCreER Apcfl/fl needed to be harvested due to signs of ill health. In contrast, VilCreER Catnblox(ex3)/+ developed signs of ill health after about 25 days which was associated with a small intestinal CPC phenotype. In the colon, there was not an obvious CPC phenotype at this stage (Figs EV2C and EV3).
Figure EV3. Rapid colonic phenotype in VilCreER Catnblox(ex3)/lox(ex3) and Apcfl/fl mice, but not in VilCreER Catnblox(ex3)/+ mice.
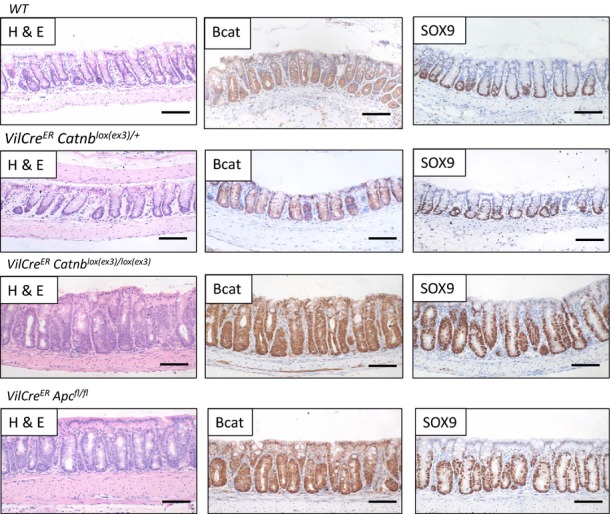
Wild-type mice show very little nuclear β-catenin, and the expression of Sox9 is restricted to the bottom of the crypt. VilCreER Catnblox(ex3)/+ show a similar phenotype. In contrast, VilCreER Catnblox(ex3)/lox(ex3) and VilCreER Apcfl/fl mice show increased nuclear β-catenin and high expression of Sox9. Scale bar, 100 μm. Mice were sampled at day 4 after induction.
Given this failure to drive a CPC phenotype, we next wanted to ask whether single copy activation of β-catenin within intestinal stem cells could lead to intestinal tumourigenesis. To do this, we interbred Catnblox(ex3)/+ to Lgr5CreER mice to allow inducible Cre-mediated recombination within the LGR5-positive intestinal stem cells. The Lgr5CreER delivers highly penetrant recombination in the small intestinal Lgr5-positive stem cell population and lower penetrant recombination in colon Lgr5-positive stem cell population. We have previously shown that targeting Apc loss to the Lgr5 compartment led to rapid small intestinal adenoma formation (Barker et al, 2009; Myant et al, 2013). Lgr5CreER Catnblox(ex3)/+ and Lgr5CreER Catnblox(ex3)/lox(ex3) mice were induced with a single intraperitoneal injection (IP) injection of tamoxifen and rapidly developed small intesti-nal tumours (Appendix Fig S1) with similar kinetics to those following Apc deletion in the Lgr5 compartment. The equal potency of Apc loss and mutation of a single β-catenin allele at transforming Lgr5-positive cells was somewhat surprising given the high frequency of APC mutations in human CRC. Therefore, we next analysed colonic lesions within Lgr5CreER Catnblox(ex3)/+, Lgr5CreER Catnblox(ex3)/lox(ex3) and Lgr5CreER Apcfl/fl mice when they were harvested due to small intestinal disease burden. We found that the majority of Lgr5CreER Catnblox(ex3)/lox(ex3) mice (88%) and Lgr5CreER Apcfl/fl mice (91%) had lesions in the colon, whereas none of the Lgr5CreER Catnblox(ex3)/+ mice (0%) had any lesions (Fig A). Colonic lesions were defined by an increase in nuclear β-catenin levels and corresponding levels of the Wnt target gene Sox9 protein (Fig 2B). Given that we observe a similar accumulation of β-catenin in the small intestine after GSK3 deletion; we predicted that Lgr5CreER Gsk3alphafl/fl Gsk3betafl/fl should develop colonic lesions. We found that mice similarly succumbed to intestinal lesions in both the small intestine and colon (Fig EV4).
Figure 2. Activation of one allele of β-catenin in Lgr5-positive stem cells does not result in colonic lesions.
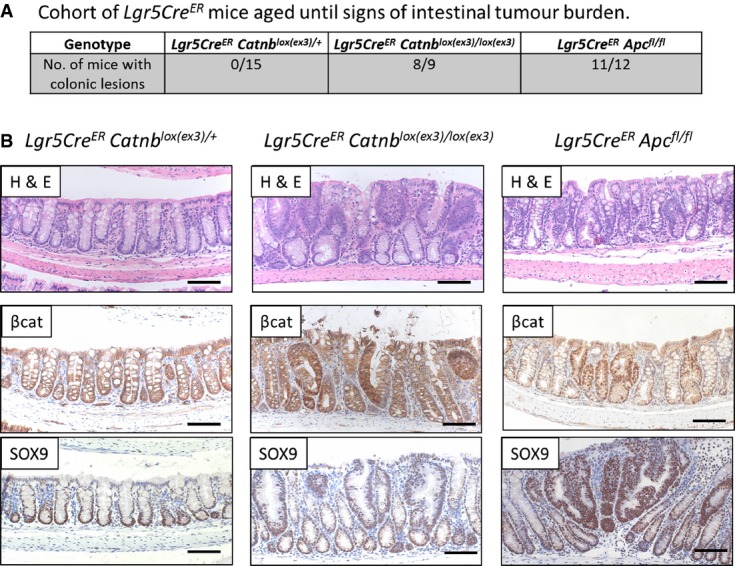
- Table shows that only homozygous activation of β-catenin (Lgr5CreER Catnblox(ex3)/lox(ex3)) or loss of Apc (Lgr5CreER Apcfl/fl) in Lgr5-positive stem cells results in colonic lesions in cohorts, sampled when signs of sickness were apparent. There were no lesions observed when only one allele of β-catenin (Lgr5CreER Catnblox(ex3)/+) was mutated (chi-squared test, P < 0.001).
- Colonic lesions were scored from H&E by identification of disorganised epithelium. Staining for β-catenin and SOX9 confirmed activation of Wnt signalling in the observed lesions. Scale bar, 100 μm.
Figure EV4. Complete loss of Gsk3 in Lgr5-positive stem cells leads to tumour formation in the small intestine and the colon.
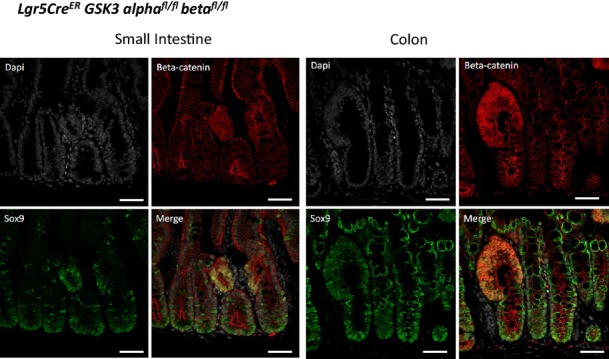
Immunofluorescence analysis shows intestinal lesions with accumulation of β-catenin and SOX9 in the small intestine and the colon, 25 days after induction. Scale bar, 50 μm.
Taken together, this showed that there are fundamental differences in the ability of a single copy of β-catenin to transform small intestinal and colorectal epithelial cells in the mouse.
E-cadherin levels are higher in the colon compared to the small intestine of wild-type mice
Although many studies have proposed that the pool of E-cadherin may limit the amount of free β-catenin, there is very little experimental evidence that this is the case. For example, E-cadherin heterozygous mice are viable and fertile with no clear deregulation of β-catenin activity (Larue et al, 1994; Riethmacher et al, 1995).
Given the clear phenotypical differences between the small and the large intestine in our VilCreER Catnblox(ex3)/+ mice, we first examined whether E-cadherin levels might be higher in the colon than the small intestine. This would suggest that E-cadherin might be playing a role in limiting the free β-catenin. Investigating expression by both qRT–PCR and immunoblotting showed markedly higher levels of E-cadherin in the colon compared to the small intestine in wild-type mice (Fig 3A–C).
Figure 3. Increased E-cadherin:β-catenin levels in colonic crypts of wild-type mice.

- Staining of small intestinal and colonic crypts for E-cadherin. Scale bar, 100 μm.
- Western blot of purified crypts from the small intestine and colon (N = 4 mice).
- qRT–PCR of whole tissue from small intestine and colon. Expression of mRNA (2(−dCt)) calculated relative to GAPDH (N = 3 mice). Statistics: one-sided Mann–Whitney U-test.
- Proximity ligation assay for E-cadherin and β-catenin. For each mouse (N = 6), at least 10 crypts per tissue were analysed to calculate the mean. Statistics: one-sided Mann–Whitney U-test; white scale bar, 50 μm.
To quantify not just the E-cadherin levels in both tissues, but rather the complexes of β-catenin and E-cadherin, we used the well-characterised proximity ligation assay (PLA). Using antibodies for β-catenin and E-cadherin allowed us to quantify the number of complexes in the small intestine and the colon of wild-type mice. Consistent with the increase in E-cadherin expression, there was a 2- to 3-fold increase in the number of E-cadherin:β-catenin complexes in the colon compared to the small intestine (Fig 3D).
Accumulation of mutated β-catenin at the adherens junctions is slower in the colon
Next, we investigated the dynamics of E-cadherin:β-catenin complexes. We were able to do this as deletion of exon 3 of β-catenin (and hence activation) produces a smaller protein which is easily detected by immunoblotting. Thus, in a VilCreER Catnblox(ex3)/lox(ex3) mouse, all newly produced β-catenin in intestinal epithelial cells will be smaller (Δex3). Using co-immunoprecipitation for E-cadherin, the relative amounts of mutant β-catenin complexed with E-cadherin can be assessed. We induced VilCreER Catnblox(ex3)/lox(ex3) mice and sampled the small intestine and the colon after 24 and 48 h. After just 24 h (Fig 4A, day 1), approximately 70% per cent of the β-catenin bound to E-cadherin is of the mutated form in the small intestine (72:28 Δex3:wt). In contrast, the majority of β-catenin in the colon bound to E-cadherin is of the WT form (35:65 Δex3:wt). By 48 h (day 2), in the small intestine (SI), approximately 90 per cent of β-catenin bound to E-cadherin is of the mutant form (90:10 Δex3:wt); similarly, the ratio in the colon shifted towards the mutant form (63:37 Δex3:wt). Thus, this shows that the E-cadherin:β-catenin complexes in the small intestine are rapidly saturated with mutant β-catenin.
Figure 4. E-cadherin saturates with mutant β-catenin over time.

- Immunoprecipitation (IP) of E-cadherin from VilCreER Catnblox(ex3)/lox(ex3) mice, 24 h (day 1, top) and 48 h (day 2, bottom) after induction. The ratio of wild-type (WT, top lane) and mutant exon 3 β-catenin (Δex3, bottom lane) bound to E-cadherin (IP:Ecad) was calculated for each tissue. Graphs show average of experiments (N = 3 mice for each genotype and time point); error bars represent s.e.m.
- IP of E-cadherin from VilCreER Catnblox(ex3)/+ mice at day 4 and day 8. Graphs show average of experiments (N = 3 mice for each genotype and time point); error bars represent s.e.m.
We next analysed the ratio in VilCreER Catnblox(ex3)/+ mice. Here, only half of the newly produced β-catenin will be of the mutant form, so we hypothesised that saturation would take longer, especially given the protracted time taken to generate the CPC phenotype. However, we would expect the mutant form to accumulate as this would not be turned over by the destruction complex. First, we examined E-cadherin:β-catenin complexes 4 days post-Cre induction. We showed that 75% of the β-catenin bound to E-cadherin is of the mutated form in the small intestine (75:25 Δex3:wt), whereas the colon has the mutant and the wild-type β-catenin in a roughly similar ratio (53:47 Δex3:wt). At day 8 after induction, approximately 85% of β-catenin bound to E-cadherin in the SI is mutated (85:15 Δex3:wt) compared to 55:45 Δex3:wt in the colon. Therefore, this relative increase in the mutant form of β-catenin compared to the wild-type protein showed that mutant β-catenin was specifically accumulating with E-cadherin at the adherens junction. This suggested that this could be acting as a sink for β-catenin, stopping the mutant β-catenin being translocated into the nucleus where it would bind TCF/LEF transcription factors and drive Wnt targets and the CPC phenotype.
Haploinsufficiency for E-cadherin is sufficient for Wnt pathway activation in the presence of single allele β-catenin mutation
If E-cadherin was acting as a sink and limiting mutant β-catenin from entering the nucleus, this would predict the following: (i) when β-catenin can be targeted for destruction, reduction in E-cadherin levels should have no phenotype, (ii) when there is mutant β-catenin (Δex), reduction in E-cadherin should cause this sink to be saturated quicker and hence lead to a translocation of β-catenin to the nucleus and a CPC phenotype.
To test this hypothesis, we intercrossed AhCreER Catnblox(ex3)/+ mice to mice carrying a conditional knockout E-cadherin allele to generate AhCreER Catnblox(ex3)/+ Cdh1fl/+ mice and controls (Boussadia et al, 2002). First, we examined the phenotype of loss of a single copy of E-cadherin. Five days following Cre induction, mice heterozygous for E-cadherin (AhCreER Cdh1fl/+ or VilCreER Cdh1fl/+) showed a reduction in E-cadherin expression to ∼60% and importantly a reduction in E-cadherin:β-catenin complexes assessed by PLA (Appendix Fig S2A and B). However, despite this reduction, intestines from these mice showed no phenotype and no increase in Wnt signalling (Fig EV5A). This is consistent with previous studies on whole body knockout E-cadherin heterozygotes where there are also no intestinal phenotypes (Larue et al, 1994; Riethmacher et al, 1995).
Figure EV5. Haploinsufficiency for E-cadherin lowers the threshold for Wnt activation of β-catenin mutation.
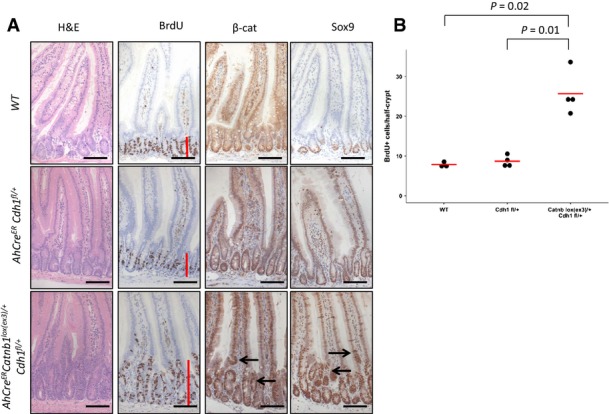
- AhCreER Cdh1fl/+ at day 5 post-induction shows no intestinal phenotype. AhCreER Catnblox(ex3)/+ Cdh1fl/+ mice show increased proliferation (BrdU, red line) and accumulation of β-catenin and Sox9 in cells higher up the crypt-villus axis (arrows). Scale bar, 100 μm.
- Increased proliferation as scored by BrdU+ cells per half-crypt. N ≥ 3, statistics: one-sided Mann–Whitney U-test.
Finally, we investigated the phenotype of the AhCreER Catnblox(ex3)/+ Cdh1fl/+ mice. In contrast to the AhCreER Catnblox(ex3)/+ 10 days post-induction, AhCreER Catnblox(ex3)/+ Cdh1fl/+ showed a CPC phenotype in both the small and large intestine with nuclear β-catenin and increased expression of the Wnt target Sox9 (Fig A and B). Indeed, a CPC phenotype with increased proliferation was already observed in the small intestine by day 5 (Fig EV5). The phenotype was confirmed using VilCreER Catnblox(ex3)/+ Cdh1fl/+ mice which had a CPC phenotype 4 days post-induction with increased expression of several Wnt target genes (Axin2, Lgr5, cMyc, CD44, Appendix Fig S3). Using the intestinal organoid system described earlier, small intestinal crypts from AhCreER Catnblox(ex3)/+ Cdh1fl/+ mice formed spheroids in an R-spondin1-independent manner 5 days post-induction (Fig 5C). This was in contrast to AhCreER Catnblox(ex3)/+ mice. Moreover, targeting a copy of E-cadherin loss in combination with a β-catenin mutation to Lgr5 ISC (Lgr5CreER Catnblox(ex3)/+ Cdh1fl/+) significantly accelerated tumourigenesis when compared to Lgr5CreER Catnblox(ex3)/+ mice. Most importantly, when these mice with reduced E-cadherin were analysed for colonic lesions, 6 of 7 mice had lesions with high nuclear β-catenin (Fig 5D).
Figure 5. Haploinsufficiency for E-cadherin in the presence of single allele β-catenin mutation leads to Wnt deregulation.

- AhCreER Catnblox(ex3)/+ compared to AhCreER Catnblox(ex3)/+ Cdh1fl/+ at day 10 post-induction. Note the presence of colonic lesions in the colon of AhCreER Catnblox(ex3)/+ Cdh1fl/+ mice (arrows). Scale bar, 100 μm; red bar indicates proliferative zone (BrdU).
- Scoring of BrdU-positive cells per half-crypt in the small intestine of wild-type, AhCreER Cdh1fl/+, AhCreER Catnblox(ex3)/+ and AhCreER Catnblox(ex3)/+ Cdh1fl/+ mice at day 10 post-induction. N ≥ 3, at least 25 crypts per mouse were scored. There was significantly higher proliferation in the AhCreER Catnblox(ex3)/+ Cdh1fl/+ mice (P = 0.028, one-sided Mann–Whitney U-test).
- In vitro growth of crypts (small intestine) from mice as indicated at day 5 post-induction without R-spo1. Quantification of sphere-forming efficiency of AhCreER Catnblox(ex3)/+ AhCreER Catnblox(ex3)/+ Cdh1fl/+, day 5 post-induction. N = 3 mice per genotype, mean of 6 technical replicates per mouse, P = 0.04 one-sided Mann–Whitney U-test.
- Survival of Lgr5CreER Catnblox(ex3)/+ and Lgr5CreER Catnblox(ex3)/+ Cdh1fl/+ shows significant acceleration (P = 0.000123, log-rank test) after E-cadherin reduction. About 85% (6/7 mice) had colonic lesions, as identified with β-catenin IHC in contrast to Lgr5CreER Catnblox(ex3)/+ mice.
It is interesting to note that when attempting to recapitulate our crypt culture findings from AhCreER Catnblox(ex3)/+ mice (Fig C) using colonic crypts, we found that the colonic crypts grew more successfully than the ones from the small intestine despite the fact that in vivo β-catenin mutation could not transform the colon (Appendix Fig S5A). A possible explanation for this is the different EDTA concentration needed to purify the crypts from the small intestine and the colon (2 and 25 mM EDTA, respectively) which is known to disrupt the E-cadherin bindings. Indeed, when we analysed wild-type crypts straight after the EDTA purification, we observe similar PLA counts for the crypts of both tissues (Appendix Fig S5B). These changes persist in vitro, when we compared intestinal crypts from VilCreER Apcfl/fl from the small intestine and the colon (Appendix Fig S6A and B).
Human cancers, characterised by β-catenin mutation, are associated with reduction in E-cadherin levels
We finally wanted to examine if these observations from the mouse might possibly translate to human tumourigenesis. Recent sequencing studies with over 200 CRC showed that mutations in β-catenin (CTNNB1) in colorectal cancer are very rare, approximately 5% (11/212). Closer analysis showed that many of these were not in exon 3 and therefore unlikely to affect β-catenin degradation. In total, only 2 of 11 mutations were in the most common exon 3 hotspot (codons 31–45; Appendix Fig S4). Comparisons of these mutations with cancers where activating mutations of CTNNB1 are common such as hepatocellular carcinoma (HCC) or solid pseudopapillary tumours of the pancreas show very different patterns of mutations (cbioportal, Cerami et al, 2012; Gao et al, 2013), indicating these maybe passenger mutations (Appendix Fig S4D).
Given almost all solid pseudopapillary tumours (SPT) have a β-catenin mutation in exon 3, this suggests deregulation of Wnt signalling as an essential step for tumourigenesis in this cancer. From our model, we would predict a change in E-cadherin levels for a β-catenin activating mutation to have the greatest transforming properties. Importantly in these SPT tumours, in addition to the nuclear accumulation of β-catenin, an aberrant localisation of E-cadherin has been reported (Chetty & Serra, 2008). We therefore analysed a tissue microarray (TMA) with normal and SPT tissue for E-cadherin:β-catenin complexes and saw a dramatic decrease in the number of complexes in SPT tumours, compared to normal tissue (Fig 6A).
Figure 6. Human cancers, characterised by β-catenin mutation, are associated with reduction in E-cadherin levels.

- Proximity ligation assay for E-cadherin:β-catenin on a tissue microarray of SPT patients. PLA dots/cells in normal and tumour tissue were counted. Each dot in the boxplot represents the average for a single patient. Staining for β-catenin confirms accumulation of nuclear β-catenin in tumour tissue. Representative pictures are shown. N = 18, statistics: Mann–Whitney U-test, P = 0.0009051. White scale bar, 50 μm.
- Correlation of several Wnt target genes with the expression of E-cadherin (CDH1) was analysed in 269 HCC patients (TCGA provisional), and linear regression line (blue) and confidence region (shaded) were added.
Finally, we examined hepatocellular carcinomas (HCC) where approximately 20% of tumours have a mutation in exon 3 of β-catenin. Analysing expression data from 269 patients (TCGA provisional), we saw a significant negative correlation of E-cadherin with several Wnt target genes (Fig 6B).
Discussion
Although APC is well established in controlling the Wnt signalling pathway, there has been controversy over the role of the activation of Wnt signalling in the initiation of CRC. Much of this debate has been due to the failure to detect nuclear β-catenin at the early stages of the disease and its relatively heterogeneous nature at later stages. Moreover, data from experiments in zebrafish have suggested other pathways to be very important for the phenotypes of Apc loss (Phelps et al, 2009). This coupled with the rarity of β-catenin mutations (after early studies suggesting much higher rates (Sparks et al, 1998)) and the lack of mutual exclusivity with APC mutations has led to many discussions on the reasons for APC mutation in CRC.
Here, we provide definitive in vivo proof (in the murine intestine) that activation of Wnt signalling by loss of the destruction complex or bi-allelic β-catenin mutation is sufficient to drive rapid intestinal transformation. In contrast, upon monoallelic deletion of exon 3, the mutant form of β-catenin takes much longer to accumulate in the small intestine and lead to a CPC phenotype. In the colon, monoallelic mutation of β-catenin did not drive a CPC phenotype (even 25 days post-induction) and did not cause lesions when targeted to the Lgr5 compartment. This suggests that the level of Wnt deregulation required to transform the colorectal epithelium is higher than in the small intestine. This idea often referred to the “just right” model has previously been associated with different levels of Wnt signalling induced by different APC mutations (Fodde & Brabletz, 2007; Buchert et al, 2010). Moreover, within the ApcMin mouse, the distribution of tumours in the small and large intestines has been suggested to reflect Wnt gradients within the tissue (Leedham et al, 2013). Differences between the colon and small intestine in Wnt signalling have been reported in mice deficient for Tcf-4. In this case, deletion of Tcf-4 stopped the formation of small intestinal crypts, but colonic crypts were still present (Korinek et al, 1998).
The differences between the small intestine and the colon correlated with higher E-cadherin levels and increased E-cadherin:β-catenin complexes in colonic crypts. We show that E-cadherin can act as a sink for mutated β-catenin and that the adherens junctions saturate over time with the mutant form of β-catenin. Importantly, a reduction in the expression of E-cadherin leads to a quicker accumulation of sufficient β-catenin to facilitate transformation (Fig 7). To our knowledge, this is the first demonstration in vivo that levels of E-cadherin can be limiting to cancer initiation. It is interesting to note that mice heterozygous for E-cadherin (Cdh1+/−) have an increased tumour burden when crossed to the Apc1638N/+ mouse model, a long latency model of Apc-loss induced tumourigenesis (Smits et al, 2000).
Figure 7. Model of single β-catenin mutation and interaction with E-cadherin in the small intestine and colon.
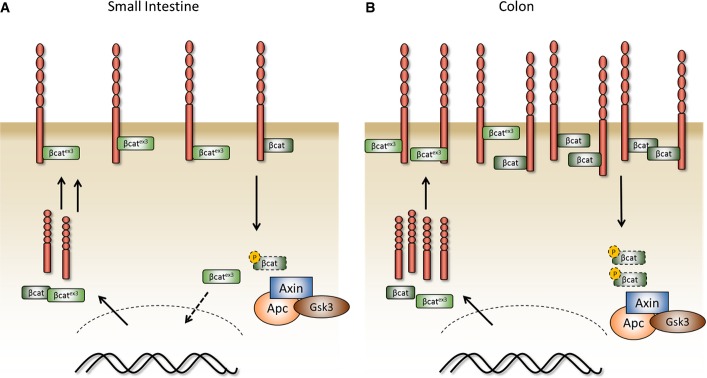
- A, B Model of a single activating β-catenin mutation in the murine small intestine and the colon. In contrast to the small intestine (A), the increased levels of E-cadherin in the colon complexed with mutant β-catenin prevent its accumulation in the nucleus (B).
When we examined solid pseudopapillary tumours of the pancreas, a tumour characterised by β-catenin mutations within exon 3, these tumours showed a strong reduction in E-cadherin:β-catenin complexes. Moreover in HCC, a tumour type which has approximately 20% β-catenin exon 3 mutations, there was a good correlation between reduction in E-cadherin and activation of Wnt signalling targets. Thus, it may be that in these cancers, E-cadherin limits the precise levels of Wnt signalling driven by β-catenin mutation. Thus, downregulation of E-cadherin in these tumours may drive tumour progression. It should also be noted that opposing patterns of Wnt signalling and E-cadherin have been shown in murine liver, with β-catenin higher in zone 3 of the liver and E-cadherin in zone 1. Therefore, one might predict that β-catenin mutations would yield a greater phenotype in hepatocytes from zone 3 of the liver versus zone 1 (Benhamouche et al, 2006). Hence, one could speculate E-cadherin levels might modulate the cell of origin of HCC that carry a β-catenin mutation.
One key question from our work is how does this translate to human cancer? Why do humans not get small intestinal cancer with β-catenin exon 3 mutations rather than CRC with APC mutations? One obvious difference is in sporadic human CRC, tumours develop over a number of years and the sequential mutations in APC may provide different selective benefits and other functions of APC may be very important in human carcinogenesis. Data exist showing that stepwise Apc loss might also induce a more dramatic phenotype (Fischer et al, 2012). Moreover, it is also possible that some of the other consequences of APC mutation may only be revealed when there are only a small number of APC-deficient cells rather than entire crypts. For example, the Wnt-independent roles of APC in microtubules, centrosomes and mitosis have been suggested to modify stem cell division and could favour retention of APC-deficient ISC. We have observed clear differences between Gsk3 and Apc deletion in the ability to respond to microtubule-stabilising drugs which show these other functions of APC are important in vivo (Radulescu et al, 2010).
One could also imagine different dietary carcinogens and microbiota leading to differential mutational spectra that might favour the observed loss of functions mutations in APC rather than the very specific activating mutations required to produce non-degradable β-catenin.
Another potential reason for the lack of β-catenin mutations in both small intestinal and colon cancer is suggested from recent work investigating the selective advantage of different tumour-promoting mutations in murine small and large intestinal epithelium. The Winton group showed that a single inactivating Apc mutation in the intestine had an advantage over wild-type stem cells (Vermeulen et al, 2013). This increased the likelihood that a stem cell carrying this mutation would be retained to repopulate the entire crypt. A second mutation in Apc further increased the fitness of the stem cell compared to wild-type and was even more likely to repopulate the entire crypt. However, it is of interest to note that despite these selective advantages, given the increase in fitness never increased the probability of repopulating crypt to 1 (WT is neutral 0.5, the Apc+/− 0.62 and Apc−/− 0.79), then on many occasions, cells carrying Apc mutations would be lost. From our work, we would suggest that as it takes a protracted time for the β-catenin exon 3 mutation to have a phenotype in the small intestine, this mutation would probably be neutral (0.5) for some time. Thus, given neutral drift within the intestine, one might predict that these mutant cells would be lost.
For many years, there has been much discussion of why ApcMin mice (a model of FAP) mainly develop small intestinal tumours rather than FAP where patients preferentially develop colon tumours, though they will eventually develop duodenal tumours. Our data show acute Apc loss manifests phenotypes in both the small intestine and colon which does not explain the tropism for small intestine in the mouse. A recent study by Tomasetti and Vogelstein (Tomasetti & Vogelstein, 2015) has suggested that differences in stem cell proliferation within colon versus small intestine could explain the tropism to colon in the human CRC.
We have also shown that within the intestine, GSK3 is only obviously limiting to the Wnt signalling pathway. Given GSK3 has many functions within the cells and is highly expressed, this suggests other kinases can compensate grossly for these activities within the intestine. It has previously been postulated that GSK3 inhibition may also be of therapeutic relevance for diabetes. One obvious concern is that long-term GSK3 inhibition might predispose to colorectal cancer. Our data here and indeed from hypomorphic mutants in Apc (Buchert et al, 2010) argue that to induce the crypt-progenitor phenotype and tumourigenesis, complete gene function has to be deleted rather than being simply inhibited. Thus, as GSK3 inhibitors would only reduce rather than ablate GSK3, it is unlikely this would predispose to cancer. Moreover, given the fact that there are 4 alleles of GSK3, mutation of a single allele would not activate the Wnt pathway much further, so there would be no selection for loss of GSK3, reinforcing the notion that GSK3 inhibition would not drive CRC. Therefore, there may be a therapeutic opportunity for GSK3 in diabetes.
In summary, we show that a single β-catenin mutation is not as potent in activating the Wnt signalling pathway as loss of Apc. Furthermore, we show that E-cadherin levels can limit the transforming potential of an activating β-catenin mutation.
Materials and Methods
Mouse experiments
All experiments were performed under the UK Home Office guidelines. The alleles used in this study were as follows: AhCreER (Kemp et al, 2004) VilCreER (el Marjou et al, 2004), AhCre (Ireland et al, 2004), Lgr5CreER (Barker et al, 2007) Catnb1lox(ex3) (Harada et al, 1999), Gsk3alphafl, Gsk3betafl (Patel et al, 2008), Apcfl (Shibata et al, 1997) and Cdh1fl (Boussadia et al, 2002). Cre induction strategies are described in the Appendix Supplementary Methods. N numbers for the animal experiments are provided on the figure legends.
Immunohistochemistry
Standard immunohistochemistry techniques were used throughout this study. The following primary antibodies were used: BrdU (1/200, #347580, BD Biosciences), pGSKB-Ser9 (1/200, #9336, Cell Signaling), β-catenin (1/50 #610154, BD Biosciences), cMyc (1/200, sc-764, Santa Cruz), Sox9 (1/500, #AB5535 Chemicon), lysozyme (1/200, DAKO #A0099) and E-cadherin (1/200, R&D Systems AF748). Staining for nuclear β-catenin was performed on tissue samples fixed at 4°C for less than 24 h in 10% formalin prior to processing.
Immunoblotting
Standard Western blot techniques were used. Crypts from the small and large intestine were purified by incubation with 2 and 25 mM EDTA/PBS (respectively) for 30 min at 4°C. Crypts were further purified by centrifugation with low speed (50 × g, 3 min). The following antibodies were used: E-cadherin (1/5,000, BD Transduction Lab, #610182); β-actin (1/5,000, Sigma A2228).
Immunoprecipitation
Small intestinal and colonic tissue (∼4 cm) was lysed in lysis buffer (200 mM NaCl, 75 mM Tris–HCl [pH 7], 7.5 mM EDTA, 7.5 mM EGTA, 0.15% [v/v] Tween-20). Lysates were clarified by centrifugation at 16,000 × g for 10 min at 4°C. Magnetic beads conjugated to anti-mouse IgG (Dynabeads) were incubated with 1 mg of protein lysates, 1 μg of either monoclonal anti-E-cadherin (BD Biosciences #610182) or IgG isotype control (Sigma) for 1 h at 4°C with constant rotation. After several washes with lysis buffer, bound proteins were eluted from the beads by boiling at 100°C for 5 min in SDS reducing buffer. Bound proteins and 5 μg of total lysates (Input) were run on 4–12% Bis–Tris Gradient SDS gel and probed for β-catenin (1/1,000 BD Biosciences, #610154).
Proximity Ligation Assay (PLA)
PLA was performed on tissue samples fixed at 4°C for < 24 h in 10% formalin prior to processing using the Duolink Detection kit (Sigma) according to the manufacturer's instructions. Briefly, after citrate buffer-mediated antigen retrieval, the slides were incubated with goat E-cadherin (1/200, R&D Systems AF748) and mouse β-catenin (1/2,000 for mouse tissue, 1/200 for human tissue, #610154, BD Biosciences) overnight. Detection was performed with PLA probes (anti-goat and anti-mouse) conjugated to oligonucleotides. After ligation, amplification detection with a fluorescent probe, slides were imaged on a Zeiss LSM confocal microscope. Z-stacks with 40× objectives were taken. PLA dots in crypts were analysed with ImageJ and either calculated as area fraction or count/nuclei.
Sphere culture
Crypts were purified from mice as previously described (Sato et al, 2009). Crypts were seeded in growth factor-reduced Matrigel (BD Biosciences) with the addition of EGF and Noggin (both Peprotech), without R-spo1 or with 50 ng/ml R-spo1 (R&D Systems). About 300 crypts were plated per 20 μl matrigel/well, and procedures were carried out within 3 h after sacrificing the mouse. Representative photographs were taken after 7 days.
Human TMA of solid papillary tumours (SPT) of the pancreas
Tissue microarray of 18 SPT patients was used as described before, and normal tissue was used as control (Serra et al, 2007). PLA was performed as described above.
qRT–PCR of mouse intestine
For detailed description and primer sequences, see Appendix Supplementary Methods.
Acknowledgments
The authors would like to thank James Woodgett for the supply of Gsk3 floxed mice and Trevor Graham for helpful comments. Grant support: supported by Cancer Research UK grant C596/A17196, and DJH is funded by the European Union Seventh Framework Programme FP7/2007–2013 under grant agreement number 278568. FS is funded by the North West Cancer Research Fund CR700. OJS is funded by an ERC investigator grant COLONCAN.
Author contributions
DJH, RAR, ARC and OJS contributed to study concept and design; DJH RAR, SR, ML, ADC, SB, SL, GM, LP, JM, FS, FC, KRR, VSM, BD and NB to acquisition of data; DJH, RAR, SR, AH, FC, BD, AW, LL and OS to analysis and interpretation of the data; DJH, RAR and OJS to drafting of the manuscript; AH and GK to statistical analysis; and AM, SS and RC to provision of reagents.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Appendix
Expanded View Figures PDF
Review Process File
References
- Ahn SM, Jang SJ, Shim JH, Kim D, Hong SM, Sung CO, Baek D, Haq F, Ansari AA, Lee SY, Chun SM, Choi S, Choi HJ, Kim J, Kim S, Hwang S, Lee YJ, Lee JE, Jung WR, Jang HY, et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology. 2014;60:1972–1982. doi: 10.1002/hep.27198. [DOI] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, Vasseur-Cognet M, Kuo CJ, Kahn A, Perret C, Colnot S. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev Cell. 2006;10:759–770. doi: 10.1016/j.devcel.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Boussadia O, Kutsch S, Hierholzer A, Delmas V, Kemler R. E-cadherin is a survival factor for the lactating mouse mammary gland. Mech Dev. 2002;115:53–62. doi: 10.1016/s0925-4773(02)00090-4. [DOI] [PubMed] [Google Scholar]
- Buchert M, Athineos D, Abud HE, Burke ZD, Faux MC, Samuel MS, Jarnicki AG, Winbanks CE, Newton IP, Meniel VS, Suzuki H, Stacker SA, Nathke IS, Tosh D, Huelsken J, Clarke AR, Heath JK, Sansom OJ, Ernst M. Genetic dissection of differential signaling threshold requirements for the Wnt/beta-catenin pathway in vivo. PLoS Genet. 2010;6:e1000816. doi: 10.1371/journal.pgen.1000816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetty R, Serra S. Membrane loss and aberrant nuclear localization of E-cadherin are consistent features of solid pseudopapillary tumour of the pancreas. An immunohistochemical study using two antibodies recognizing different domains of the E-cadherin molecule. Histopathology. 2008;52:325–330. doi: 10.1111/j.1365-2559.2007.02949.x. [DOI] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Cui J, Zhou X, Liu Y, Tang Z, Romeih M. Wnt signaling in hepatocellular carcinoma: analysis of mutation and expression of beta-catenin, T-cell factor-4 and glycogen synthase kinase 3-beta genes. J Gastroenterol Hepatol. 2003;18:280–287. doi: 10.1046/j.1440-1746.2003.02973.x. [DOI] [PubMed] [Google Scholar]
- Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12:957–971. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edamoto Y, Hara A, Biernat W, Terracciano L, Cathomas G, Riehle HM, Matsuda M, Fujii H, Scoazec JY, Ohgaki H. Alterations of RB1, p53 and Wnt pathways in hepatocellular carcinomas associated with hepatitis C, hepatitis B and alcoholic liver cirrhosis. Int J Cancer. 2003;106:334–341. doi: 10.1002/ijc.11254. [DOI] [PubMed] [Google Scholar]
- Fischer JM, Miller AJ, Shibata D, Liskay RM. Different phenotypic consequences of simultaneous versus stepwise Apc loss. Oncogene. 2012;31:2028–2038. doi: 10.1038/onc.2011.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–158. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, Cho KR, Fearon ER. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007;67:9721–9730. doi: 10.1158/0008-5472.CAN-07-2735. [DOI] [PubMed] [Google Scholar]
- Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ, Winton DJ. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of beta-catenin. Gastroenterology. 2004;126:1236–1246. doi: 10.1053/j.gastro.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Kemp R, Ireland H, Clayton E, Houghton C, Howard L, Winton DJ. Elimination of background recombination: somatic induction of Cre by combined transcriptional regulation and hormone binding affinity. Nucleic Acids Res. 2004;32:e92. doi: 10.1093/nar/gnh090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, Clevers H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet. 1998;19:379–383. doi: 10.1038/1270. [DOI] [PubMed] [Google Scholar]
- Larue L, Ohsugi M, Hirchenhain J, Kemler R. E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci USA. 1994;91:8263–8267. doi: 10.1073/pnas.91.17.8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leedham SJ, Rodenas-Cuadrado P, Howarth K, Lewis A, Mallappa S, Segditsas S, Davis H, Jeffery R, Rodriguez-Justo M, Keshav S, Travis SP, Graham TA, East J, Clark S, Tomlinson IP. A basal gradient of Wnt and stem-cell number influences regional tumour distribution in human and mouse intestinal tracts. Gut. 2013;62:83–93. doi: 10.1136/gutjnl-2011-301601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB, Schwitalla S, Kalna G, Ogg EL, Athineos D, Timpson P, Vidal M, Murray GI, Greten FR, Anderson KI, Sansom OJ. ROS production and NF-kappaB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 2013;12:761–773. doi: 10.1016/j.stem.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathke IS. The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium. Annu Rev Cell Dev Biol. 2004;20:337–366. doi: 10.1146/annurev.cellbio.20.012103.094541. [DOI] [PubMed] [Google Scholar]
- Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol. 2008;28:6314–6328. doi: 10.1128/MCB.00763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW, Jones DA. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009;137:623–634. doi: 10.1016/j.cell.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulescu S, Ridgway RA, Appleton P, Kroboth K, Patel S, Woodgett J, Taylor S, Nathke IS, Sansom OJ. Defining the role of APC in the mitotic spindle checkpoint in vivo: APC-deficient cells are resistant to Taxol. Oncogene. 2010;29:6418–6427. doi: 10.1038/onc.2010.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethmacher D, Brinkmann V, Birchmeier C. A targeted mutation in the mouse E-cadherin gene results in defective preimplantation development. Proc Natl Acad Sci USA. 1995;92:855–859. doi: 10.1073/pnas.92.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, Vass JK, Athineos D, Clevers H, Clarke AR. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–679. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, Batlle E, Simon-Assmann P, Clevers H, Nathke IS, Clarke AR, Winton DJ. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385–1390. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- Serra S, Salahshor S, Fagih M, Niakosari F, Radhi JM, Chetty R. Nuclear expression of E-cadherin in solid pseudopapillary tumors of the pancreas. JOP. 2007;8:296–303. [PubMed] [Google Scholar]
- Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, Matsumoto H, Takano H, Akiyama T, Toyoshima K, Kanamaru R, Kanegae Y, Saito I, Nakamura Y, Shiba K, Noda T. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 1997;278:120–123. doi: 10.1126/science.278.5335.120. [DOI] [PubMed] [Google Scholar]
- Smits R, Ruiz P, Diaz-Cano S, Luz A, Jagmohan-Changur S, Breukel C, Birchmeier C, Birchmeier W, Fodde R. E-cadherin and adenomatous polyposis coli mutations are synergistic in intestinal tumor initiation in mice. Gastroenterology. 2000;119:1045–1053. doi: 10.1053/gast.2000.18162. [DOI] [PubMed] [Google Scholar]
- Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58:1130–1134. [PubMed] [Google Scholar]
- TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78–81. doi: 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen L, Morrissey E, van der Heijden M, Nicholson AM, Sottoriva A, Buczacki S, Kemp R, Tavare S, Winton DJ. Defining stem cell dynamics in models of intestinal tumor initiation. Science. 2013;342:995–998. doi: 10.1126/science.1243148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File