

Abstract
The EsxB protein from Bacillus anthracis belongs to the WXG100 family, a group of proteins secreted by a specialized secretion system. We have determined the crystal structures of recombinant EsxB and discovered that the small protein (∼10 kDa), comprised of a helix-loop-helix (HLH) hairpin, is capable of associating into two different helical bundles. The two basic quaternary assemblies of EsxB are an antiparallel (AP) dimer and a rarely observed bisecting U (BU) dimer. This structural duality of EsxB is believed to originate from the heptad repeat sequence diversity of the first helix of its HLH hairpin, which allows for two alternative helix packing. The flexibility of EsxB and the ability to form alternative helical bundles underscore the possibility that this protein can serve as an adaptor in secretion and can form hetero-oligomeric helix bundle(s) with other secreted members of the WXG100 family, such as EsxW. The highly conserved WXG motif is located within the loop of the HLH hairpin and is mostly buried within the helix bundle suggesting that its role is mainly structural. The exact functions of the motif, including a proposed role as a secretion signal, remain unknown.
Keywords: type VII secretion system, ESAT-6 like secretion system, WXG family, EsxB, helix bundle, antiparallel dimer, bisecting U dimer, tetramer
Introduction
Bacterial pathogens transport virulence factors through their diverse secretion systems to the extracellular matrix or directly into the host cell. The recently characterized type VII secretion system (T7SS) was first discovered in Mycobacterium tuberculosis.1 T7SSs secrete a family of small proteins, which commonly encompass about 100 residues and carry no canonical secretory signal peptide(s). Because of their low sequence identities, except for the conserved Trp-Xaa-Gly (WXG) motif, these secreted proteins are classified as members of the WXG100 superfamily2 (Supporting Information Fig. S1). These proteins have also been identified in the genomes of many Gram-positive bacteria.3 Experimental evidence supported the notion that many Gram-positive bacteria encode a T7-like secretion system often referred to as the ESAT-6 like secretion system or WXG100 secretion system because of the distinct genetic features in these organisms as compared to acid-fast stainable species.4–6 Based on existing structures and bioinformatics analyses, WXG100 proteins are believed to be all α-helical and form oligomers.2,7 The association/dissociation of WXG100 proteins seems to play a crucial role in their secretion, although a lot of details remain unclear.
EsxB is one of six WXG100 proteins (EsxB, EsxL, EsxP, EsxQ, EsxW, and EsxV) encoded by the genome of Bacillus anthracis, the causative agent of anthrax.8 It was shown that under laboratory conditions, EsxW and EsxB are actively secreted. However, EsxB is indispensible for the secretion of EsxW, whereas the deletion of the esxW gene has no effect on EsxB secretion. This led to the hypothesis that EsxB acts as a transporter or chaperone for other WXG100 proteins. The secretion of EsxB is not affected by fusing ubiquitin to its N- and C-termini, or by truncating its first and last five residues. But secretion can be abolished by excessive truncation at either termini or insertion of ubiquitin in the middle of EsxB, both of which presumably disrupt its structure.8 Thus, it was proposed that the secretion behavior of EsxB could be controlled by its three-dimensional structure.8
Here, recombinant EsxB has been purified and characterized to determine its three-dimensional structure. The EsxB molecule with a His6 affinity tag attached to its C-terminus (WT-EsxB-His6) unexpectedly forms a rarely observed bisecting U (BU) dimeric assembly of a helix-loop-helix (HLH) hairpin (Fig. 1). Notable examples of this form of helical dimer include a mutant form (A31P) of repressor of primer (Rop),9 the N-terminal domain of an adaptor molecule containing PH and SH2 domains (APS)10 and a native-like de novo designed protein11; all three proteins represent truncated or point mutant constructs. The BU form of APS is believed to be a stable dimer with a phenylalanine zipper. The BU form of the Rop mutant dimer is expected to be less stable than the antiparallel (AP) form of the wild-type Rop dimer or another syn-parallel Rop mutant dimer as predicted by energy-landscape theory.12
Figure 1.
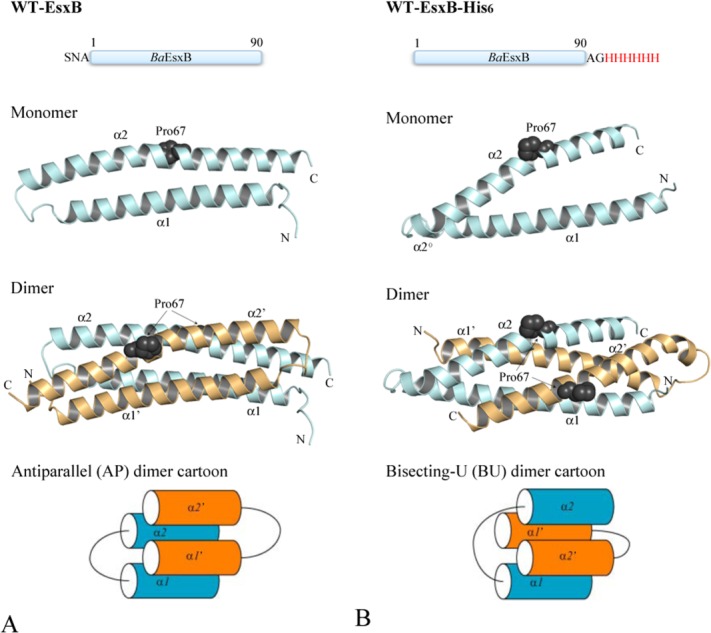
The crystal structures and schematic diagrams of the antiparallel (AP, Panel A) and bisecting-U (BU, Panel B) topologies of EsxB. Top panels show the linear sequence and tags included in the recombinant proteins used for crystallization. Middle panels show the ribbon diagrams of the structure of EsxB monomers and dimers with Pro67 drawn in sphere representation to highlight the kink in α2. Monomers A and B are in pale cyan and light orange, respectively. The helical motifs within the inter-helices links of the BU dimer are labeled as α2° (see text for details). The lower panels show the schematic diagrams of the AP and BU dimers. Figures 1–4 are prepared with the program PyMOL (http://www.PyMOL.org).
Considering that two other WXG100 protein structures, the CFP-10/ESAT-6 heterodimer from M. tuberculosis7 and the EsxA homodimer from Staphylococcus aureus13 adopt the AP topology, we investigated whether the presence of the C-terminal His6-tag might have contributed to the BU dimerization of B. anthracis EsxB. It has long been recognized that poly-histidine appendages confer distinctive oligomerization properties on tagged proteins.14,15 We re-cloned the gene and expressed EsxB with a cleavable N-terminal His6-tag. After cleaving the tag, the N-terminus is left with only three extra residues, SNA. Interestingly, this EsxB construct crystallized as an AP form of the HLH hairpin dimer (Fig. 1). For the convenience of the following discussion, we refer to this EsxB construct as WT-EsxB.
We further analyzed the sequence of the WXG100 family and identified the possible basis of the EsxB structural dichotomy. Based on these predictions, several mutants were subsequently designed and purified and one mutation, Y65F, resulted in a switch from an AP WT-EsxB dimer assembly to a BU WT-EsxB dimer. Oligomeric states and stability of these recombinant EsxB proteins were also examined. The ability of EsxB to form different helix bundles and oligomeric states, either with itself or with other WXG100 protein(s) indicates that EsxB may serve as an adaptor/chaperone protein for other B. anthracis proteins and assist their secretion across the envelope.
Results
We have determined the high-resolution crystal structures of several EsxB protein constructs (WT-EsxB, WT-EsxB-His6, and point mutants P65A and Y67F). The structures of WT-EsxB and WT-EsxB-His6 were obtained using SeMet-labeled and native protein crystals. Because labeled and native protein structures are essentially identical (Supporting Information Table I), though the crystals were grown under different crystallization conditions, only the structures determined and refined at higher resolutions are presented in the Results and the following Discussion.
Table I.
Crystallographic Statistics
| Data collection | SeMet WT-EsxB | SeMet WT-EsxB-His6 | WT-EsxB Y65F | WT-EsxB P67A |
|---|---|---|---|---|
| Space group | P212121 | P31 | P3121 | P212121 |
| Unit cell (Å, °) | a = 33.08, b = 38.39, c = 130.75, α = β = γ = 90 | a = b = 77.34, c = 71.23, α = β = 90, γ = 120 | a = b = 76.22, c = 71.38, α = β = 90, γ = 120 | a = 45.51, b = 96.20, c = 134.37, α = β = γ = 90 |
| MW Da (residue) | 10,362 (93)a | 11,040 (98)b | 10,346 (93)a | 10,336 (93)a |
| Mol (AU) | 2 | 4 | 2 | 2 |
| SeMet (AU) | 2 | 4 | 0 | 0 |
| Wavelength(Å) | 0.9793 (peak) | 0.9793 (peak) | 0.9792 | 0.9792 |
| Resolution(Å) | 1.44–50 | 1.88–50 | 1.28–50 | 1.52–50 |
| Number of unique reflections | 30,476c | 38,708c | 61,931 | 89,122 |
| Redundancy | 6.8 (5.0)d | 4.5 (4.0)e | 6.5 (5.2)f | 4.5 (2.5)g |
| Completeness (%) | 97.9 (85.2)d | 100.0 (99.9)e | 99.8 (99.9)f | 97.5 (80.4)g |
| Rmerge | 0.078 (0.440)d | 0.095 (0.616)e | 0.042 (0.657)f | 0.049 (0.373)g |
| I/σ(I) | 46.10 (4.22)d | 18.9 (2.18)e | 40.6 (2.15)f | 36.9 (2.57)g |
| Solvent content (%) | 38.0 | 55.5 | 57.4 | 48.1 |
| Phasing | ||||
| Range (Å) | 1.44–50 | 1.88–50 | 3.00–50h | 3.00–50h |
| RCullis (anomalous) | 0.66 | 0.90 | ||
| Figure of merit | 0.296 | 0.171 | ||
| Figure of merit after DM | 0.787 | 0.869 | ||
| Correlation coefficienth | 0.607 | 0.0.322 | ||
| Refinement | ||||
| Resolution | 1.44–29.0 | 1.88–48.8 | 1.28–33.62 | 1.52–40.60 |
| Reflections (work/test) | 28,829/1542 | 36,743/1939 | 58,759/3139 | 84,560/4457 |
| Rcrystal/Rfree (%) | 17.9/20.4 | 14.1/16.2 | 16.3/17.4 | 18.1/20.3 |
| RMS deviation from ideal geometry; bond length (Å)/angle (°) | 0.006/0.976 | 0.006/0.806 | 0.005/0.969 | 0.005/0.802 |
| No. of atoms (protein/HETATM) | 1371/198 | 2504/253 | 1304/161 | 4004/517 |
| Mean B value (Å2); (mainchain/sidechain) | 22.80/29.46 | 18.20/24.16 | 16.31/21.95 | 26.07/33.18 |
| Ramachandran favored (%)i; Ramachandran outliers (%)i | 100.0; 0.0 | 98.7; 0.0 | 98.8; 0.7 | 100.0; 0.0 |
| Rotamer outliers (%)i | 1.4 | 4.3 | 0.7 | 2.5 |
| Clashscorei | 6.67 | 12.43 | 3.78 | 7.34 |
| PDB deposit | 4J10 | 4IYI | 4J42 | 4J41 |
Including three N-terminal vector-derived residues, SNA.
Including eight C-terminal His-tag residues, AGHHHHHH.
Including Bijvoet pairs.
Last resolution bin, 1.44–1.46 Å.
Last resolution bin, 1.88–1.91 Å.
Last resolution bin, 1.28–1.30 Å.
Last resolution bin, 1.52–1.55 Å.
Molecular replacement.18
Defined by MolProbity.19
Antiparallel (AP) dimer of WT-EsxB
The crystal structure of WT-EsxB has been determined at a resolution of 1.44 Å. In one asymmetric unit there are two hairpin monomers, which forms a pseudo two-fold AP homodimer [Fig. 1(A)]. Each monomer comprises two helices, α1 and α2, in a common form of the HLH hairpin. α1 and α2 span residues Glu9 to Leu39 and Ala46 to Phe83, respectively. While α1 is mostly straight, α2 has a 14° kink at Gln63, which is located at one turn ahead of Pro67 [Fig. 1(A)]. The loop that connects α1 and α2 consists of six residues, from Glu40 to Gly45, including a 310 helix motif before the conserved WXG motif, Trp43-Ala44-Gly45. The electron densities for the loop region are excellent and very well defined for both helices, suggesting limited mobility of the inter-helical link. The highly conserved Trp43, which is buried inside a small hydrophobic pocket, seemingly acts as an anchor for the entire loop. The hydrophobic pocket, also observed in the structures of other WXG100 proteins,7,13 is lined by residues from both subunits that include Leu39 and Phe51 for subunit A and Leu11’, Leu76’ and Ile79’ for subunit B (prime indicates residues of the second subunit) [Fig. 2(A)]. Except for one hydrogen bond formed between the side chains of Trp43 and Gln48, there are no other hydrogen bonds between backbone atoms either within the loop or between the loop and the two helices.
Figure 2.
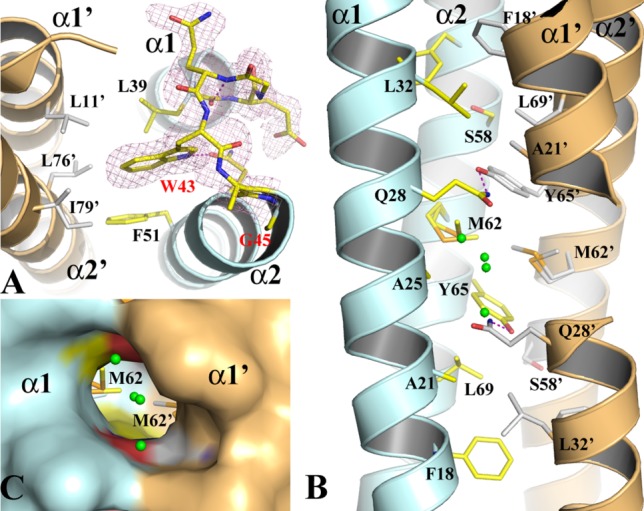
The structure of the WT-EsxB AP dimer. (A) The structure of the inter-helix link drawn in stick format to show the conformation of the conserved WXG motif from monomer A of the AP dimer. The 2FoFc electron density map drawn in magenta mesh is contoured at 1σ. (B) The packing of the core of the helical bundle of the AP dimer. Monomers A and B are in pale cyan and light orange, respectively. Key residues contributing to the interaction across the dimer interface are drawn in stick format. Primes on labels for residues or secondary structures refer to those from monomer B. A pair of hydrogen bonds between Q28 and Y67’ are drawn in magenta dash line. Water molecules are drawn in small green spheres. (C) A molecular surface representation of part of the three-way tunnel within the core of the helical bundle of the AP dimer. Two other smaller entrances to the tunnel are on the two sides between α1 and α2, α1’ and α2’, respectively.
In the EsxB AP four-helix bundle, the two helices from the same monomer are adjacent to each other and each of them is antiparallel to both neighboring helices [Fig. 1(A)]. The size of the EsxB dimer interface is about 1614 Å2 based on a PISA (Protein, Interface, Structures and Assemblies) analysis16 and the majority of the residues across the interface are hydrophobic. The exception is a pair of hydrogen bonds between Tyr65 and Gln28’ of the subunits and within the hydrophobic core [Fig. 2(B)]. A hydrophobic layer is also formed by the symmetric residues Met62/Met62’ and Ala25/Ala25’ [Fig. 2(B)]. The short side chains of the alanines create a cavity that measures around 300 Å3 in the middle of the helical bundle with three openings to the protein surface. The cavity calculation was performed with the CASTp server.17 The major opening between both α1 helices is large enough for solvent to enter the cavity and four ordered water molecules are observed near the entrance and inside [Fig. 2(B,C)].
Bisecting U (BU) dimer of WT-EsxB-His6 and its possible tetramer
The 1.88 Å structure of WT-EsxB-His6 was first solved in space group P3121 from a twinned crystal [Fig. 1(B)]. The structure refinement was performed in space group P31 by utilizing a twin operator (Table I). There are four EsxB monomers in one asymmetric unit, which form two nearly identical BU hairpin dimers [Fig. 3(A)]. Electron densities for these EsxB molecules are excellent except for a few terminal residues and the His6 affinity tags [Fig. 3(B)]. Therefore, in the crystal the His6-tags are disordered and do not contribute to the structure. For the following description of the WT-EsxB-His6 structure, only one BU dimer will be referenced.
Figure 3.
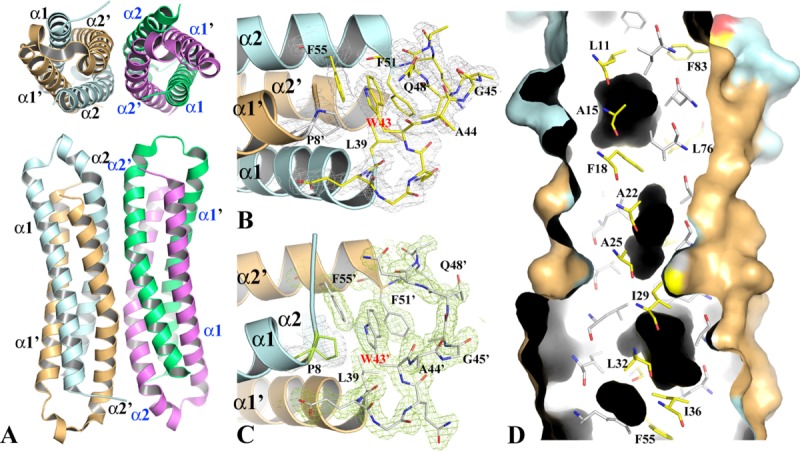
The structure of WT-EsxB-His6 BU dimer and tetramer. (A) The top and side views of the two WT-EsxB-His6 BU dimers in asymmetric unit. The possible tetramer assembly was deduced by PISA16 analysis. The dimer on the left side, formed by monomers A (in pale cyan) and B (in light orange) is used as a model for the detailed structural description provided in the text. (B) and (C) The structures of the inter-helix links of monomers A and B, respectively. The links from different dimers drawn in stick format show their different conformations. The link from monomer A contains a one-turn helix while link B contains two β-turns. The 2FoFc electron density maps of both link regions are contoured at 1σ. (D) Molecular surface representation and ribbon diagram of a portion of the BU dimer. The dimer is cross-sectioned from one side to show the packing of the core of the helical bundle as well as cavities (in black) resulted from the packing. Key residues contributing to the packing are drawn in stick format. Primes on secondary structures refer to those from monomer B. For clarity, only residues from monomer A are labeled. The corresponding residues from monomer B are related to them by a twofold symmetry.
With an acute angle of 30° between the α1 and α2 helices (calculated with anglebetweenhelices scripts in PyMol), the conformation of each WT-EsxB-His6 monomer is somewhat different from the WT-EsxB monomer described for the AP dimer [Fig. 1(B)]. In each monomer the length of helix α1 (residues Glu9 to Leu39) is the same as in the AP dimer. The structural alignment of the AP and BU EsxB forms shows Cα root mean square deviation (RMSD) 0.9 Å for helix α1, suggesting that the α1 helices are structurally very similar. However, α2 in the BU dimer is shorter and starts from Phe51 instead of Ala46 as seen in the AP dimer, which makes the inter-helical linker of the BU dimer five residues longer. For the structural alignment of helix α2, the Cα RMSD is larger (1.6 Å), mainly due to an increased kink angle from 14° to 23° and a shift of the kink itself from Gln63 to Pro67. In the BU dimer the inter-helical linker is quite different in each monomer. In monomer A, the linker contains one complete turn of helix, from Ala44 to Lys50 [Fig. 3(B)]; in monomer B, the linker contains two β-turns [Fig. 3(C)]. The side chain of the conserved Trp43 makes fewer nonpolar contacts, including those with residues Gln48 (hydrophobic part of this side chain), Phe55, Pro8’ and Leu39’ and the resulting cavity is more solvent accessible.
In the EsxB BU helical bundle, the two helices from the same monomer occupy the two diagonal positions and each of them is antiparallel to one of its neighboring helix and parallel to the other. The hydrophobic core of the BU helical bundle is quite different to that of the AP helical bundle in terms of packing hydrophobic residues [Fig. 3(D)]. As a result, there are several cavities formed within the core of the helical bundle with the volumes of the three largest being 191.4, 33.4, and 29.6 Å3 (calculated with the CASTp server17). Interestingly, a PISA analysis16 of WT-EsxB-His6 suggests that the two BU dimers in the asymmetric unit could form a stable tetrameric assembly [Fig. 3(A)]. The dimer-dimer interface is formed by packing of four antiparallel α2 helices. A close examination of the dimer-dimer interface reveals several hydrophobic residues (Y65 and I68) and hydrogen-bond forming residues (Q60, Q64, and Y65) contributing to the formation of this tetrameric assembly.
EsxB variants
To understand the basis of the AP and BU dimer formation, several variants of untagged WT-EsxB were designed and produced. Some mutations, such as G53A and E54Q, have no notable impact on the EsxB assembly structure and associate only as AP dimers (Supporting Information Table SI). Two mutations Y65F and P67A, however, resulted in remarkable structural changes.
In the AP dimer of WT-EsxB, residue Y65 forms a hydrogen bond with Q28’ in the center of the dimer interface [Fig. 2(B)]. Such hydrogen bonds are often found in the hydrophobic cores of helical bundles. In the BU dimer of WT-EsxB-His6, both Y65 and Q28 move away from the center of the dimer as elaborated later in the Discussion section. Replacement of Y65 by a phenylalanine obliterates the hydrogen bond and repositions the polar residue, Q28, along the hydrophobic core, which presumably destabilizes the AP dimer. Therefore, BU dimer formation appears to be favored by the Y65F variant [Fig. 4(A)]. A structural alignment between Y65F and WT-EsxB-His6 results in a RMSD of only 0.52 Å suggesting that the two BU dimers are essentially the same. Interestingly, a quaternary assembly analysis using PISA16 also predicts that the Y65F BU dimer in the asymmetric unit could also form a stable tetramer with a symmetry-related BU dimer using the same four antiparallel α2 helices [Fig. 4(A)]. The dimer–dimer tetramer is similar to the tetramer described for WT-EsxB-His6 [Fig. 3(A)]. An alignment between these two tetramers shows a RMSD of 0.64 Å. The three cavities identified in the WT-EsxB-His6 structure are also present in the Y65F mutant structure with similar dimensions.
Figure 4.
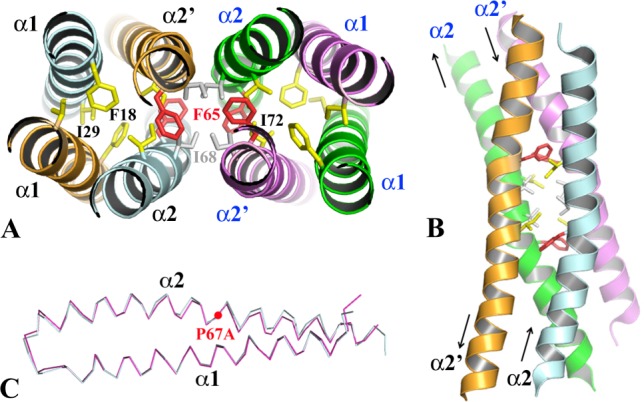
The structures of WT-EsxB Y65F and P67A mutants. (A) The hydrophobic core of the dimer–dimer interface of the Y65F tetramer and its connection to the helical bundle cores of two contributing BU dimers. Residue I68 (in grey stick) and substituting residue F65 (in red stick) play important roles in tetramer formation. In the similar WT-EsxB-His6 tetramer, the hydroxyl group of Y65 is also involved in a hydrogen-bond network. Some residues from the hydrophobic core of the BU dimer are highlighted in yellow stick format. (B) The four antiparallel α2 helices form the dimer–dimer interface. (C) A structural alignment of AP dimers of WT-EsxB and P67A mutant. To show the conformational change of the α2 helix due to the mutagenesis, only α1 was used in least-square calculation for the alignment.
The tetramer form of either the Y65F untagged EsxB variant or WT-EsxB-His6 can be considered as a building unit of their crystals (data not shown). The packing of the tetramer is contributed by four antiparallel α2 helices [Fig. 4(A,B)]. In each BU dimer, two α2 helices are antiparallel [Fig. 1(B)]. The dimer-dimer interaction resembles that of a common helical hairpin–hairpin packing [Fig. 4(B)]. Residues I68, F65 (in Y65F EsxB) or Y65 (in WT-EsxB-His6) from each α2 helix form a hydrophobic core across the dimer-dimer interface. The hydrophobic core is also connected to the hydrophobic center of each contributing dimer.
Residue P67 causes a kink in the helix α2, a feature shared by both the AP and BU dimers (Fig. 1). This kink is believed to contribute to the formation of several cavities in the AP and BU dimers (Figs. 2 and 3). Replacing the proline residue is likely to reduce opening(s) within dimer interfaces. The crystal structure of P67A EsxB shows three nearly identical AP dimers in one asymmetric unit. Each AP dimer is similar to that of the WT-EsxB dimer albeit that the α2 helix shows a small conformation change in the C-terminal portion for the mutant [Fig. 4(C)]. A structural alignment by SSM between the two dimers yields a RMSD of 0.56 Å. The point mutation results in a tighter AP dimer. In the P67A mutant the volume of the largest cavity found within the core of the WT-EsxB dimer is reduced by nearly half in volume to 165 Å3 [Fig. 2(C)]. A few water molecules can be seen inside the cavity (data not shown). A PISA quaternary assembly analysis16 finds no stable higher-order oligomer for the P67A EsxB variant despite several different ways of helical packing inside the crystal.
EsxB oligomers in solution
Size-exclusion chromatography (SEC) was performed to investigate the oligomeric states of WT-EsxB and isogenic variants as well as WT-EsxB-His6. At pH 8.0, all proteins with exception of tagged WT-EsxB-His6 eluted unexpectedly at the position corresponding to an EsxB tetramer [Fig. 5(A); ∼40 kDa]. In contrast, the His6-tagged protein (WT-EsxB-His6) migrated to an apparent size similar to an EsxB monomer [Fig. 5(A); ∼10 kDa]. Because WT-EsxB-His6 and the Y65F mutant show similar BU dimers and potentially stable tetramers in their crystal structures, the SEC tetramer and monomer profiles of Y65F and WT-EsxB-His6, respectively, suggest that the presence of the polyhistidine tag in WT-EsxB-His6 is the sole cause of the significant increase of the retention time. Meanwhile, the SEC profiles of WT-EsxB and the Y67A mutant also suggest a preferred tetramer formation in solution that was not observed in the crystals where the proteins selectively crystallized as dimers.
Figure 5.
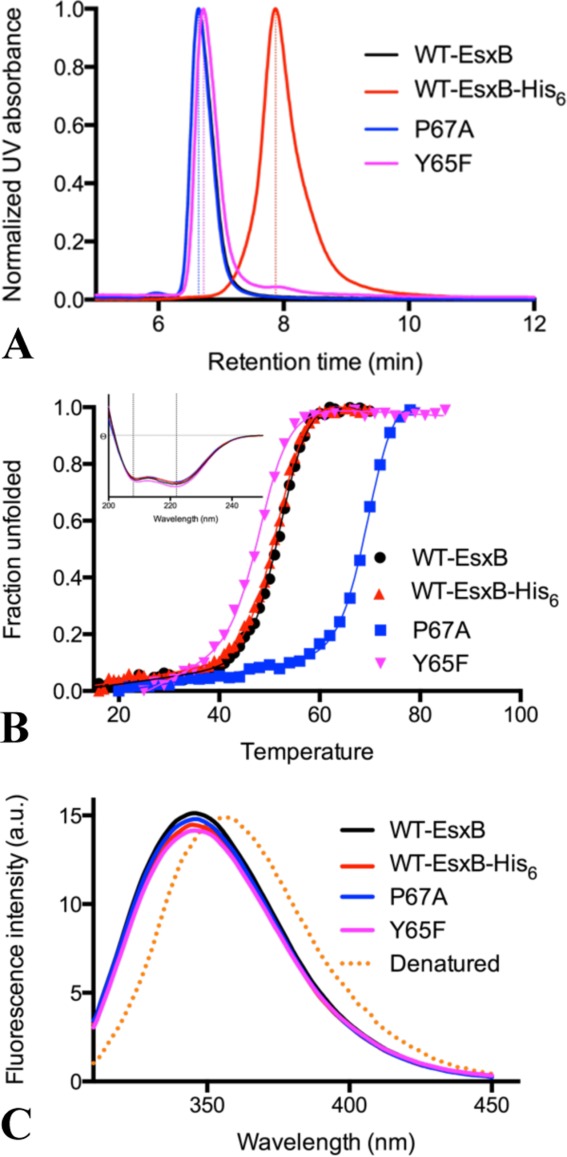
Size-exclusion chromatography, thermal denaturation and intrinsic fluorescence of EsxB proteins. (A) Size-exclusion chromatograph profiles of WT-EsxB-His6, Y65F and P67A WT-EsxB mutants. Based on calibration, all three WT-EsxB proteins eluted at a position closer to an EsxB tetramer while WT-EsxB-His6 eluted at a position corresponding approximately to a monomer. (B) Thermal denaturation profiles of various forms of EsxB. The insert at the upper left corner shows the far-UV CD spectra of these proteins. The spectra have strong peaks at 222 and 208 nm, and Θ222nm/Θ208nm>1, characteristic of a coiled-coil structure. Thermal denaturation was monitored by CD signal at 222 nm, with all curves being sigmoidal. (C) Intrinsic fluorescence of various constructs of EsxB. All spectra are very similar, with fluorescence emission peaks at 346 nm. The fluorescence of the fully denatured WT-EsxB, with maximum value at 356 nm, is also shown as reference (orange dotted line). The coloring scheme is as follows: black for WT-EsxB; red for WT-EsxB-His6; blue for P67A and purple for Y65F.
Thermal denaturation of EsxB proteins
Thermal denaturation of EsxB proteins was performed and monitored using circular dichroism (CD) spectra in the far-UV region at pH 7.4. The WT-EsxB and WT His6-EsxB proteins share similar properties in terms of CD spectra profiles and denaturation temperatures [Fig. 5(B)]. Their CD spectra, with features at 208 and 222 nm, are characteristic of proteins composed of α-helices. Their thermal denaturation curves, as monitored by the CD signal at 222 nm, are indicative of cooperative unfolding. Their denaturation temperatures are similar and with only a marginally higher melting temperature (Tm) for WT-EsxB as compared to WT-EsxB-His6 [Fig. 5(B); ∼0.6°C higher]. This suggests that WT-EsxB and WT-EsxB-His6 have very similar helical bundle structures in solution. Distinct denaturation temperatures and melting profiles would have been expected for the monomer and tetramer of EsxB, reinforcing the notion that the increased SEC retention profile of WT-EsxB-His6 is caused by the histidine tag.
In contrast, the P67A mutant is more stable with a dramatically increased Tm [Fig. 5(B); ∼17.6° higher than WT-EsxB]. The increased stability can be explained by the improved packing of side chains and reduction in the volume of cavities within the core of the helical bundle since no major openings were observed within the AP dimer. Although the P67A mutant eluted as a tetramer in solution, tetramers were not observed in the crystal structure. Possibly, as with WT-EsxB, dimers of P67A are selectively crystallized. The helical content of the Y65F mutant is in between that of WT-EsxB and WT-EsxB-His6. Its Tm decreases more than 4 degrees as compared to WT-EsxB [Fig. 5(B)]. The lower stability of this mutant may be caused by the unfavorable exposure of the hydrophobic side chain of the substituting phenylalanine, elimination of the hydrogen bond and repositioning of Q28 along the hydrophobic core.
Fluorescence spectra of EsxB proteins
The conformational states of the various forms of EsxB proteins in solution were further evaluated by fluorescence spectroscopy. When excited at 295 nm, the intrinsic fluorescence of EsxB proteins predominantly comes from Trp43, which is the only tryptophan residue in EsxB. We discovered that despite the different crystal structures, all forms of EsxB proteins have very similar fluorescence spectra with maximum emission at 346 nm. On the other hand, fully denatured EsxB protein has a fluorescence maximum at a longer wavelength of 356 nm [Fig. 5(C)]. These data indicate that Trp43 in all EsxB proteins are in a similar nonpolar environment, consistent with the crystal structures.
Discussion
We have determined the crystal structures of recombinant EsxB in two different forms as well as several point mutants. We have also examined their oligomeric states and thermal stabilities (Table II). Our data suggest that a small modification of the EsxB sequence, such as the addition of a His6-tag to its C-terminal terminus or a point mutation could change the way the protein dimerizes. Further analysis indicates that the structural basis for the alternate helical assembly seems to be associated with the unique duality of the helical heptad repeats of the EsxB sequence [Fig. 6(A)]. For the helical wheels of α1 and α2, the a and d positions of α2 can be clearly and easily identified. However, in α1, assignments of the a and d positions are based on the packing of the AP dimer. The a position of the α1 helical wheel is hydrophobic whereas the d position shows low hydrophobicity and includes both polar, Q38, and charged, D35 residues. In the AP dimer, Q38 forms a hydrogen bond with Y65 on the d position of the α2 helical wheel and contributes a layer of side chain packing within the core of the helical bundle [Fig. 6(B)]. D35 points outward and does not contribute to the layering of the hydrophobic core of the helical bundle. Therefore the AP dimer, formed by a typical packing from the residues of the a and d positions, may not represent a stable arrangement. On the other side, the e position of the α1 helical wheel has elevated hydrophobicity. The two consecutive alanines (A15 and A22) do not contribute to the layering of the hydrophobic core but rather provide an alternative way for helical bundle formation. It is α1 that could use the d and e positions instead of a and d positions to pack with the a and d positions of α2 to form a new helical bundle [Fig. 6(C)]. The BU dimer represents one such possible alternative packing.
Table II.
Summary of Experimental Measurements
| EsxB protein | WT-EsxB | WT-EsxB-His6 | WT-EsxB Y65F | WT-EsxB P67A |
|---|---|---|---|---|
| Dimer form in crystal | AP | BU | BU | AP |
| Recognizable quaternary assembly in crystal | Dimer | Dimer/tetramer | Dimer/tetramer | Dimer |
| Apparent oligomer in size-exclusion chromatography | Tetramer | Monomer | Tetramer | Tetramer |
| Thermal denaturation temperature (Tm C) | 51.3 | 50.7 | 47.0 | 68.9 |
Figure 6.
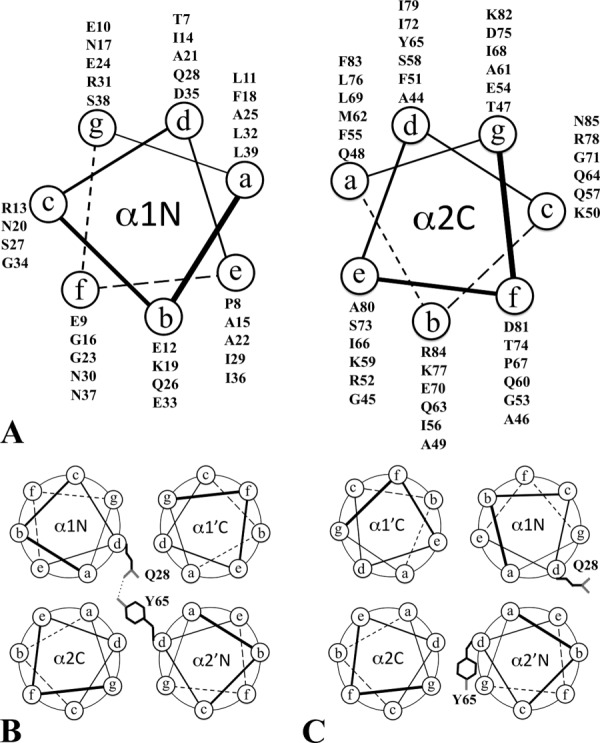
Helical wheels of EsxB and its alternative packing. (A) Helical wheel diagrams for the α1 and α2 helices of EsxB. The label α1N on the left side helix indicates the orientation of the α1 helix with its N-terminus pointing to the reader. The label α2C on the right side helix indicates the orientation of the α2 helix with its C-terminus pointing to the reader. (B) Packing of the AP dimer as observed in WT-EsxB and P67A mutant. Both α1 and α2 helices use their a and d positions for the helical bundle packing. One of two pairs of Q28-Y65 hydrogen bonds within the core of the dimer is highlighted. (C) Packing of the BU dimer as observed in WT-EsxB-His6 and Y65F mutant. The α1 helix uses its a and e positions instead of a and d positions for helical packing. One of two Q28 residues and one of Y65 residues from the dimer were highlighted for their new positions in the WT-EsxB-His6 BU dimer. In Y65F, Y65 is replaced by a phenylalanine.
This structural duality indicates the potential of EsxB to form not only homo-oligomeric helix bundles but also hetero-oligomeric helix bundles with other proteins, especially with WXG family members. Our previous studies have shown that both WT-EsxB and WT-EsxB-His6 are effectively secreted by bacilli and when produced from a plasmid, either one of these proteins restores the secretion of EsxW.8 Thus, secretion mediated by EsxB in B. anthracis is accomplished regardless of the helix hairpin packing in the structure of the dimer, supporting the notion that EsxB acts as a very flexible adaptor/chaperone molecule.
Recombinant WT-EsxB used in this study is likely to represent the naturally occurring EsxB variant in structure and function. Based on SEC data, the protein eluted as a tetramer. WT-EsxB crystallized in a wide range of pHs exclusively as the AP hairpin dimer form. However, the observation of dimers in crystal structures does not exclude the possibility of a tetramer protein in solution. Similar dimer-tetramer transitions between crystalline and solution states have been observed for other proteins.20–22 Because the tetramer protein exists in equilibrium with the dimer, either the tetramer or dimer forms can preferentially crystallize.
Recombinant WT-EsxB-His6 with a C-terminally appended histidine tag eluted as a monomer based on the SEC profile. However, the elution profile is quite broad indicating very likely non-specific interactions with the SEC resin. On the basis of its nearly identical thermal denaturation profile with WT-EsxB that behaves as a tetramer both in solution and in the crystal, we conclude that WT-EsxB-His6 is a tetramer in solution. Moreover, the BU dimer forming Y65F mutant shares a similar tetrameric crystal structure as WT-EsxB-His6 and elutes as a tetramer. This is further corroborated by the fluorescence measurements. The intrinsic fluorescence spectrum of WT-EsxB-His6 is virtually identical to that of WT-EsxB, and is indicative of a conformational state in which Trp43 is folded into the hydrophobic core. If WT-EsxB-His6 were to exist as a monomer in solution, its Trp43 residue would be exposed to solvent and give rise to a fluorescence spectrum resembling that of denatured EsxB. Therefore, our observations are consistent with the increased interaction of WT-EsxB-His6 with the column matrix and delayed elution profile caused by the four poly-histidine tags in a small protein tetramer.
The preferred AP dimer conformation observed for EsxB can be switched to the BU dimer by particular changes in amino acid sequence, for example by substitution of Y65 with a phenylalanine. Interestingly, other members of the WXG100 family have Tyr, Phe, Leu or another hydrophobic residue. If this position is responsible for the switch, more members of the family may form BU-like dimers. It is not clear why C-terminal tagging favors BU dimer formation but possibly it points to a low association energy for AP dimers and a shallow energy barrier between the two dimers. The switch from an AP to a BU dimer caused by the Y65F point mutation can be explained by unfavorable interactions of the polar residue Q28 left inside the hydrophobic core that is likely to destabilize the AP dimer. Changes in the stability of various forms of EsxB seem to also be correlated with changes in the volume of the hydrophobic cavities. The Y65F mutation does not significantly change the cavities’ volume in the BU dimer structure, and its Tm is just 4° lower than WT-EsxB-His6. The P67A mutant, on the other hand, decreased the largest cavity volume by 140 Å3, and exhibits remarkably enhanced thermal stability, with a Tm nearly 18° higher than the wild-type protein. It is interesting to note that although the P67A mutation reduces the kink angle and the cavity side, it does not completely eliminate the cavity, suggesting that Pro67 is not solely responsible for the imperfect packing of the hydrophobic core of WT-EsxB. In fact, the residues in this position are highly variable in other members of the WXG100 family (Supporting Information Fig. S1). Our data suggest that B. anthracis EsxB is not optimized for the formation of a homodimer, in either AP or BU forms, but rather is designed to preserve flexibility in interactions with multiple proteins of the WXG100 family and promote their secretion. Furthermore, since the structural flexibility of B. anthracis EsxB is mostly attributed to its heptad dichotomy, a trait that is shared among other WXG100 family members (Supporting Information Fig. S1 and annotations therein), we speculate that the dynamics of their helices provide a common mechanism that allows WXG100 proteins to undergo biologically important conformational changes.
Materials and Methods
Protein cloning, expression, and purification
The cloning of B. anthracis WT-EsxB-His6 was performed as described previously.8 For WT-EsxB, the same esxB gene was cloned into the pMCSG7 vector.23 The proteins were expressed in E. coli BL21(DE3)Magic cells. To obtain seleno-methionine (SeMet)-labeled EsxB proteins for structure determination, the cells were grown at 37°C in enriched M9 medium containing SeMet under conditions known to inhibit methionine biosynthesis.24 For native protein expression, the cells were grown in LB containing 40 mM K2HPO4. All proteins were purified using Ni-affinity chromatography.25 The proteins were concentrated using an Amicon Ultra 3K centrifugal filter device (Millipore) in 20 mM HEPES pH 8, 250 mM NaCl, and 2 mM dithiothreitol (DTT). Site-directed mutations of Y65F, P67A, E54Q, and G53A were created by the PIPE cloning method26 and verified by sequencing. Briefly, plasmid encoding WT-EsxB was used as the template, primers were added to a final concentration of 0.4 µM, and mutagenesis was performed in a final volume of 50 µL using PfuUltra Hotstart PCR Master Mix (Stratagene). Subsequent procedures from gene cloning and expression, and protein purification were as described above for WT-EsxB.
Size exclusion chromatography
Size exclusion chromatography of WT-EsxB, WT-EsxB-His6, and mutants Y65F EsxB and P67A EsxB were performed on an SRT SEC-150 column (Sepax Technologies) connected to a Dionex HPLC equipped with a temperature-controlled autosampler housing two 96-well plate sample racks, a GP50 gradient pump, and a PDA-100 photodiode array detector (Thermo Scientific). The column was pre-equilibrated with crystallization buffer (20 mM HEPES pH 8, 250 mM NaCl, 2 mM DTT) and calibrated with premixed protein standards, which included ribonuclease A (13.7 kDa), carbonic anhydrase (29 kDa), conalbumin (75 kDa), aldolase (158 kDa), ferritin (440 kDa), and blue dextran (2,000 kDa). The chromatography runs were carried out at room temperature with a flow rate of 1 mL min−1 using a 30 µL protein sample at a concentration of 5 mg mL−1.
Protein crystallization
All EsxB proteins were screened for crystallization conditions with the help of a Mosquito nanoliter liquid handler (TTP LabTech) using the sitting drop vapor diffusion technique in 96-well CrystalQuick plates (Greiner). For each condition, 0.4 µL of protein and 0.4 µL of crystallization formulation were mixed; the mixture was equilibrated against 140 µl of the crystallization solution in each reservoir well. The crystallization screens used were MCSG-1–4 at 4 and 24°C. Diffraction quality crystals appeared under conditions including: (1) SeMet-labeled WT-EsxB-His6: 0.1 M Na2HPO4:citric acid, 2.0 M ammonium sulfate, pH 4.2; (2) WT-EsxB-His6: 0.1 M sodium acetate, 2.0 M ammonium sulfate, pH 4.5; (3) SeMet-labeled WT-EsxB: 0.1 M phosphate citrate, 40% (v/v) PEG 300, pH 4.2; (4) WT-EsxB: 0.2 M NaCl, 0.1 M Tris:HCl, 30% (w/v) PEG 3000, pH 7.0; (5) Y65F: 0.1 M Bis-Tris propane, 2 M di-ammonium hydrogen citrate, pH 7.0; (6) P67A: 0.1 M sodium citrate:citrate acid, 40% (v/v) PEG 600, pH 5.5; (7) G53A: 0.1 M sodium acetate:HCl, 25% (w/v) PEG 3350, pH 4.5; (8) E54Q: 0.17 M ammonium acetate, 0.085 M sodium acetate:HCl, 25.5% (w/v) PEG 4000, 15% glycerol, pH 4.6. Prior to data collection, the crystals were treated with cryoprotectants and cryocooled directly in liquid nitrogen.
X-ray diffraction and structure determination
A set of single-wavelength diffraction data was collected near the selenium absorption peak (12.66 keV) at 100 K from each type of EsxB crystal. All data were obtained at the 19-ID beamline of the Structural Biology Center at the Advanced Photon Source at Argonne National Laboratory using the program SBCcollect.27 The intensities of each data set were integrated, scaled and merged with the HKL3000 program suite28 (Table I). For SeMet-labeled protein crystals, single-wavelength anomalous diffraction (SAD) method was applied. Se sites were located using the program SHELXD29 and they were used for phasing with the program MLPHARE.30 After density modification,30 partial models were built in three cycles of Arp/Warp model building.31 All of the above programs are integrated within the program suite HKL3000.28 The models were then completed manually using the program COOT.32 The structures of native EsxB crystals were determined using the molecular replacement method.18 The search templates used were structures of either SeMet WT-EsxB-His6 or SeMet WT-EsxB, which were solved with the SAD phasing approach mentioned above. Subsequent model rebuilding for each structure was performed using the program COOT.32 All final models were refined using the program Phenix.refine33 (Table I).
Circular dichroism spectroscopy assay
Circular dichroism (CD) spectroscopy was performed on a J-810 CD spectropolarimeter (Jasco). The cell path length was 1 mm and the protein was diluted in 50 mM phosphate buffer pH 7.4 to a final concentration of 50 μM. Each CD spectrum acquired represents an average of three scans collected from 200 to 260 nm with a step size of 0.1 nm at a rate of 50 nm min−1 and a bandwidth of 1 nm. For the thermal stability experiment, one spectrum was measured every 5° from 25 to 90°C. At each step, the temperature was increased to 0.5°C min−1 and stabilized for 3 min before data acquisition. The baseline of each spectrum was corrected by subtracting the spectrum of a buffer blank obtained at 25°C. The results were converted to per residue molar absorption units, Δε (M−1 cm−1). The data were analyzed using the program GraphPad Prism (GraphPad Software).
Fluorescence spectroscopy
Fluorescence spectra of protein samples were acquired on a PerkinElmer LS50B spectrofluorometer. The protein intrinsic fluorescence spectra were recorded at 20°C with excitation at 295 nm and emission monitored from 310 to 450 nm. The final spectra were the average of five accumulations that were collected at a scan rate of 150 nm min−1. The protein samples were diluted in 50 mM phosphate buffer at pH 7.4 with 100 mM NaCl to a final concentration between 3 and 5 μM. For spectrum comparison, denatured WT-EsxB sample was also prepared with 6 M guadinium hydrochloride and was incubated at room temperature for 1 h before running fluorescence assays.
Acknowledgments
The authors wish to thank members of the Structural Biology Center at Argonne National Laboratory for their help with data collection at the 19-ID beamline.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- 1.Stanley SA, Raghavan S, Hwang WW, Cox JS. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci USA. 2003;100:13001–13006. doi: 10.1073/pnas.2235593100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poulsen C, Panjikar S, Holton SJ, Wilmanns M, Song YH. WXG100 protein superfamily consists of three subfamilies and exhibits an alpha-helical C-terminal conserved residue pattern. PLoS One. 2014;9:e89313. doi: 10.1371/journal.pone.0089313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pallen MJ. The ESAT-6/WXG100 superfamily—and a new Gram-positive secretion system? Trends Microbiol. 2002;10:209–212. doi: 10.1016/s0966-842x(02)02345-4. [DOI] [PubMed] [Google Scholar]
- 4.Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke-Grauls CM, Appelmelk BJ, Bitter W. Type VII secretion–mycobacteria show the way. Nat Rev Microbiol. 2007;5:883–891. doi: 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- 5.Desvaux M, Hebraud M, Talon R, Henderson IR. Outer membrane translocation: numerical protein secretion nomenclature in question in mycobacteria. Trends Microbiol. 2009;17:338–340. doi: 10.1016/j.tim.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Schneewind O, Missiakas DM. Protein secretion and surface display in Gram-positive bacteria. Philos Trans R Soc Lond Ser B Biol Sci. 2012;367:1123–1139. doi: 10.1098/rstb.2011.0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Renshaw PS, Lightbody KL, Veverka V, Muskett FW, Kelly G, Frenkiel TA, Gordon SV, Hewinson RG, Burke B, Norman J, Williamson RA, Carr MD. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J. 2005;24:2491–2498. doi: 10.1038/sj.emboj.7600732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garufi G, Butler E, Missiakas D. ESAT-6-like protein secretion in Bacillus anthracis. J Bacteriol. 2008;190:7004–7011. doi: 10.1128/JB.00458-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glykos NM, Cesareni G, Kokkinidis M. Protein plasticity to the extreme: changing the topology of a 4-alpha-helical bundle with a single amino acid substitution. Structure. 1999;7:597–603. doi: 10.1016/s0969-2126(99)80081-1. [DOI] [PubMed] [Google Scholar]
- 10.Dhe-Paganon S, Werner ED, Nishi M, Hansen L, Chi YI, Shoelson SE. A phenylalanine zipper mediates APS dimerization. Nat Struct Mol Biol. 2004;11:968–974. doi: 10.1038/nsmb829. [DOI] [PubMed] [Google Scholar]
- 11.Hill RB, DeGrado WF. Solution Structure of α2D, a Nativelike de novo designed protein. J Am Chem Soc. 1998;120:1138–1145. [Google Scholar]
- 12.Levy Y, Cho SS, Shen T, Onuchic JN, Wolynes PG. Symmetry and frustration in protein energy landscapes: a near degeneracy resolves the Rop dimer-folding mystery. Proc Natl Acad Sci USA. 2005;102:2373–2378. doi: 10.1073/pnas.0409572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sundaramoorthy R, Fyfe PK, Hunter WN. Structure of Staphylococcus aureus EsxA suggests a contribution to virulence by action as a transport chaperone and/or adaptor protein. J Mol Biol. 2008;383:603–614. doi: 10.1016/j.jmb.2008.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu J, Filutowicz M. Hexahistidine (His6)-tag dependent protein dimerization: a cautionary tale. Acta Biochim Polym. 1999;46:591–599. [PubMed] [Google Scholar]
- 15.Amor-Mahjoub M, Suppini JP, Gomez-Vrielyunck N, Ladjimi M. The effect of the hexahistidine-tag in the oligomerization of HSC70 constructs. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;844:328–334. doi: 10.1016/j.jchromb.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 16.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 17.Binkowski TA, Naghibzadeh S, Liang J. CASTp: computed atlas of surface topography of proteins. Nucleic Acids Res. 2003;31:3352–3355. doi: 10.1093/nar/gkg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 19.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D, Biological Crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pazy Y, Eisenberg-Domovich Y, Laitinen OH, Kulomaa MS, Bayer EA, Wilchek M, Livnah O. Dimer-tetramer transition between solution and crystalline states of streptavidin and avidin mutants. J Bacteriol. 2003;185:4050–4056. doi: 10.1128/JB.185.14.4050-4056.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferreira-da-Silva F, Pereira PJ, Gales L, Roessle M, Svergun DI, Moradas-Ferreira P, Damas AM. The crystal and solution structures of glyceraldehyde-3-phosphate dehydrogenase reveal different quaternary structures. J Biol Chem. 2006;281:33433–33440. doi: 10.1074/jbc.M605267200. [DOI] [PubMed] [Google Scholar]
- 22.Arai S, Yonezawa Y, Okazaki N, Matsumoto F, Tamada T, Tokunaga H, Ishibashi M, Blaber M, Tokunaga M, Kuroki R. A structural mechanism for dimeric to tetrameric oligomer conversion in Halomonas sp. nucleoside diphosphate kinase. Protein Sci. 2012;21:498–510. doi: 10.1002/pro.2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eschenfeldt WH, Makowska-Grzyska M, Stols L, Donnelly MI, Jedrzejczak R, Joachimiak A. New LIC vectors for production of proteins from genes containing rare codons. J Struct Funct Genom. 2013;14:135–144. doi: 10.1007/s10969-013-9163-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 25.Kim Y, Dementieva I, Zhou M, Wu R, Lezondra L, Quartey P, Joachimiak G, Korolev O, Li H, Joachimiak A. Automation of protein purification for structural genomics. J Struct Funct Genomics. 2004;5:111–118. doi: 10.1023/B:JSFG.0000029206.07778.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klock HE, Lesley SA. The polymerase incomplete primer extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol Biol. 2009;498:91–103. doi: 10.1007/978-1-59745-196-3_6. [DOI] [PubMed] [Google Scholar]
- 27.Rosenbaum G, Alkire RW, Evans G, Rotella FJ, Lazarski K, Zhang RG, Ginell SL, Duke N, Naday I, Lazarz J, Molitsky MJ, Keefe L, Gonczy J, Rock L, Sanishvili R, Walsh MA, Westbrook E, Joachimiak A. The structural biology center 19ID undulator beamline: facility specifications and protein crystallographic results. J Synchrotron Radiat. 2006;13:30–45. doi: 10.1107/S0909049505036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr Sect D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 29.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr Sect D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 30.30. CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr Sect D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 31.Cohen SX, Morris RJ, Fernandez FJ, Ben Jelloul M, Kakaris M, Parthasarathy V, Lamzin VS, Kleywegt GJ, Perrakis A. Towards complete validated models in the next generation of ARP/wARP. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:2222–2229. doi: 10.1107/S0907444904027556. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr Sect D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information