

Abstract
Recombinant human α-galactosidase A (rhαGal) is a homodimeric glycoprotein deficient in Fabry disease, a lysosomal storage disorder. In this study, each cysteine residue in rhαGal was replaced with serine to understand the role each cysteine plays in the enzyme structure, function, and stability. Conditioned media from transfected HEK293 cells were assayed for rhαGal expression and enzymatic activity. Activity was only detected in the wild type control and in mutants substituting the free cysteine residues (C90S, C174S, and the C90S/C174S). Cysteine-to-serine substitutions at the other sites lead to the loss of expression and/or activity, consistent with their involvement in the disulfide bonds found in the crystal structure. Purification and further characterization confirmed that the C90S, C174S, and the C90S/C174S mutants are enzymatically active, structurally intact and thermodynamically stable as measured by circular dichroism and thermal denaturation. The purified inactive C142S mutant appeared to have lost part of its alpha-helix secondary structure and had a lower apparent melting temperature. Saturation mutagenesis study on Cys90 and Cys174 resulted in partial loss of activity for Cys174 mutants but multiple mutants at Cys90 with up to 87% higher enzymatic activity (C90T) compared to wild type, suggesting that the two free cysteines play differential roles and that the activity of the enzyme can be modulated by side chain interactions of the free Cys residues. These results enhanced our understanding of rhαGal structure and function, particularly the critical roles that cysteines play in structure, stability, and enzymatic activity.
Keywords: α-galactosidase A, site-directed mutagenesis, cysteine variants, structure–function, enzymes, activity, stability, fabry disease
Introduction
The lysosomal enzyme α-galactosidase A (αGal) catalyzes the removal of the terminal α-galactose from glycosphingolipids, primarily globotriosylceramide. In humans, the absence of functional αGal causes accumulation of the globotriosylceramide substrate in tissues, leading to Fabry disease, an X-linked lysosomal storage disorder.1 Human αGal (hαGal) is a homodimeric glycoprotein, consisting of two identical subunits of approximately 50kD each. It also has mannose-6-phosphate containing oligosaccharides which are required for the molecule to traffic to the lysosome via the mannose-6-phosphate receptor pathway.2
The structures of recombinant human αGal (rhαGal) and the complex of rhαGal with its catalytic substrate, a covalent intermediate, and galactose product have been determined by X-ray crystallography.3–5 The crystal structures reveal that each monomer consists of two domains: a (β/α)8 barrel containing the active site and an antiparallel β-sheet domain. The catalytic mechanism of the enzyme involves two critical aspartic acid residues Asp170 and Asp231, which act as a nucleophile and an acid/base, respectively.4 To date, more than 600 mutations have been identified in the hαGal gene, including more than 300 missense mutations (Human Gene Mutation Data Base http://www.hgmd.cf.ac.uk5–9). As most patients with Fabry disease have a single point mutation in their hαGal gene, the mutations have been mapped in the three-dimensional structure and grouped into primarily two categories: those near the active site, and those buried residues distant from the active site that adversely affect the folding state of the hαGal molecule.5,10 While active site mutations may directly perturb the catalytic mechanism of the mutant protein leading to loss of function and corresponding substrate accumulation, the pathologic effect of mutations disparate from the active site are less obvious. In fact, these non-catalytic Fabry-associated mutations represent the majority of the Fabry cases and are pervasive throughout the structure of the protein. For example, over half of the residues in the protein have been found to have changes in patients with Fabry disease and most of these genetic mutations lead to disruption of the hydrophobic core of the protein. Specifically, as has been pointed out by Garman and Garboczi,5 28% of the residues in domain 2 that are at least 25 Å away from the active site, have been found mutated in Fabry patients. Alteration of these residues likely leads to the disruption of the hydrophobic core of the protein and thus be intolerant to even conservative amino acid substitutions.5 For example, replacement of the completely buried Met187 residue with the chemically similar Val leads to loss of enzymatic activity and the development of Fabry disease symptoms.11 It is concluded, therefore, that Fabry disease is primarily a disease of protein folding.7
Given the complex nature of the hαGal structure–function relationship in the context of Fabry disease and the paucity of data yielding direct insight into the molecular nature of this relationship, research into the basis of hαGal structural stability is warranted. There have been multiple recent studies investigating the functional properties of the Fabry variant enzymes by expressing them recombinantly in COS-1, COS-7, and HEK293 cells.9,12,13 In the present study, we performed a systematic examination of recombinant Cys variants to study their impact on hαGal structure, function, and stability.
There are 12 cysteines in each monomer of the rhαGal protein, ten form disulfide bonds within monomeric subunits and two contain free sulfhydryl (Fig. 1). Interestingly, mutants leading to development of Fabry disease have been identified at all of the disulfide bond-involving cysteines (Human Gene Mutation Data Base http://www.hgmd.cf.ac.uk5–9). However, mutations have not been found on the two free cysteines until a recent report of a novel C174R mutation in a Fabry patient.14 Since the five pairs of disulfide bonds are distributed in different secondary structures and tertiary folding of the protein with only one pair (Cys142/Cys172) making direct contribution to the active site,5 it is not clear whether mutations at different Cys positions contribute to structural destabilization by different mechanisms.
Figure 1.
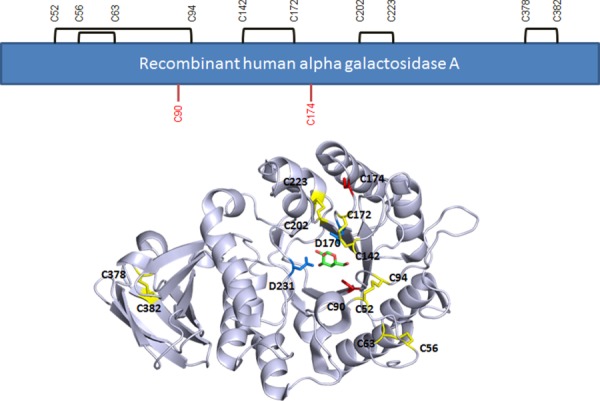
Disulfide assignment and positions of the two free cysteines in rhαGal. Disulfides are in yellow. Free cysteines C90 and C174 are in red. Catalytic residues D170 and D231 are in blue. The galactose product in the crystal is shown in green and red stick presentation. Structure based on PDB:3HG5.4
In general, free cysteines have been found to play a variety of roles in protein structure and function, including dimerization, metal coordination, enzyme catalysis, redox regulation and thermal stability.15 The function of the cysteine residue appears to largely depend on its chemical environment within the folded protein structure16,17 and has been studied particularly for enzymes involved in lysosomal storage diseases.18,19 In rhαGal, the two free cysteines occupy different structural environments. Cys90 is completely buried inside the structure while Cys174 is partially exposed to solvent at the surface of the molecule. In the present study, we sought to investigate the roles Cys residues, particularly the two free Cys residues, on rhαGal structure and activity as well as the impact when replaced with other amino acids.
Results
Expression and activity of Cys to Ser mutants
Twelve single point mutants, converting each cysteine residue to serine, were created to evaluate rhαGal disulfide bonds and their structure–function relationships. The enzymatic activity of the mutants in the conditioned media after transient transfection was analyzed using a 4-MU activity assay. Only the wild type protein, the C90S and C174S mutants, and the C90S/C174S double mutant were found to be active (Fig. 2). These two Cys correspond to the positions of free cysteines determined from the crystal structure.5
Figure 2.
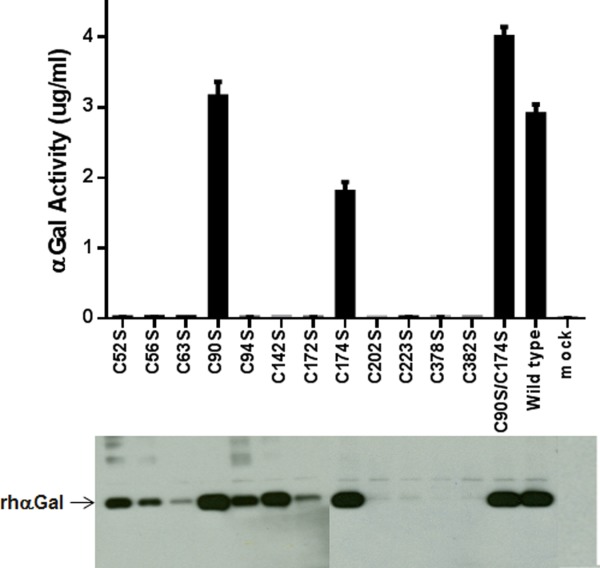
Western blot and activity assay of Cys-to-Ser mutants at all Cys positions. Upper panel: 4-MU assay of αGal activity in conditioned media. Media from mock transfection was included as a control. Bottom panel: Conditioned media expressing various mutants were run on 4–20% Tris Glycine gel and probed with a biotinylated polyclonal anti-αGal antibody.
Western blot analysis was used to study the protein expression level in the conditioned media. The result showed bands of varying intensities for the twelve single mutants and the double mutant. As expected from the activity assay, the C90S, C174S and the C90S/C174S mutant were expressed at a level similar to the wild type. Four of the ten inactive Cys-to-Ser mutants were found to have reduced expression level (C52S, C56S, C94S, and C142S). The other six (C63S, C172S, C202S, C223S, C378S, and C382S) showed little or no expression (Fig. 2). These results indicate that only the free cysteines, Cys90 and Cys174, can be mutated to Ser individually without completely abolishing structural/functional integrity.
The impact of mutagenesis on the expression levels of the two respective participating Cys in the same disulfide-bond pairs seems to vary. No expression was detected with any single mutants Cys202/Cys223 and Cys378/Cys382 pairs. C52S and C94S mutants were both expressed at a reduced level. Two single mutants in the same pair were expressed at different levels for the Cys56/Cys 63 and Cys142/Cys172 pairs. For example, C142S was expressed at a level similar to wild type, while C172S expression was much lower (Fig. 2).
Five double mutants were prepared, each simultaneously replacing two Cys involved in the same disulfide bond to explore the possible disulfide mismatch in single Cys-to-Ser mutants and, if so, whether the activity and expression could be restored by these double mutants. 4-MU activity assay of the conditioned media detected no activity in any double mutant, except for the C142S/C172S mutant where a low level (<5%) of the activity was observed compared to the wild type control. Therefore disulfide shuffling may not be the main cause for the loss of activity or expression by the single Cys mutants. Western blotting indicated that three of the five double mutants were expressed in the conditioned media: C52S/C94S, C142S/C172S, and C56S/C63S. No expression was detected in the conditioned media for the other two double mutants, C202S/C223S and C378S/C382S.
Structural and functional characterization of purified mutants
The wild type control and the three active free Cys mutants (C90S, C174S, and C90S/C174S) were produced with a scaled up transfection that yielded about 500 mL conditioned media. In addition, four of the inactive disulfide mutants that demonstrated low expression levels (C52S, C56S, C94S, and C142S) were also produced. A two-step purification process using a phenyl column followed by an ANX column was used. The three active mutants were purified close to homogeneity (≥95%, Fig. 3). However, attempts to purify the inactive mutants using the same procedure revealed mixed results. The purification was unsuccessful for three out of the four inactive mutants (C52S, C56S, and C94S). The reason for this failure is not clear but it may be caused by different protein-column matrix interaction by the three inactive mutants, possibly due to conformational changes or partial unfolding. Interestingly, the inactive C142S mutant could be purified with this purification method, although with a reduced purity (about 80%). An additional purification step using size exclusion chromatography (SEC) was performed to increase the purity of C142S to the same level as the other mutants (≥95%) for further characterization. The final purified mutants contained less than 2% aggregation by SEC-HPLC analysis (Fig. 3). The elution times of the mutants are consistent with a homo-dimeric protein.
Figure 3.
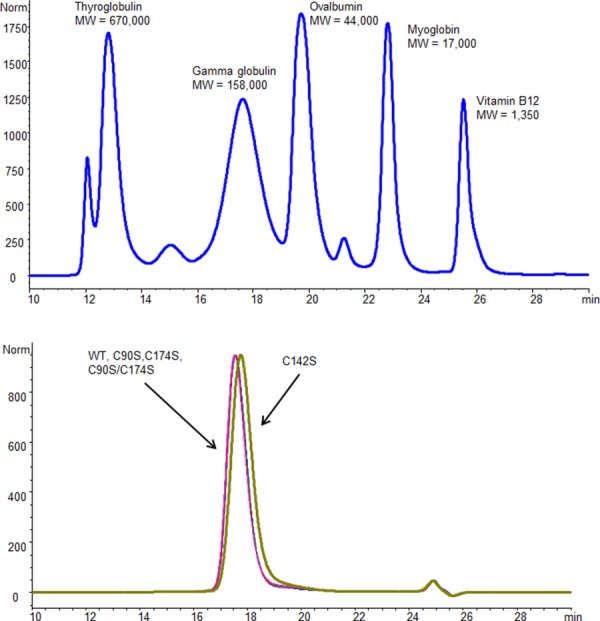
SEC-HPLC analysis of purified rhαGal mutants. The samples were analyzed on a G3000SWXL column for 35 min with a flow rate of 0.5 mL/min of phosphate buffered saline pH 7.0. Molecular weight standard mix was used to calibrate the molecular weight (up panel). The size of the wild type and purified mutants is consistent with that of a homodimeric rhαGal protein.
The 4-MU activity assay was performed on the purified proteins. The results (Table I) confirmed the data obtained using the conditioned media. The specific activities of the mutants replacing the two free Cys in rhαGal were comparable to or greater than the wild type. The activity of C90S, for example, was 19% greater than the wild type (2.19 vs. 1.84 units). As expected, the C142S mutant showed little (approximately 2%) activity compared to the wild type.
Table I.
Enzymatic Activity and Aggregation Characterization of Purified Mutants
| Sample | Specific activitya (mmol/hr/mg rhAGAL) | % of wild type | % Aggregationb |
|---|---|---|---|
| WT | 1.84 ± 0.15 | 100% | 1.2 |
| C90S | 2.19 ± 0.21 | 119% | 0.9 |
| C90A | 2.82 ± 0.21 | 153% | ND |
| C90T | 3.45 ± 0.31 | 187% | ND |
| C90V | 3.34 ± 0.15 | 181% | ND |
| C174S | 1.69 ± 0.13 | 92% | 1.1 |
| C90S/C174S | 1.81 ± 0.12 | 98% | 1 |
| C142S | 0.04 ± 0.01 | 2% | 0.8 |
Activity is reported as the mean ± the standard deviation.
Aggregation percentage was determined from SEC-HPLC.
ND = below detection.
Far-UV circular dichroism (far-UV CD) was used to probe protein secondary structure and characterize the purified mutants. The results [Fig. 4(A)] indicate similar spectra for the wild type rhαGal and the three active Cys-to-Ser mutants (C90S, C174S, and C90S/C174S), suggesting comparable secondary structures. The purified inactive mutant (C142S) appears to have reduced secondary structure content, as indicated by the reduced negative ellipticity in the 208 nm and 222 nm spectral bands that are attributed mainly to α-helix.20
Figure 4.
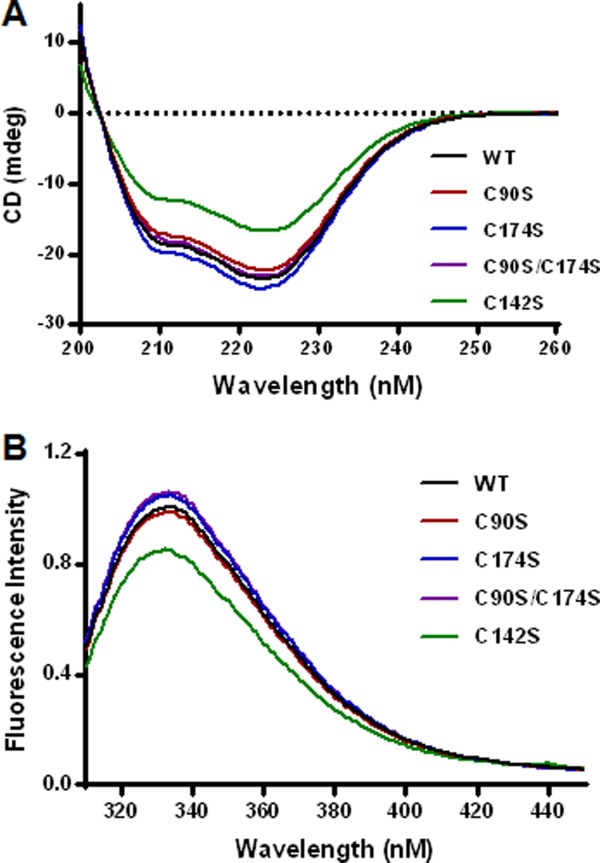
Circular Dichroism (A) and fluorescence analysis (B) of purified rhαGal mutants. The reduced negative ellipticity in the 208 nm and 222 nm spectral bands was used to assess loss of secondary structure attributed mainly to α-helix. The intensity of the fluorescence emission spectra of the mutants were examined as an indication of the tertiary structure perturbation involving changes in the local environment of tryptophan residues.
The fluorescence emission spectra of the mutants were examined as an indication of the tertiary structure perturbation involving changes in the local environment of tryptophan residues. Emission spectra in the range 310–450 nm, excited at 295 nm, were collected and are indicated in Figure 4(B). The results are consistent with those of far-UV CD in that the three active Cys mutants demonstrate comparable spectra to the wild type control, whereas the inactive C142S mutant demonstrates significant reduction in peak fluorescence intensity.
Intrinsic fluorescence-based thermal denaturation was performed to study the impact of Cys mutants on the thermal stability of rhαGal. The results (Fig. 5) indicate reduced apparent melting temperature compared to the wild type control for substitutions at the free Cys residues (Cys90 and Cys174) and more so at the disulfide-bonding Cys142. The thermal stability can be ranked in the following order: Wild type > C174S > C90S > C90S/C174S ≫ C142S.
Figure 5.
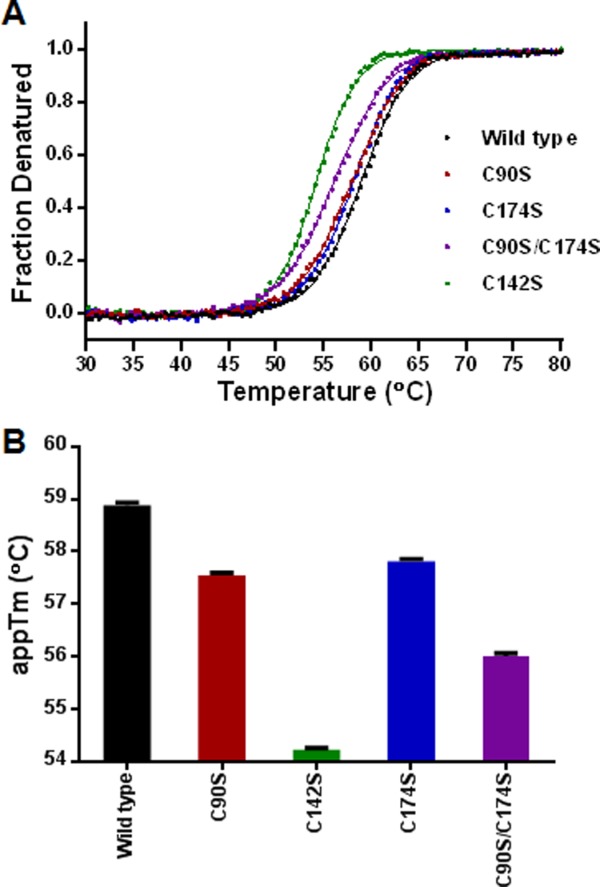
Thermal denaturation analysis of the wild type and mutant rhαGal. (A) Fluorescence-based thermal denaturation was used to study the thermal stability by measuring the intrinsic tryptophan fluorescence emission scans at excitation wavelength of 295 nm. The temperature range of 25–95°C was used. (B) The apparent melting temperature of wild type and mutant proteins were plotted for comparisons.
Saturation mutagenesis at Cys90 and Cys174
One of the Cys-to-Ser mutants, C90S, was found to have 19% higher activity compared to wild type (Table I). Since Ser substitution at positions 90 and 174 induced only minor perturbation to rhαGal structure and stability, we hypothesized that these sites may be flexible enough to accommodate non-conservative amino acid substitutions. Saturation mutagenesis was performed to replace each of the two free Cys with the nineteen other possible amino acids to determine if positive modulators of rhαGal function could be identified. The enzymatic activity in the conditioned media was tested and protein expression was determined by Western blot (Fig. 6). For Cys90 substitutions, 17 of the 19 mutants (with the exception of C90P and Cys90G) were expressed at various levels. Several mutants seem to have higher activity than the wild-type. The activity in the media could be ranked as C90T > C90A ≅ C90S > C90V > wild type. Other mutants were either inactive (C90W, C90K, C90Y, C90F, and C90R), or had significantly reduced activity, below 30% of wild type (C90E, C90M, C90Q, C90D, C90H, C90N, C90I, and C90L). The four Cys90 mutants with enhanced activity were purified, evaluated again with the 4-MU activity assay and compared with wild type (Table I and Fig. 7). They all demonstrated significantly higher enzymatic activity, with C90T having the highest activity, about 187% compared to wild type.
Figure 6.
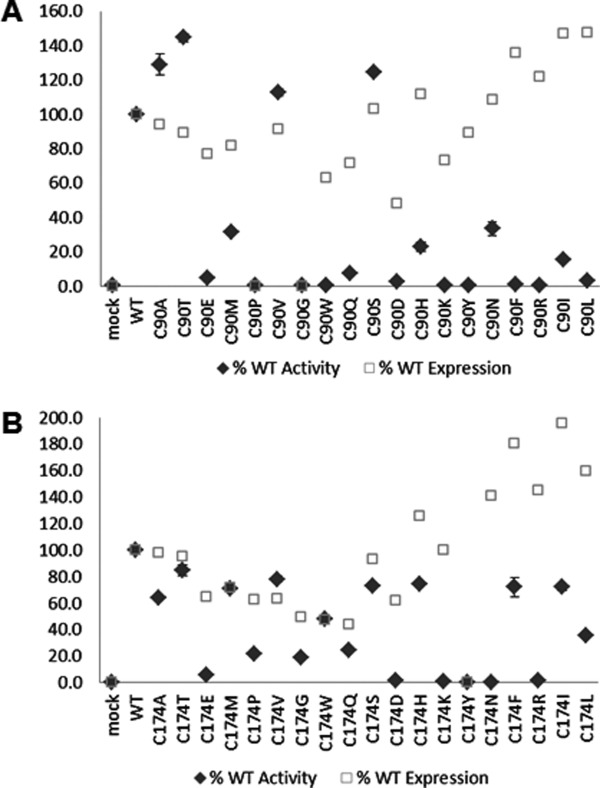
Expression and activity analysis of saturation mutants made at the two free Cys positions. Conditioned media were analyzed by Western blot (open square) and 4-MU activity assay (filled diamond) for Cys90 mutants (Panel A) and Cys174 mutants (Panel B). Error bars represent standard errors from three assays.
Figure 7.
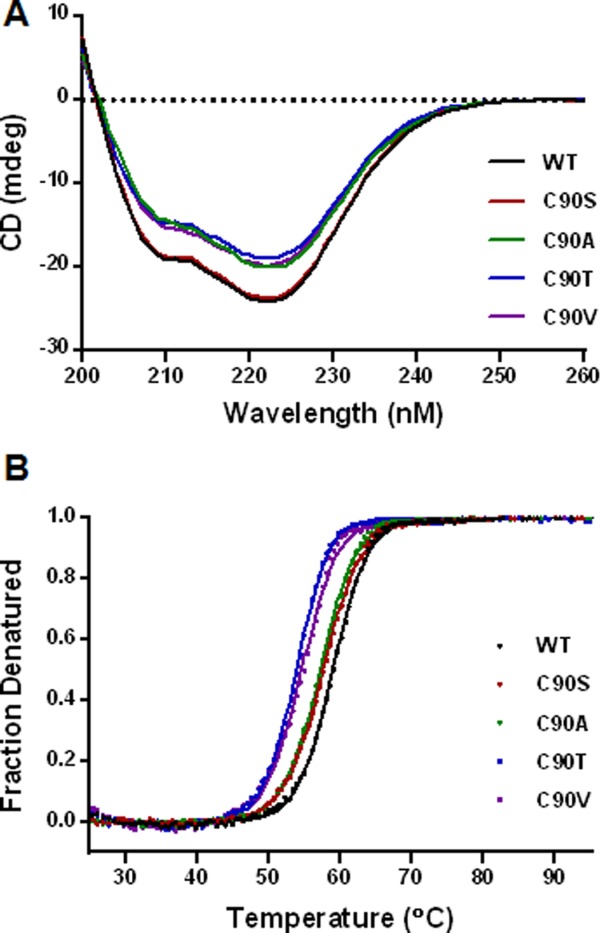
Far-UV CD analysis (A) and thermal denaturation study (B) of Cys90 mutants. The reduced negative ellipticity was used to assess loss of secondary structures. Fluorescence-based thermal denaturation was performed with samples diluted to 0.1 mg/mL in 50 mM sodium phosphate, pH 7.0. Fluorescence intensity data were acquired as the temperature increase from 25°C to 95°C to determine fractions denatured and apparent melting temperatures.
Most of the Cys174 mutants were expressed in conditioned media but all of them demonstrated lower enzymatic activity compared to wild type [Fig. 6(B)]. Two mutants (C174D and C174Y) had very low expression. Several mutants were expressed well but had no or very low enzymatic activity (C174E, C174P, C174K, C174N, and C174R).
Discussion
Cysteine has unique chemical properties due to its side chain thiol group and can participate in a variety of functional roles such as disulfide bonding, hydrogen bonding and redox regulation.15 While disulfide bonds play important roles for protein structure and stability, functional roles of free cysteines often depend on their positions and local environment in the protein, and whether it is interior or solvent exposed.17 We performed a systematic study in this work by replacing each Cys in the rhαGal protein, including the two free Cys, to examine their impact on protein expression, structure, activity and stability.
Replacing each of the ten disulfide bonding Cys in rhαGal with a Ser resulted in a loss of enzymatic activity in the conditioned media. In most cases this is due to absence of proteins in the conditioned media as a result of protein misfolding (Fig. 2). This is consistent with the important roles disulfide bonds play in protein folding. The result from double mutants of the same disulfide-bond pair suggests that replacing any Cys involved in disulfide bonding will inactivate the rhαGal, and it is not due to disulfide shuffling. The effect on expression also seems to depend on the positions of the cysteine. Mutants at some positions are expressed, while others completely abolished protein expression. There is also the formal possibility that some mutants are expressed but not soluble due to misfolding. We did see some mutant protein present in the detergent solubilized cell lysate by Western analysis. However, the relative signal intensities were similar in general to the level detected in conditioned media.
A few inactive mutants did express at high levels but they could not be purified using the same chromatographic procedures, likely as a result of altered protein folding and conformation. The only mutant that was adequately purified (C142S) was confirmed to be inactive and found to be conformationally perturbed with reduced thermostability (Figs. 2, 4, and 5). These results suggest that the disulfide bonds are critical for rhαGal folding and support the conclusion based on structural studies that Fabry disease is primarily a disease of protein folding.5,7 This group of inactive mutants can be potentially followed up with affinity purification such as His-tagged rhαGal expression vector as reported by Guce et al.4
Replacing the two free cysteines in rhαGal (Cys90 and Cys174) with the chemically similar serine, either separately or simultaneously, does not substantially influence the expression, activity, or structure of the protein (Figs. 2, 4, and 5). The only detectable difference observed was a small decrease (1–2 degree) in the apparent melting temperature measured by intrinsic tryptophan fluorescence-detected thermal denaturation. These results suggest that these two free Cys only play relatively minor structural roles within the protein, which has been found to be insensitive to conservative substitutions such as Ser.
We did observe major changes in activity and expression when these two Cys were mutated to other amino acids by site-saturation mutagenesis. Cys90 appears to tolerate only a few conservative mutations with small side chains, namely C90S, C90A, C90T and C90V [Fig. 6(A)]. This probably can be explained by the location of this free Cys, completely buried in the interior of the protein surrounded by relatively large hydrophobic and aromatic residues L45, Y88, L166, and F295, and capped by K168 [Fig. 8(A)]. Small polar residues, similar to Cys, such as Ser and Thr [Figs. 8(B,C)], or small hydrophobic residues Ala and Val [Figs. 8(D,E)], seem to be well tolerated at this position in structure modeling. Bulky and charged side chain substitutions, such as Tyr [Fig. 8(F)] at this Cys residue would either create steric clashes or disrupt the local hydrophobic environment, and destabilize the three-dimensional structure of αGAL. Indeed, even the conservative C90S mutation caused minor loss of thermostability (Figs. 5 and 7). This is consistent with studies by Lee and Blaber on a buried and conserved Cys in the fibroblast growth factor family (FGF).16,21 They found that, even though modeling studies suggested that conservative residues Ala, Ser, Thr, and Val are acceptable mutations at the FGF Cys83 site, none of the amino acids in this set can substitute for Cys without incurring a stability penalty. Interestingly, the C90S, C90A, and C90T rhαGal mutants all resulted in a higher enzymatic activity [Fig. 6(A) and Table I]. One explanation could be that the subtle changes to the interior non-covalent interactions introduced by these mutations leads to enhanced conformational flexibility and concomitant increase in substrate binding or catalytic activity at the expense of stability. All in all Cys 90 mutant behavior is consistent with the structural environment of Cys90.
Figure 8.
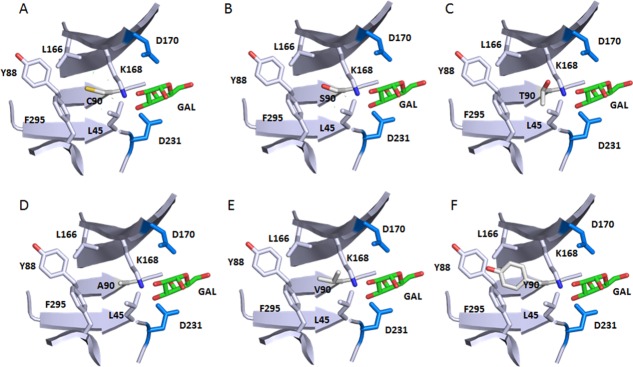
Local environment of Cys90 and the effect of selective mutations. Only part of the backbone are shown for simplifications. The atom coloring scheme is same as in Figure 1 except for what is specified for each panel. Catalytic residues D170 and D231 and bound product GAL are shown too. Structure is from 3HG5.4 Residues surrounding residues of C90 form a hydrophobic pocket. (A) WT Cys90 sulfhydryl sulfur is shown in yellow. (B) C90S mutation with the Serine hydroxyl oxygen in red. (C) C90T mutation with the Threonine hydroxyl oxygen in red. (D) C90A. (E) C90V. (F) C90Y with the Tyrosine hydroxyl oxygen in red. C90Y clashes with nearby Y88.
The expression and activity results from substituting the other free Cys in rhαGal, Cys174, is rather interesting and unexpected for some mutants. Although Cys174 mutants were expressed in the media (except C174Y), they all had lower enzymatic activities than the wild type control. Several mutants were found to have undetectable activity (C174K, C174Y, and C174N). The crystal structure of rhαGal indicates that Cys174 is at the surface of the protein with the side chain sulfhydryl partially buried and in close proximity with Leu180 of the neighboring α-helix and Val137 [Fig. 8(B)]. The loss of activity in the Cys174 mutants suggests that the free sulfhydryl may be involved in non-covalent interactions within the protein to stabilize its conformation. The loss of activity in those mutants could be caused by the local structural perturbation as a result of Cys substitution. Cys174 is only one residue away from the disulfide pair Cys142-Cys172, which in turn constitute part of the active site, while the Cα of Cys174 is 10 Å away from the catalytic residue Asp170 side chain carbonyl. Therefore, one can easily imagine that introducing a large polar or charged residue at this position could push it away from Leu180 and in turn alter the position of the Cys142-Cys172 disulfide pair, affecting the active site [Fig. 9(A)]. This explains why small residues such as Thr and Val substitution could still retain approximately 75% of the WT activity [Figs. 6(B) and 9(B,C)], but mutations with large polar [Tyr, Fig. 9(D)], charged [Lys, Fig. 9(E)] side chains, lead to lose of activity or even expression.
Figure 9.

Local environment of Cys174 and the effect of selective mutations. Only part of the backbone are shown for simplifications. The atom coloring scheme is same as in Figure 1 except for what is specified for each panel. Catalytic residues D170 and D231 and bound product GAL are shown too. The neighboring C142/C172 disulfide is also shown. Structure is from 3HG5.4 (A) WT Cys174 sulfhydryl sulfur in yellow. (B) C174T mutation with the Threonine hydroxyl oxygen in red. (C) C174V. (D) C174Y with the Tyrosine hydroxyl oxygen in red. C174Y would not fit in this position. (E) C174K with the Lysine amine nitrogen in blue. C174K clashes with the neighboring α-helix. (F) C174N with the amide oxygen in red, and nitrogen in blue. C174N can be potentially glycosylated as it introduces a consensus glycosylation NXS/T sequence and clashes with the neighboring α-helix.
Our experimental results corroborate very well with the human genome mutation database (HGMD). In the HGMD, all ten cysteines in disulfide bonds are found as point mutations in Fabry disease, and there is only a single instance of mutation (C174R) in two unpaired cysteines leading to Fabry disease. This strengthens our result that the C174R variant has no enzyme activity and thus would lead to Fabry disease. It is interesting to note that the R118C variant, recurrently described in large Fabry disease screening studies of high-risk patients based on the theoretical arguments about the chemistry of the cysteine residue, has been recently demonstrated to not cause a Fabry disease phenotype.22
In summary, the ten Cys in the disulfide bonds are critical in maintaining the proper folding of the protein required for catalytic activity. Replacing any of the ten Cys leads to protein misfolding and loss of enzymatic activity. The two free Cys are also essential for rhaGal expression and activity. However, these two residues, particularly Cys at position 90 are more permissive to chemical change. Amino acid substitution can even enhance enzymatic activity by almost 2-fold. These results enhanced our understanding of rhαGal structure and function, particularly the roles the cysteines play and the impact when replaced with other amino acids.
Materials and Methods
Materials
Gateway cloning reagents and transfection reagents were obtained from Life Technologies (Grand Island, NY). HiTrap Phenyl FF (hi sub) and HiTrap ANX FF (hi sub) columns were purchased from GE Healthcare (Piscataway, NJ). Other reagents were purchased from Sigma (St Louis, Missouri) unless otherwise stated.
Generation of rhαGal Cys to Ser mutants
DNAs encoding hαGal genes (including its signal peptides) were synthesized and cloned into a pENTR221 Gateway entry vector (from GeneArt/Life Technologies). Oligonucleotide primers containing single Cys to Ser mutants were used to introduce Cys mutations via QuikChange Multi Lightning kit (Agilent). The resulting wild type and mutant vectors were put into expression vectors pCEP4(-E+I)Dest via Gateway cloning, and transiently expressed using HEK293 adhesion or Expi293F suspension cells obtained from Life Technologies.
Saturation mutagenesis at selected cysteine residues
Cys90 and Cys174 were mutated to 19 other amino acids by saturation mutagenesis. NNK degenerate primers (N=A, G, C, or T; K=G or T) were synthesised and used as the PCR primer for site-directed mutagenesis using QuikChange Lightning Multi Site-Directed Mutagenesis kit. Plasmid DNAs from 96 colonies were prepared with QIAprep 96 Turbo Miniprep kit (Qiagen) and analyzed by DNA sequencing. A few point mutations missing after this degenerate mutagenesis procedure were introduced with individual mutagenic primers using QuikChange Lightning Site-Directed Mutagenesis the same kit to get a complete set of 19 amino acids. The mutants were subcloned into pCEP4(-E+I)Dest expression vector by Gateway cloning, and transiently expressed using HEK293 adhesion or Expi293F suspension cells obtained from Life Technologies.
Purification of rhαGal Cys mutants
The conditioned media obtained from transient transfection was used to purify the wild type and mutant rhαGal protein. Ammonium sulfate was added to the media to a final concentration of 50 mM before loading to a 5 mL HiTrap Phenyl FF (hi sub) column equilibrated with 25 mM sodium phosphate, 50 mM ammonium sulfate pH 7.0. The column was washed with 25 mM sodium phosphate, 50 mM ammonium sulfate pH 7.0 for 5 column volumes before eluting with 5 column volumes of 25 mM sodium phosphate, 50% ethylene glycol (w/v), pH 6.5. The pH of the phenyl eluate was adjusted to 7.0 using 0.3M sodium phosphate dibasic, and then loaded to a 5 mL HiTrap ANX (hi sub) column pre-equilibrated with 2 column volumes of 200 mM sodium phosphate pH 7.0 and four column volumes of 25 mM sodium phosphate pH 7.0. The ANX column was washed with four column volumes of 25 mM sodium phosphate, pH 7.0, and eluted with five column volumes of 10 mM sodium phosphate, 200 mM sodium chloride, pH 7.0. An additional SEC-HPLC step was used to further purify selected mutants on a Superdex 200 10/300GL column (GE Healthcare), and with a flow rate of 0.4 mL/min of phosphate buffered saline pH 7.0 for 60 min.
SDS-PAGE and Western blot
Novex 4−20% Tris Glycine gel (Life Technologies, NY) was used for electrophoretic analysis with Tris glycine SDS running buffer (Life Technologies, NY), and stained with coomassie blue. Conditioned media (10 µL) was subjected to SDS PAGE, transferred to PVDF membranes using the iBlot Dry Blotting System (Life Technologies). Immunoblots were probed with 1:1000 dilution of biotinylated polyclonal anti- hαGal antibody overnight at 4°C. After washing the blots were incubated with a 1:2000 dilution of streptavidin HRP and then developed with enhanced chemiluminescent reagents and imaged on a Kodak Image Station.
HPLC purity analysis
Purified rhαGal Cys mutants were analyzed on a YMC Octyl C8 column (2.0 mm × 100 mm × 5 µm, 300Å, Waters, MA) at 215 nm using an Agilent 1200 HPLC system with a multiple wavelength detector (Agilent Technologies, CA). The column was equilibrated with mobile phase A (0.1% w/v TFA in water) with 5% mobile phase B (0.084% (w/v) TFA in Acetonitrile). The sample was eluted using a linear gradient starting with 5% mobile phase B (0.084% (w/v) TFA in Acetonitrile) up to 95% mobile phase B over 61 minutes at 0.25 mL/min. Mutants were also analyzed by SEC-HPLC on a G3000SWXL column (7.8 mm × 30 cm × 5 µm, Tosoh, PA) with a TSK-Gel Guard column with a flow rate of 0.5 mL/min of phosphate buffered saline pH 7.0 for 35 min.
Far-UV CD
Samples were diluted to 0.2 mg/mL in 50 mM sodium phosphate, pH 7.0 and concentrations were determined by spectrophotometry (E280 = 2.54 L g−1 cm−1). CD data were collected on a Jasco J-810 spectropolarimeter using a 1-mm pathlength cuvette. All spectra were acquired at 25°C over the wavelength range of 195–260 nm. Buffer blank spectra were subtracted from raw sample spectra and the signal units in mdeg were converted to elipticity units in deg cm2 dmol−1 using the measured concentrations and mean residue weight of 114.2.
Trp fluorescence emission scan
Samples were diluted to 0.1 mg/mL in 50 mM sodium phosphate, pH 7.0, and loaded into 1.0 cm quartz fluorescence cuvettes. A Jasco J-815 spectropolarimeter with 90° fluorescence module was used for analysis. Samples were held at 25°C for 10 min and then intrinsic tryptophan fluorescence emission scans were acquired at excitation = 295 nm, and emission range 310–450 nm. Raw emission spectra were compared qualitatively to assess comparability.
Thermal denaturation
Fluorescence-based thermal denaturation was performed using a Jasco J-815 spectropolarimeter with 90° fluorescence module. The samples were diluted to 0.1 mg/mL in 50 mM sodium phosphate, pH 7.0 and loaded into a 1.0 cm pathlength quartz fluorescence cuvette. Samples were held at 25°C for 10 min and the temperature was increased to 95°C at a rate of 6°C/minute, during which fluorescence intensity data were acquired at excitation and emission wavelengths of 295 nm and 332 nm, respectively. Apparent melting temperatures (appTm) were determined by nonlinear least squares fitting of the denaturation profiles to a model-free Boltzmann sigmoidal function with fitted pre- and post-transition baselines:
Where the observed signal (YOBS) is given as a function of the pre- and post-transition baselines (YPRE and YPOST) assuming linearity with temperature (Yi = mi × T + yi), ΔT is the difference between the apparent transition midpoint temperature and experimental temperature at each coordinate (appTm–T), and k is an arbitrary transition shape factor. Raw profiles were converted to fraction denatured profiles using the fitted parameters to facilitate visual comparison.
4-MU activity assay
Purified rhαGal Cys mutants were diluted in assay buffer (27 mM citric acid, 46 mM sodium phosphate dibasic, 0.5% BSA pH4.4). Diluted samples (20 µL) were added to 100 µL of working substrate (27 mM citric acid, 46 mM sodium phosphate dibasic, 0.5% BSA, 12 mM 4-Methylumbelliferyl α-D-galactopyranoside, N-acetyl-D-galactosamine, pH4.4) and incubated for 20 minutes at 37°C. Reactions were stopped by the addition of 0.5M Glycine, pH10.8. Fluorescence intensity was read at 465nm with an excitation at 365 nm on a Molecular Devices plate reader. A 4-methylumbellifone (4-MU) standard curve ranging from 0.03 nm to 4 nm was run with each assay occasion. Activity for the purified samples is expressed as mean ± the standard deviation of three to four separate assay occasions.
Acknowledgments
The authors thank Lihui Hou for assistance in purification of some rhαGal mutants, Julie Bird for her help in initial protein quantitation using Octet, Lee Sherman for discussions on protein purification methods, Bernard Benichou and Karen Mittleman for critical reading of the manuscript. All authors received personal compensation as employees of Sanofi at the time the work described in this publication was performed.
Glossary
- 4-MU
4-methylumbellifone
- CD
circular dichroism
- FGF
fibroblast growth factor family
- HGMD
human genome mutation database
- MES
2-(N-morpholino) ethanesulfonic acid
- PBS
phosphate buffered saline
- rhαGal
recombinant human α-galactosidase A
- SEC
size exclusion chromatography
- SEC
size exclusion chromatography.
References
- 1.Mahmud HM. Fabry's disease—a comprehensive review on pathogenesis, diagnosis and treatment. J Pakistan Med Assoc. 2014;64:189–194. [PubMed] [Google Scholar]
- 2.Lee K, Jin X, Zhang K, Copertino L, Andrews L, Baker-Malcolm J, Geagan L, Qiu H, Seiger K, Barngrover D, McPherson JM, Edmunds T. A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. Glycobiology. 2003;13:305–313. doi: 10.1093/glycob/cwg034. [DOI] [PubMed] [Google Scholar]
- 3.Lieberman RL, D'Aquino JA, Ringe D, Petsko GA. Effects of pH and iminosugar pharmacological chaperones on lysosomal glycosidase structure and stability. Biochemistry. 2009;48:4816–4827. doi: 10.1021/bi9002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guce AI, Clark NE, Salgado EN, Ivanen DR, Kulminskaya AA, Brumer H, Garman SC., 3rd Catalytic mechanism of human alpha-galactosidase. J Biol Chem. 2010;285:3625–3632. doi: 10.1074/jbc.M109.060145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human alpha-galactosidase. J Mol Biol. 2004;337:319–335. doi: 10.1016/j.jmb.2004.01.035. [DOI] [PubMed] [Google Scholar]
- 6.Shabbeer J, Yasuda M, Benson SD, Desnick RJ. Fabry disease: identification of 50 novel alpha-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Hum Genomics. 2006;2:297–309. doi: 10.1186/1479-7364-2-5-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garman SC. Structure-function relationships in alpha-galactosidase A. Acta Paediatr (Oslo, Norway: 1992) 2007;96:6–16. doi: 10.1111/j.1651-2227.2007.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuzawa F, Aikawa S, Doi H, Okumiya T, Sakuraba H. Fabry disease: correlation between structural changes in alpha-galactosidase, and clinical and biochemical phenotypes. Hum Genet. 2005;117:317–328. doi: 10.1007/s00439-005-1300-5. [DOI] [PubMed] [Google Scholar]
- 9.Ebrahim HY, Baker RJ, Mehta AB, Hughes DA. Functional analysis of variant lysosomal acid glycosidases of Anderson-Fabry and Pompe disease in a human embryonic kidney epithelial cell line (HEK 293 T) J Inherit Metabol Dis. 2012;35:325–334. doi: 10.1007/s10545-011-9395-4. [DOI] [PubMed] [Google Scholar]
- 10.Garman SC, Hannick L, Zhu A, Garboczi DN. The 1.9 A structure of alpha-N-acetylgalactosaminidase: molecular basis of glycosidase deficiency diseases. Structure. 2002;10:425–434. doi: 10.1016/s0969-2126(02)00726-8. [DOI] [PubMed] [Google Scholar]
- 11.Ashton-Prolla P, Tong B, Shabbeer J, Astrin KH, Eng CM, Desnick RJ. Fabry disease: twenty-two novel mutations in the alpha-galactosidase A gene and genotype/phenotype correlations in severely and mildly affected hemizygotes and heterozygotes. J Invest Med. 2000;48:227–235. [PubMed] [Google Scholar]
- 12.Filoni C, Caciotti A, Carraresi L, Cavicchi C, Parini R, Antuzzi D, Zampetti A, Feriozzi S, Poisetti P, Garman SC, Guerrini R, Zammarchi E, Donati MA, Morrone A. Functional studies of new GLA gene mutations leading to conformational Fabry disease. Biochim Biophys Acta. 2010;1802:247–252. doi: 10.1016/j.bbadis.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yasuda M, Shabbeer J, Benson SD, Maire I, Burnett RM, Desnick RJ. Fabry disease: characterization of alpha-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum Mutat. 2003;22:486–492. doi: 10.1002/humu.10275. [DOI] [PubMed] [Google Scholar]
- 14.Meng Y, Zhang WM, Shi HP, Wei M, Huang SZ. Clinical manifestations and mutation study in 16 Chinese patients with Fabry disease. Zhonghua yi xue za zhi. 2010;90:551–554. [PubMed] [Google Scholar]
- 15.Fomenko DE, Marino SM, Gladyshev VN. Functional diversity of cysteine residues in proteins and unique features of catalytic redox-active cysteines in thiol oxidoreductases. Mol Cells. 2008;26:228–235. [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J, Blaber M. Structural basis of conserved cysteine in the fibroblast growth factor family: evidence for a vestigial half-cystine. J Mol Biol. 2009;393:128–139. doi: 10.1016/j.jmb.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 17.Nakaniwa T, Fukada H, Inoue T, Gouda M, Nakai R, Kirii Y, Adachi M, Tamada T, Segawa S, Kuroki R, Tada T, Kinoshita T. Seven cysteine-deficient mutants depict the interplay between thermal and chemical stabilities of individual cysteine residues in mitogen-activated protein kinase c-Jun N-terminal kinase 1. Biochemistry. 2012;51:8410–8421. doi: 10.1021/bi300918w. [DOI] [PubMed] [Google Scholar]
- 18.Moharram R, Maynard D, Wang ES, Makusky A, Murray GJ, Martin BM. Reexamination of the cysteine residues in glucocerebrosidase. FEBS Lett. 2006;580:3391–3394. doi: 10.1016/j.febslet.2006.04.096. [DOI] [PubMed] [Google Scholar]
- 19.Qiu H, Edmunds T, Baker-Malcolm J, Karey KP, Estes S, Schwarz C, Hughes H, Van Patten SM. Activation of human acid sphingomyelinase through modification or deletion of C-terminal cysteine. J Biol Chem. 2003;278:32744–32752. doi: 10.1074/jbc.M303022200. [DOI] [PubMed] [Google Scholar]
- 20.Pelton JT, McLean LR. Spectroscopic methods for analysis of protein secondary structure. Anal Biochem. 2000;277:167–176. doi: 10.1006/abio.1999.4320. [DOI] [PubMed] [Google Scholar]
- 21.Lee J, Blaber M. The interaction between thermodynamic stability and buried free cysteines in regulating the functional half-life of fibroblast growth factor-1. J Mol Biol. 2009;393:113–127. doi: 10.1016/j.jmb.2009.08.026. [DOI] [PubMed] [Google Scholar]
- 22.Ferreira S, Ortiz A, Germain DP, Viana-Baptista M, Caldeira-Gomes A, Camprecios M, Fenollar-Cortes M, Gallegos-Villalobos A, Garcia D, Garcia-Robles JA, Egido J, Gutierrez-Rivas E, Herrero JA, Mas S, Oancea R, Peres P, Salazar-Martin LM, Solera-Garcia J, Alves H, Garman SC, Oliveira JP. The alpha-galactosidase A p.Arg118Cys variant does not cause a Fabry disease phenotype: data from individual patients and family studies. Mol Genet Metab. 2015;114:248–258. doi: 10.1016/j.ymgme.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]