

Abstract
The development of accurate protein function annotation methods has emerged as a major unsolved biological problem. Protein similarity networks, one approach to function annotation via annotation transfer, group proteins into similarity-based clusters. An underlying assumption is that the edge metric used to identify such clusters correlates with functional information. In this contribution, this assumption is evaluated by observing topologies in similarity networks using three different edge metrics: sequence (BLAST), structure (TM-Align), and active site similarity (active site profiling, implemented in DASP). Network topologies for four well-studied protein superfamilies (enolase, peroxiredoxin (Prx), glutathione transferase (GST), and crotonase) were compared with curated functional hierarchies and structure. As expected, network topology differs, depending on edge metric; comparison of topologies provides valuable information on structure/function relationships. Subnetworks based on active site similarity correlate with known functional hierarchies at a single edge threshold more often than sequence- or structure-based networks. Sequence- and structure-based networks are useful for identifying sequence and domain similarities and differences; therefore, it is important to consider the clustering goal before deciding appropriate edge metric. Further, conserved active site residues identified in enolase and GST active site subnetworks correspond with published functionally important residues. Extension of this analysis yields predictions of functionally determinant residues for GST subgroups. These results support the hypothesis that active site similarity-based networks reveal clusters that share functional details and lay the foundation for capturing functionally relevant hierarchies using an approach that is both automatable and can deliver greater precision in function annotation than current similarity-based methods.
Keywords: active site profiling, similarity-based clustering, network-based clustering, protein similarity network analysis, Structure-Function Linkage Database (SFLD), protein function annotation, function annotation transfer
Introduction
As high-throughput sequencing has become faster and easier to accomplish, protein sequences have accumulated at an astounding rate.1 Structure characterization methods lag far behind the efficiency of sequencing, but even structure determination lies well ahead of the speed and cost of experimental function characterization. A key to effective utilization of the massive sequence databases is understanding the function of the encoded proteins; however, in 2005 it was estimated that less than 5% of functions had been experimentally determined.2 As the rate of sequencing continues to increase, this issue becomes more acute as function annotation is essential to understanding the underlying biology. The need for accurate protein function annotation using automated approaches has become critical, as large-scale experimental function determination is infeasible at the level of detail useful to understanding biological mechanism.
Of major concern is that automated approaches to functional annotation are prone to misannotation,3,4 sometimes at “alarmingly high levels.”5 A major contributor is “over-annotation,” or assigning functional detail to a protein without sufficient supporting evidence. Often, over-annotation is due to function annotation transfer from a known protein to one of unknown function based on pairwise sequence similarity comparisons. As pairwise comparisons are typically based on overall sequence similarity without regard to residue motifs that may distinguish their different reaction specificities, the predicted annotation lacks the informative larger context obtained from placing the unknown sequence in a multiple sequence alignment or phylogenetic tree. To identify molecular functional details that better distinguish specific functions among distantly related proteins, it is essential to move beyond simple comparisons of full-length sequences,6,7 but execution remains a significant challenge with the speed and scale required.
“Protein similarity networks” [conceptually illustrated in Fig. 1(A)] have recently been used to study and visualize sequence, structure, and function relationships on a large-scale.8–11 Most typically used with sequence data,10,12–16 these networks can be associated with many different types of functional information for interactive exploration using programs such as Cytoscape.17 As they can realistically handle many thousands of homologous proteins (or hundreds of structures), similarity networks offer a powerfully useful context by which functional properties of uncharacterized proteins can be inferred or hypothesized from those of experimentally characterized proteins with which they cluster. One starts with a fully connected network, in which each edge represents a quantitative pairwise comparison between each pair of nodes (proteins) in the network. As the threshold of the edge metric is increased [illustrated in Fig. 1(A,B)] edges that fall below the threshold are removed, resulting in smaller clusters whose nodes share more similarity among themselves than with other clusters or singleton nodes.
Figure 1.

Conceptual illustration of network-based clustering, which, ideally, would produce a functional hierarchy matching the SFLD. A. A similarity network is composed of nodes (proteins) connected to one another with edges (pairwise similarity scores). As the edge threshold is increased, all scores below that threshold are removed, producing distinct clusters. B. An actual similarity network hierarchy for the enolase superfamily. Each protein structure is represented by a node; pairwise ASP scores are represented by edges. This network is clustered into groups roughly mimicking subgroup annotations (border color and node shape) at a threshold of 0.31, but begins to break into specific families (fill color) and smaller groups at a threshold of 0.6. C. The SFLD defines a functional hierarchy, with superfamilies defined as sets of proteins which share a mechanistic step and the most detailed level, families, defined as sets of proteins which share entire mechanisms. Subgroups are illustrated with border color and families are illustrated with fill color.
In this work, we have generated networks and clustered proteins using three different similarity features: sequence, three dimensional structure, and active site microenvironments. Accuracy of functionally relevant clustering was evaluated based on comparison to the expert manual curation provided by the Structure-Function Linkage Database (SFLD)18 for four different enzyme superfamilies (Table I) that exhibit a diversity of structural and functional relationships: enolase, peroxiredoxin (Prx), glutathione transferase (GST), and crotonase. SFLD curation has previously been used as a gold standard19 to evaluate methods for clustering sequence or structural data into functionally relevant groups.12,13,16,20,21
Table I.
Four Functionally Diverse Superfamilies were used to Create Protein Networks
| Superfamily | Subgroup (no. representative proteins) |
|---|---|
| Enolase (159) | Enolase (20), GalD (1), GlucD (16), MR (56), ManD (17), MAL (3), MLE (46) |
| Peroxiredoxin (Prx) (47) | AhpC-Prx1 (17), AhpE (1), BCP-PrxQ (6), Prx5 (7), Prx6 (3), Tpx (9), uncharacterized (4) |
| Glutathione Transferase (GST) (127) | AMPS (42), Main 1 (13), Main 2 (9), Main 3 (7), Main 4 (14), Main 5 (3), Main 6 (1), Main 7 (1), Main 8 (7), Main 9 (6), Main 10 (4), Main Remainder (11), Remainder (5), Xi (4) |
| Crotonase (88) | Crotonase Like (70), Retro-Claisenase-like (2), uncharacterized (16) |
ManD: mannonate dehydratase; GlucD: glucarate dehydratase; GalD: galactarate dehydratase; MAL: methylaspartate ammonia lyase; MR: mandelate racemase; MLE: muconate cycloisomerase.
The SFLD defines a hierarchical classification system of protein function in enzyme superfamilies.22 Each level within the hierarchy corresponds to a level of molecular functional detail [Fig. 1(C)].18 Protein sets are grouped most broadly into superfamilies, composed of proteins that share a common partial reaction step or other chemical capability, typically catalyzed by a core group of well-conserved key residues. Superfamilies are divided into subgroups, distinguished by sequence information and sometimes differences in domain structure or major inserts; proteins in each subgroup have more shared functional features than the superfamily as a whole. Finally, subgroups are divided into families, which are sets of proteins that perform the same chemical reaction with a similar mechanism. This hierarchical classification scheme defines molecular function at various levels of detail [Fig. 1(C)]. Comparison with the similarity networks presented here allows us to evaluate how network topology at each edge metric and threshold compares to levels of functional detail in this hierarchy [Fig. 1(B)].
Here, we compare networks created using three different edge metrics to each of the SFLD hierarchical levels of superfamily, subgroup, and family. Edge metrics evaluated are: full-length sequence similarity, overall structure similarity, and active site motif similarity (hereinafter called “full sequence,” “structure,” and “active site profiling (ASP),” respectively). To distinguish functionally relevant clusters for each superfamily, all-by-all pairwise comparisons of each feature type were generated and visualized as networks [illustrated for the enolases in Fig. 1(B)]. BLAST scores23 were used as the edge metric for full sequence-based networks, and TM-Align scores24 as the edge metric for structure-based networks. Both of these methods have previously been used as metrics for sequence25–27 and structural28–30 comparison, respectively. For active site comparison, we used active site profiling, a method previously developed to identify and quantitatively compare active site microenvironments.31 Active site features are captured from sequence fragments in the structural vicinity of the defined functional site; these fragments are then aligned into a continuous sequence called an active site signature (Fig. 2). Active site similarity between two proteins is quantified by calculating a pairwise active site profile (ASP) score which takes into account residue identity, strong similarity, weak similarity, and gaps in the alignment between two active site signatures.31 Using the pairwise ASP score between two signatures as the edge metric, these networks can be created in a manner analogous to those generated using full sequence or structure similarity scores.
Figure 2.
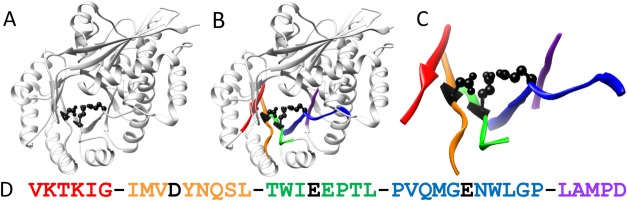
Representation of active site profiling to generate an active site signature. The protein structure is used to define the microenvironment surrounding an enzyme's active site. Multiple user-identified key residues (A) define the structural location of the active site. All residues within 10 Å of one of the key residues are considered to be part of the active site microenvironment (B). These residues are extracted from the protein sequence (C) and aligned N-C terminus to create the active site signature (D). Dashes and text color separate the non-contiguous fragments in this example (PDB: 1MDR).
The results indicate that networks based on active site microenvironment features often identify similarity groupings that are more consistent with known functional groups. Sequence- and structure-based networks identify overall sequence similarity or major structural rearrangements, respectively. They may define functional similarity at the superfamily and, sometimes, subgroup level, but they may lose the details of molecular function at the subgroup and family level. Active site comparisons are useful in capturing detailed functional differences. This work lays the foundation for identifying functionally relevant hierarchies of annotation detail on a large scale using an approach that can deliver greater precision than current sequence-based methods and that is amenable to automated application.
Results and Discussion
Similarities and differences in clustering are observed using sequence-based, structure-based, and signature-based networks
Three network series were created for proteins of known structure in each superfamily: enolase, peroxiredoxin (Prx), glutathione transferase (GST), and crotonase (Table I). Each series was created using a different edge metric—pairwise BLAST scores,23 TM-Align scores,24 or active site profiling (ASP) scores31—for the sequence-, structure-, and signature-based networks, respectively (see Methods). To create the fully-connected networks, proteins were compared in an all-by-all pairwise manner. Edges were filtered at increasingly stringent thresholds [Fig. 1(A)], eliminating the weakest relationships, forming clusters with relatively higher scoring edges and, thus, proteins more “closely related” (with “relationship” defined by the edge metric), as illustrated for the enolases [Fig. 1(B)]. Select examples from the network series for the enolase superfamily are shown in Figure 3. More complete network series for each superfamily are in Supporting Information Figures 1 to 4.
Figure 3.
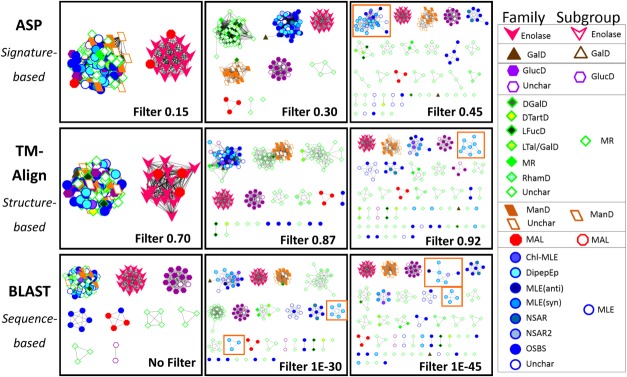
Representative signature-, structure-, and sequence-based network series for the enolase superfamily. The signature-, structure-, and sequence-based networks are shown at the top, middle, and bottom of the figure, respectively. From left to right, increasingly stringent edge thresholds for each network type, resulting in clusters that share more similarity (with similarity defined based on the edge metric). SFLD subgroup designations are indicated with both node shape and node border color, and the SFLD family designations are indicated with node fill color according to the legend. Filter levels (or edge thresholds) for each network were chosen in a qualitative fashion such that the number and size of protein clusters were relatively consistent between the three networks for a superfamily. More comprehensive networks for all four superfamilies are presented in Supporting Information Figures 1 to 4. The meaning of “no filter” for the initial sequence-based network is described in Supporting Information Figure 5.
Some subgroups cluster consistently across networks and score thresholds. For instance, the enolase subgroup of the enolase superfamily clusters together consistently by all three metrics, even at stringent edge thresholds (Fig. 3 and Supporting Information Fig. 1, pink vees). The GlucD subgroup is also clustered consistently (Fig. 3 and Supporting Information Fig. 1, purple hexagons).
Other subgroups and families show significant differences in clustering, even at the least stringent edge thresholds. The MLE subgroup (Fig. 3 and Supporting Information Fig. 1, blue outlined circles) is extremely difficult to annotate at a detailed level;32,33 network analysis shows clustering variation between the three edge metrics even at the least stringent edge thresholds (Fig. 3, left panels). At the most relaxed thresholds, the MLE subgroup clusters with the MR and ManD subgroups in the structure- and sequence-based networks, while ManD becomes its own cluster early in the signature-based network series (Supporting Information Fig. 1). Detailed observation of families within the MLE subgroup shows differing clustering patterns between the networks. For instance, the DipepEp family (Fig. 3 and Supporting Information Fig. 1, cyan blue circles) forms different clusters in each of the three networks at these more stringent thresholds (Fig. 3 and Supporting Information Fig. 1, orange boxes). In the signature-based networks, DipepEp proteins cluster together with several other families until an edge threshold of 0.65, at which time they break into two groups of three and several singlets. In structure-based networks, these proteins also cluster with other MLE families until an edge threshold of 0.92, at which point it breaks into a cluster of eight and multiple singlets; at the final threshold of 0.95, this family “disintegrates” into singlets and doublets. In the sequence-based networks, this family clusters with other MLE families, except at the relatively relaxed threshold of 1e-20 a separate triplet is formed. At a threshold of 1e-30, an additional cluster of five proteins split into their own group, while the remaining four proteins remain clustered with other proteins from other MLE families. Likewise, the enoyl CoA hydratase (subsequently referred to as enoyl) family of the crotonase superfamily forms different clusters in each network series (Supporting Information Fig. 4, royal blue circles), consistent with previous data suggesting that this family may be mechanistically diverse and difficult to annotate.34,35 In the structure- and sequence-based networks, an enoyl triplet forms at relatively relaxed thresholds of 0.50 and 1e-15, respectively (Supporting Information Fig. 4, red circles) while the remaining enoyl proteins cluster with many other families (Supporting Information Fig. 4, blue circles). Conversely, in the signature-based network, most of the enoyl proteins stay in one large cluster up through filter 0.45 (Supporting Information Fig. 4, black circle) before separating at filter 0.50. These three network series demonstrate that subgroups and families separate into smaller clusters at different thresholds depending on the edge metric used in creation of the network and how similar the subgroup or family is compared with other subgroups and families in the superfamily.
These specific examples prompted us to attempt to quantify the observations further. Thus, we counted the number of SFLD-defined subgroups (enolase, Prx, GST) or families (crotonase) that form distinct, all-inclusive clusters at each edge threshold for each of the three network types, and the threshold with the highest count was identified for each network (Supporting Information Figs. 1–4, blue stars). In two of the four superfamilies (enolase and crotonase), the ASP-based networks clustered more functional groups (subgroups and families, respectively) with 100% accuracy than either sequence- or structure-based networks. Five enolase subgroups are identified distinctly in the ASP-based networks at the best threshold, while only three are identified distinctly in both the sequence- and structure-based networks at the best threshold (Supporting Information Fig. 1, blue stars). For the crotonases, the ASP-based networks identified six families distinctly while the structure- and sequence-based networks identified four and five, respectively (Supporting Information Fig. 4, blue stars). For the Prx and GST superfamilies, the ASP-based networks and sequence-based networks each clustered four SFLD subgroups with 100% accuracy, while the structure-based network only identified either two (Prx) or three (GST) subgroups distinctly at the best threshold. It is important to note that while other thresholds could have been chosen for use in this analysis, we do not believe that would significantly affect the conclusions drawn. Therefore, we hypothesize that subnetworks identified by comparing features in the active site microenvironment, as done in the signature-based networks, can be used to identify molecular functional details more accurately than sequence- and structure-based comparisons. This hypothesis is explored in more detail in subsequent sections.
Signature-based networks suggest overall subgroup active site similarity not captured in sequence-based networks
A quantitative example demonstrates the specific details that distinguish sequence- and signature-based networks. In the enolase superfamily, the MR subgroup (green diamonds) clusters with the MLE subgroup (blue circles) in the signature-based networks [Fig. 4(A), green arrow], but clusters with the ManD subgroup (orange parallelograms) in the sequence-based networks [Fig. 4(A), blue arrow].
Figure 4.

Analysis of the ASPs identifies why MR, MLE, and ManD proteins cluster differently in signature- and sequence-based networks. A. In the signature-based network (A, left) the MR (green diamond) and MLE (blue circle) subgroups cluster together at the 0.25 edge threshold (green arrow) while the ManD subgroup (orange parallelogram) is a distinct subnetwork. Conversely, in the sequence-based network (A, right), the MR and ManD subgroups cluster together at the 1E-20 edge threshold (blue arrow) while MLE proteins fall in different subnetworks. B. Signature logos of the MR, MLE, and ManD signature-based subnetworks (edge threshold 0.30) show similarities and differences within the subgroup active sites (yellow arrows). C. Structures of the MR, MLE, and ManD active sites (top to bottom) shown in PDBs 2HNE, 1NU5, and 2QJJ, respectively. The residues highlighted with yellow arrows in B are represented with black side chains in the structures.
To illustrate the origin of the sequence-based network topology, multiple sequence alignments (MSAs) were constructed for the MR/ManD and MR/MLE clusters (Supporting Information File 2). Across all sequences in the MR/MLE cluster, just one position was completely conserved and one showed weak similarity. Conversely, for the MR/ManD cluster, the MSA contained six completely conserved, four strongly similar, and three weakly similar positions. These observations demonstrate quantitatively why the MR and ManD subgroups cluster together in the sequence-based network.
Active site similarity for the three subgroups was compared by creating active site signature logos from the signature-based network clusters (Supporting Information File 3) at a threshold roughly corresponding to the SFLD-defined subgroups (score threshold of 0.30). (The threshold with the greatest number of distinctly identified subgroups (score threshold of 0.35; Supporting Information Fig. 1, blue star) was not used for this analysis as the MR subgroup is separated into multiple subnetworks at that threshold rendering the signature logo useless.) Three residues (other than the key residues) are quite similar between the MR and MLE signatures and, thus, likely causing much of the division of the signature-based clusters [yellow arrows, Fig. 4(B), black side chains, Fig. 4(C)]. For example, Lys in active site signature position 2 is observed in the MR and MLE proteins (K218 in 2HNE and K163 in 1NU5, respectively); whereas, Arg is observed in the ManD proteins (R147 in 2QJJ). In MR and MLE proteins, this conserved Lys is the electrophilic residue interacting with a carboxylate oxygen. The conserved Arg in this position of ManD proteins forms a dyad with Tyr 159 (in 2QJJ) and acts as the base.36 Interestingly, the analogous ManD electrophilic residue to the MLE and MR Lys is Arg 283 (in 2QJJ) which is highly conserved in logo position 37 [Fig. 4(B), yellow arrow].36 Other differences highlighted in the signature logos may play structural roles important for preserving specific reaction steps. Despite the higher overall sequence similarity observed between the MR and ManD subgroups, the MR and MLE subgroups cluster together in the signature-based networks because their active sites share more similar features than either does with the ManD site. These common features are part of common MR and MLE mechanistic details, as previously reported,36 suggesting the utility of signature-based networks in identifying such features.
Sequence-based networks, however, are incredibly useful in identifying evolutionary relationships, as demonstrated previously in the literature.37,38 Further, when evolutionary relationships correlate with molecular function, sequence-based networks mimic functional relationships.39–41 It is critical to note, however, that evolutionary relationships do not always correlate with functional relationships, especially at the detailed level of molecular function,42 and therefore one must be cautious when interpreting sequence-based clustering patterns.
Signature-based networks highlight active site similarity despite differences in domain composition
One common issue observed in annotation transfer is that of “extra domains.” Domains may be added to proteins in a modular fashion,43 sometimes bringing a new molecular function, often without affecting the original domain's molecular function.44 We evaluated how domain structure impacted network topology in sequence-, structure-, and signature-based networks.
Prx5 is a subgroup of the Prx superfamily.45 All seven proteins in the Prx5 subgroup form a distinct subnetwork in the signature-based network at an edge threshold where the subgroup is distinctly defined [Fig. 5(A) and Supporting Information Fig. 2, black circle]. However, only six Prx5 proteins cluster in the structure-based network at a similar threshold [Fig. 5(A), blue circle], leaving 1NM3 as a singlet [Fig. 5(A), red circle]. A closer look reveals that the 1NM3 active site is essentially identical to other Prx5 active sites [Fig. 5(A), bottom left], explaining why all Prx5 proteins cluster in the signature-based network. On the other hand, 1NM3 is an outlier in the structure-based networks because it contains a glutaredoxin domain covalently attached to its C-terminus [Fig. 5(A), bottom right, red domain].
Figure 5.
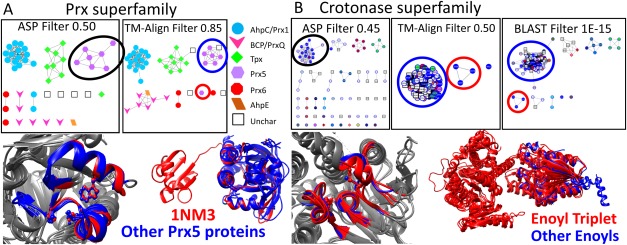
Signature-based networks cluster proteins based on active site similarity, while structure-based and sequence-based networks identify domain differences. A. In the Prx signature-based network (top left), all seven members of the Prx5 subgroup cluster together at edge threshold 0.50 (black circle). In the structure-based network (top right), six Prx5 proteins cluster together at edge threshold 0.85 (blue circle) while 1NM3 becomes a singlet in its own subnetwork (red circle). The node color and shape is based on SFLD subgroup annotation, as indicated in the legend. A close up view of the active site environment is shown for all Prx5 subgroup members (1NM3 in red and the other six proteins in blue; bottom left). Colored portions represent the active site signature fragments for each protein, and the three key residues used to define the active site are shown as ball and stick models. The complete protein structure for all Prx5 proteins (bottom right) shows that 1NM3 (red) contains an extra glutaredoxin-like domain attached to the protein's C terminus compared with the other six Prx5 proteins (blue). B. A triplet of enoyl family proteins cluster together in the structure- and sequence-based networks (red circles) while the other enoyl proteins remain in one cluster (blue circles). In the signature-based network, however, the triplet is part of the large cluster of enoyl proteins (black circle). A close up view of the active site environment is shown for the enoyl triplet (red) and a representative from the large enoyl cluster (blue) (bottom left). Colored portions represent the active site signature fragments for each protein, and the three key residues used to define the active site are shown as ball and stick models. The complete protein structure for these same proteins (bottom right) shows that the enoyl triplet proteins (red) contain an extra domain compared with the representative enoyl protein (blue).
A similar example is seen in the crotonase superfamily. In the signature-based network, the enoyl family forms one subnetwork at a score threshold of 0.45 [Fig. 5(B) and Supporting Information Fig. 4, black circle]. Conversely, in both the structure-based network [Fig. 5(B), top middle] and the sequence-based network [Fig. 5(B), top right], an enoyl triplet [Fig. 5(B), red circles] forms at less stringent score thresholds, while the remaining enoyl proteins cluster in one subnetwork [Fig. 5(B), blue circles]. Closer inspection of the active site microenvironment [Fig. 5(B), bottom left] reveals significant similarity between the enoyl triplet (red) and a representative enoyl from the larger cluster (blue). However, an examination of full protein structure [Fig. 5(B), bottom right] indicates the enoyl triplet proteins (red) contain an additional domain, causing this triplet to form a distinct subnetwork in both the structure- and sequence-based networks [Fig. 5(B), red circles].
Both results indicate that structure-based networks identify structural relationships including additional or missing domains, without regard to functional implications. Signature-based networks are less sensitive to such modifications (unless they would impact the functional site) and, instead, focus on functional site features. These examples demonstrate the value in comparing the three networks because each network provides unique information not encompassed by the other networks. It also points out the importance of understanding the desired goal in using similarity-based clustering, as the edge metric impacts what is learned from the network topologies.
Active site signature logos highlight previously identified mechanistically important residues
The SFLD hierarchical function classification organizes proteins based on functional site details,18 thus providing information about mechanistically important residues. However, years of work by expert curators were required to produce this detail on each superfamily—more efficient and rapid methods are essential. While networks have emerged as an efficient method to cluster proteins based on similarity, the protein clusters identified from networks must be functionally relevant to be accurately utilized in identifying mechanistic determinants. Examples in Figures 4 and 5 demonstrate that signature-based networks cluster proteins with similar functional site features; consequently, we explore using residue conservation in each cluster's active site profile to identify mechanistically important residues for that cluster.
Previously, this approach identified functional site residues in subgroups of the Prx superfamily. ASPs identified subgroup-specific functionally important residues, including a potentially important Glu in the AhpC/Prx1 subgroup.45 Subsequent combined molecular dynamics and electrostatics calculations indicate that this residue does, indeed, play an important functional role.46 In the current network analysis, the AhpC/Prx1/Prx6 subgroup is distinct and the Glu is well conserved in signature logos for this subgroup [Supporting Fig. 5(A), pink arrow].
This previous work on the Prxs and the network topology observations described herein suggest a generalizable and efficient strategy for producing hypotheses about the mechanistically determinant residues in each signature-based cluster. We first further validate this approach by comparison to experiment in the enolase and GST superfamilies, and then utilize the proposed approach to hypothesize novel functionally important residues in clusters of the GST superfamily. (With the limited data in the structure database, the crotonase clusters are too small to draw meaningful conclusions [Supporting Information Fig. 5(B)]).
Rakus et al. annotated subgroup-specific residues at the ends of seven structurally conserved beta strands (β2–β8) in the enolase active site.36 The active site signatures for all enolase superfamily proteins contain strands β2 through β6 [Fig. 6(A), green strands], while β7 and β8 [Fig. 6(A), magenta strands] lie outside the 10 Å signature radius (see Methods). The residues near the end of strands β3, β4, and β5 were the key residues used to define active site signatures [Fig. 6(A), black ball and stick]; these correspond to positions 10, 19, and 29 in the signature logos [Fig. 6(B), black stars; residues D198, E224, and D249 in 1BKH].
Figure 6.
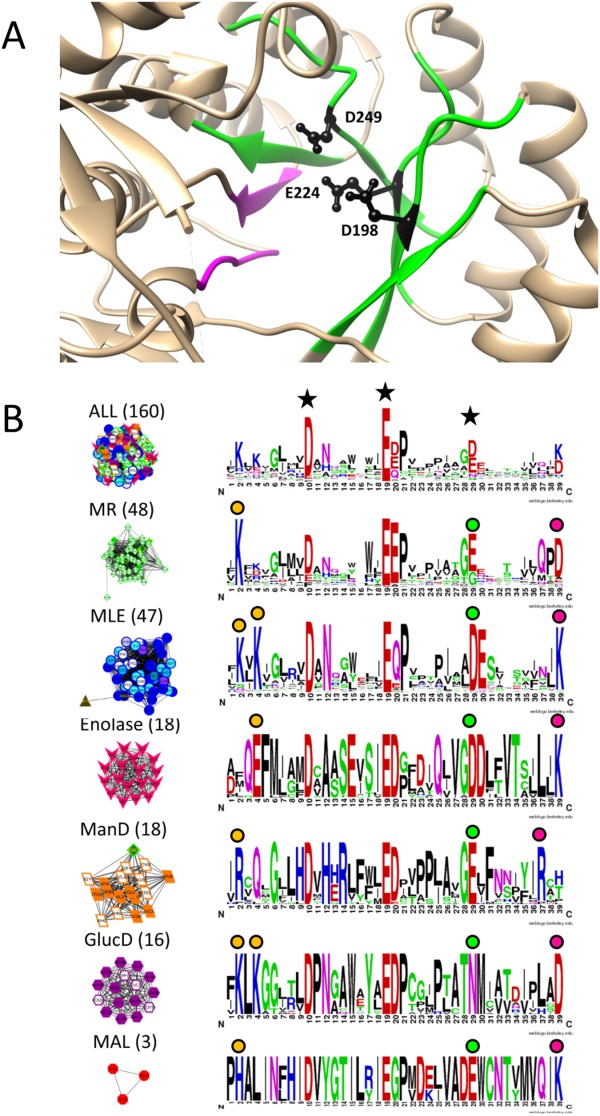
Signature logos highlight residues known to be functionally important in the enolase superfamily. A. Green ribbons represent regions included in the active site signature (β2–β6) and magenta ribbons represent β7 and β8 which are not included in the active site signature but were analyzed by Rakus et al.36 Tan areas represent the remaining protein structure. The three key residues defining the active site region (black side chains) are located on strands β3, β4, and β5, respectively. B. Signature logos were created for the entire enolase superfamily (top) and the six major groups identified as signature-based subnetworks at edge score threshold of 0.30, a cutoff chosen to correlate with the SFLD subgroups [see Fig. 2(A) and Supporting Information Fig. 2 for the network series]. Node color is based on SFLD subgroup and family designation (color key in Fig. 2). Clusters are labeled with the dominant subgroup and the number of proteins in the cluster. The key residues used to create active site signatures are labeled with black stars in the top signature logo. Functionally relevant residues in each subgroup at the ends of β2, β5, and β6 as reported by Rakus et al. are identified with orange circles, green circles, and fuchsia circles, respectively. The residues in positions 10 (Asp) and 19 (Glu) of the figure were invariant throughout the superfamily and, therefore, not labeled with colored circles in each subgroup. Note that the MAL subgroup only contains three nonredundant structures so signature conservation analysis in this subgroup should be considered preliminary. Representative examples of the orange, green, and fuchsia conserved residues, respectively, in each subgroup are the following: MR (1MDR)—K164, E247, D270; MLE (1BKH)—K167, K169, D249, K273; enolase (1E9I)—E167, D316, K341; ManD (2QJJ)—R147, E262, R283; GlucD (1BQG)—K211, K213, N295, D319; MAL (1KD0)—H194, D307, K331.
Though the threshold with the greatest number of distinctly identified subgroups in the enolase superfamily is 0.35 (Supporting Information Fig. 1, blue stars), the MR subgroup is more complete at threshold 0.30 and only two proteins are incorrectly grouped (brown triangle in MLE cluster and green diamond in ManD cluster) so the clusters at this threshold were used in the analysis. We wanted to compare residue conservation in active site profiles with previously published results; therefore, Weblogos47 were created for the signature-based clusters (herein referred to as signature logos) at an edge threshold of 0.30 [Fig. 6(B)]. We compared the residues reported by Rakus et al. to those conserved in each signature-based cluster. All functionally important residues noted by Rakus et al. near the ends of β2–β6 are easily identified in the logos created from active site signature-based subnetworks of the enolase superfamily [Fig. 6(B), circles above signature logos]. Residues near the β3 and β4 termini are well conserved throughout the superfamily [Fig. 6(B), large D and E, top logo], while the residues at the β2, β5, and β6 termini distinguish each subgroup. According to Rakus et al., the MLE, MR, and GlucD subgroups share a conserved Lys residue at the β2 terminus, which is well conserved in signature logo position 2 [Fig. 6(B), orange circles]. By comparison, residues at the β2 terminus for the enolase, MAL and ManD subgroups are Glu, His, and Arg, respectively. Similarly, the enolase, MAL, and MLE subgroups share an Asp residue near the β5 terminus [Fig. 6(B), green circles], while Glu is found in that position in the MR and ManD subgroups, and Asn in the GlucD subgroup. Finally, at the β6 terminus, the enolase, MAL, and MLE subgroups contain a Lys, the MR and GlucD subgroups share an Asp, and the ManD proteins contain an Arg [Fig. 6(B), fuchsia circles]. This analysis demonstrates that ASPs can efficiently identify known mechanistically important residues in the enolase subgroups, similar to previous results for the Prxs, suggesting an efficient and automatable strategy for hypothesizing residues that distinguish functional subgroups in other superfamilies.
As has been noted previously,31 functional residue identification using this approach is not necessarily complete, as some residues (such as those on β7 and β8 in the enolases) may fall outside of the 10 Å signature radius. Despite this lack of completeness, the similarity network clustering based on active site signatures was still able to separate the enolase subgroups, identifying a larger number of distinct subgroups at a single threshold than either sequence- or structure-based clustering (Supporting Information Fig. 1, blue stars). Further, recent work by Petrey et al. suggests using small areas of protein structures rather than full sequence and structure comparisons is more appropriate for relating protein function.48
Relationships derived from active site signature networks lead to hypotheses regarding functionally important residues
This same approach was used to predict functionally relevant residues in the GSTs (Fig. 7). SFLD-identified GST subgroups cluster distinctly into subnetworks at an edge threshold of 0.35 (Supporting Information Fig. 3, blue star); thus, subnetworks at this threshold were analyzed with signature logos (Fig. 7, residues discussed are marked with stars and circles). The six largest subnetworks were used in this analysis, including two distinct AMPS clusters (AMPS.A and AMPS.B). Several smaller subnetworks are observed at this threshold, but are not analyzed as each is composed of too few proteins to confidently identify conserved residues.
Figure 7.
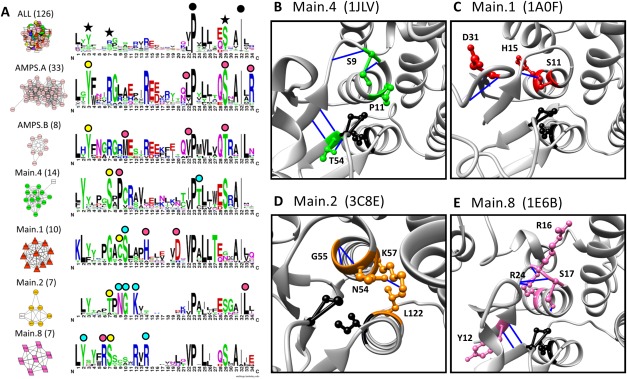
Active site signature logos for subnetworks of the GST superfamily highlight possible mechanistically determinant residues. A. Signature logos were created for the entire GST superfamily (top logo) and the six largest subnetworks identified at an edge threshold of 0.35 in the signature-based network. Black stars (top logo) indicate the three key residues used to create the active site signatures. Black circles (top logo) indicate residues, displayed as black side chains in B–E, well conserved throughout the superfamily. Colored circles indicate residues conserved within a subnetwork, but not conserved throughout the superfamily—these are represented with subgroup-colored side chains in B–E. Yellow circles indicate residues reported as functionally relevant in the literature. Blue circles indicate residues discussed in detail in the text. Pink circles indicate well-conserved residues not discussed in the text. Node colors are based on SFLD subgroup (key in Supporting Information Fig. 1). Subnetworks are labeled with the dominant SFLD subgroup and the number of proteins. B–E. Residues displayed in green, red, orange, and pink are conserved in Main.4 (1JLV), Main.1 (1A0F), Main.2 (3C8E), and Main.8 subgroup (1E6B), respectively. Hydrogen bonds are represented by blue lines in all structures.
Some residues are conserved throughout the superfamily that have been previously identified. The cis-Pro (P53 in 1JLV) and the Ile (I68 in 1JLV) are both well conserved throughout the superfamily [Fig. 7(A), black circles]; the Pro is reportedly required for binding site formation,49 while the Ile is important for structural stability of the active site.50
Additionally, some subgroup-specific residues easily identified in the GST signature logos have been previously identified [Fig. 7(A), yellow circles]. For example, a Ser in logo position 7, previously shown to activate the bound glutathione (GSH),51 is highly conserved in Main.4 (S9 in 1JLV) and Main.8 (S17 in 1E6B) subgroups. Similarly, the conserved Thr in that same position in the Main.2 subgroup (T52 in 3C8E) plays a role in ligand binding.52 In the AMPS subgroup, the Tyr in signature position 3 (Y6 in 1C72) has been reported to hydrogen bond to the sulfur of the GSH ligand, stabilizing a nucleophilic thiolate.51,53
Other residues are well conserved in one or more subgroups, but have not been previously reported [Fig. 7(A), blue circles]. For instance, a Thr in logo position 24 (T54 in 1JLV) is well conserved in the Main.4 subnetwork. Hydrogen bonding in the immediate area [Fig. 7(B), blue lines] indicates this Thr may play a role in stabilizing P53 and I68, which, as noted above, are important for ligand binding. In the Main.1 subnetwork, a well-conserved Cys in logo position 9 (C10 in 1A0F) has been previously reported to bind GSH.51 The complete conservation of Ser in position 10 of this subgroup (S11 in 1A0F) suggests it may play an important role through binding stabilization as well, possibly through hydrogen bonding to the nearby beta sheet [Fig. 7(C), blue lines]. Similarly, the well conserved NGXK motif in the Main.2 subgroup [Fig. 7(A), blue circles; residues 54–57 in 3C8E] has not been previously reported as functionally relevant. Hydrogen bonding between N54 and K57 [Fig. 7(C), blue line] and the structural proximity to the ligand-binding Thr [Fig. 7(A), yellow circle] suggests the hypothesis that this motif plays a role in protein stability and ligand binding. In the Main.8 subnetwork, a Tyr (Y12 in 1E6B) and an Arg (R24 in 1E6B) are both highly conserved [Fig. 7(A), blue circles] and may play a supporting role to the previously identified Ser [Fig. 7(A), yellow circle] that is crucial for ligand binding.51 Both Tyr and Arg participate in hydrogen bonding within the active site [Fig. 7(E), blue lines], supporting this hypothesis. There are many other subnetwork-specific residues identified in these signature logos that are conserved in the active site region [Fig. 7(A), pink circles] and may play a role in ligand binding and catalytic activity.
The rapid and efficient identification of residues specific to signature-based subnetworks illustrates a useful and automatable approach to identifying mechanistically important residues that distinguish the functionally relevant subgroups in a protein superfamily.
Role of edge metric threshold in identifying the topologically-based and functionally relevant subnetworks and their hierarchical relationships
As described above, functionally relevant clusters can be identified from the signature-based subnetworks and signature logos built from these subnetworks highlight well-conserved residues that play important functional roles within each cluster. However, such data are only advantageous if the edge metric used to define the subnetworks correlates with functional relevance because network topology and, thus, subnetworks (functionally relevant clusters) vary with the edge metric threshold [Fig. 1(A,B)]. A nontrivial question is how to determine the “correct” edge or score threshold that identifies functionally relevant clusters. We note that the difficulty of defining a useful threshold reflects complications of evolution—every superfamily has evolved at different rates and under different constraints on folding, structure and function.
In the results described in this manuscript, we used the edge threshold that most closely resembles SFLD subgroups (families for the crotonases) while keeping the subgroups (and families) as intact as possible. This technique is analogous to that used by Mashiyama et al. when determining the GST subgroup designations based on current experimental annotations.52 In the enolase superfamily, a threshold of 0.30 produced clusters that matched the six SFLD subgroups almost perfectly [Fig. 6(B)]. Likewise, a threshold of 0.35 in the GST signature-based network separated all subgroups into distinct clusters and was, therefore, used to define groups for the signature logos [Fig. 7(A)]. Signature logos were also created for the Prx and crotonase superfamilies at thresholds of 0.35 and 0.40 (Supporting Information Fig. 5), respectively, scores at which the subnetworks correlate with the SFLD subgroups (Prx) and families (crotonase) (Supporting Information Figs. 2 and 4).
We note that these chosen thresholds do not always match with the threshold identifying the most subgroups (or families) distinctly and completely (Supporting Information Figs. 4, blue stars). The blue stars provide a quantitative comparison between the network series—and the approach optimizes for smaller, homogeneous clusters. On the other hand, residue conservation was compared with literature-described subgroups and, thus, threshold was chosen to optimize comparison to literature-identified clusters. We chose to use clusters containing one or two extra proteins that did not belong rather than using clusters that contained just half of the subgroup or family of interest, which would provide less statistical significance to the residue conservation analysis.
Further investigation of functionally relevant hierarchies by exploring network topologies at multiple edge thresholds
While it is illuminating to identify and compare functionally relevant clusters at a single edge threshold, one benefit of analyzing similarity networks is the potential to identify functional hierarchies (Fig. 1). Can thresholds that reproduce the SFLD functional hierarchy (or other functionally relevant hierarchy), as illustrated in Figure 1, be identified?
A key goal is to develop a process that would identify the appropriate score thresholds to cluster the protein sequence universe into functionally relevant hierarchies (Fig. 1). Thus, network topology at increasingly stringent edge thresholds should identify hierarchical relationships within a superfamily (Fig. 1). We evaluate the counts of edge thresholds at which the most subgroups and families are identified (stars in Supporting Information Figs. 1–4) to determine if such a hierarchy can be observed in the results presented here. Subgroups are identified in the Prx, GST and enolase families at edge thresholds of 0.3 to 0.4 in the signature-based networks, while the crotonase families (a finer level of functional detail) are most readily identified at an edge threshold of 0.5. Therefore, in the signature-based networks for this very limited set of superfamilies, the threshold for identification of families is more stringent than subgroups, correlating with a required hierarchy. On the other hand, the subgroup and family thresholds overlap in the sequence- and structure-based networks: subgroups are identified at thresholds of 0.84 to 0.92 (structure-based networks) and 1e-20-1e-35 (sequence-based networks), while families are identified at 0.92 (structure-based networks) and 1e-35 (signature-based networks). All observations are taken from a very limited data set of four superfamilies. If this observation holds for additional superfamilies and edge thresholds in signature-based networks can distinguish levels of functional hierarchies (as we propose would be ideal, Fig. 1), these results open the pathway for developing an automated method for clustering the universe of protein sequences.
Given the results presented here, we ask if an example of such hierarchy can be observed in the current results. While most protein subgroups have too few structures to hypothesize functional hierarchies, the AMPS subgroup in the GST superfamily contains over forty structures that include the literature-described subfamilies of Alpha, Mu, Pi and Sigma, which are not distinguished in SFLD. Topology of the signature-based network splits this subgroup into two clusters at an edge threshold of 0.35, AMPS.A and AMPS.B [Fig. 8(A), blue and red circles]. Further, a more stringent threshold of 0.45 produces a topology of three distinct subnetworks created from AMPS.A (AMPS.A.1, AMPS.A.2, and AMPS.A.3), creating four total AMPS clusters [Fig. 8(B), blue and red circles]. Between edge thresholds of 0.35 and 0.45, all other subgroup clusters are largely unchanged. Signature logos created for the three largest AMPS groups [Fig. 8(C)] identify major differences between the clusters, residues which we hypothesize as mechanistically significant [Fig. 8(D)].
Figure 8.
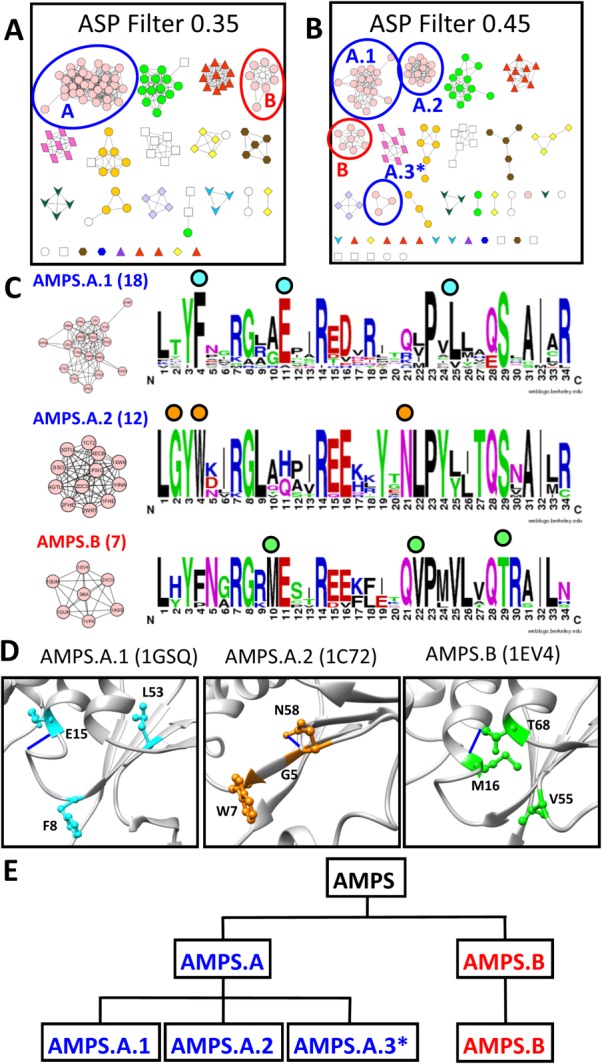
AMPS subgroup signature-based clustering suggests hierarchical organization of function. A. At the 0.35 edge threshold in the signature-based network, the AMPS subgroups splits into two distinct clusters; A (blue circle) and B (red circle). B. At the 0.45 edge threshold in the signature-based network, the AMPS.A cluster splits into three smaller clusters (blue circles) while the AMPS.B cluster only loses one protein (red circle). C. Signature logos were created for the three largest AMPS clusters at edge threshold 0.45 of the signature-based GST network. Clusters are labeled with the number of proteins comprising the cluster. Colored circles above the logos indicate residues, represented with similar-colored side chains in D, conserved within a subnetwork but not conserved throughout the entire subgroup. D. Residues distinctly conserved within the AMPS.A.1, AMPS.A.2, and AMPS.B subnetworks are mapped onto representatives from each cluster (1GSQ—cyan, 1C72—orange, 1EV4—green, respectively). E. A functional hierarchy for the AMPS subgroup is defined from the signature-based network clustering. AMPS.A and AMPS.B are the two main AMPS clusters at a 0.35 edge threshold. At the 0.45 edge threshold, the AMPS.A cluster breaks into three smaller clusters while the AMPS.B cluster remains mostly the same. *The cluster is too small for detailed active site analysis.
The AMPS.A.1 cluster contains three well-conserved residues [Fig. 8(C), cyan circles] at logo positions 4 (F8 in 1GSQ), 11 (E15 in 1GSQ), and 25 (L53 in 1GSQ), suggesting that this cluster may be comprised of the Pi and Sigma families of the AMPS subgroup.51 Likewise, conserved residues in the AMPS.A.2 cluster [Fig. 8(C), orange circles], such as Gly in logo position 2 (G5 in 1C72), Trp in position 4 (W7 in 1C72), and Asn in position 21 (N58 in 1C72), suggest these proteins belong to the Mu family of the AMPS subgroup.51 Finally, positions 10 (M16 in 1EV4), 22 (V55 in 1EV4) and 29 (T68 in 1EV4) in the AMPS.B cluster are well-conserved and unique [Fig. 8(C), green circles] suggesting that this cluster contains the Alpha family of the AMPS subgroup.51
This result suggests the intriguing possibility of identifying functional hierarchies in each protein superfamily, as suggested by the analysis of the AMPS subgroup [Fig. 8(E)]. Such hierarchies could be defined at multiple levels of functional detail, depending on the needs of the investigator. Clearly further work is needed in additional superfamilies across sequence (not just structure) space to identify edge thresholds that divide superfamilies into these functionally relevant hierarchies; such work is ongoing.
Conclusion
In computational assignment of protein function, sequence similarity is often used to transfer function annotation from one protein to another. An underlying assumption in this approach is that proteins with full sequence similarity share similar function. In this manuscript, we explored what can be learned about protein function assignment based on similarity clustering using a method in which we identify clusters from network topologies produced using three different similarity measures as edge metrics. We compared with pologies of sequence-based, structure-based, and signature-based networks to determine how well subnetworks compared with the functional hierarchy defined by SFLD.
As has been shown in the extensive literature, sequence-based networks can identify evolutionary relationships;37,38 these networks also identify functional relationships when evolution correlates with function, which is very often the case at the superfamily level and, as shown here, is frequently (but not always) the case at the subgroup level. Structure-based networks identify structure-based differences, including domain additions, which would identify the potential of additional functional sites within the protein. Signature-based networks often identify more detailed molecular functional relationships and, importantly, can identify functional hierarchies relating subgroups and families. Additionally, mechanistically important residues can be identified from the conserved residues in the signature-based subnetworks.
The results show that signature-based similarity networks introduce an efficient, accurate way to define functionally relevant groups, an approach suggested by Zhang et al.,11 and identify mechanistically important residues in those groups. This approach can be applied systematically and on a large-scale, which would contribute significantly to guiding manual curation efforts. Finally, this analysis provides a foundation for development of semi- and fully automated tools that can use sequence, structure, and active site data to group proteins in functionally relevant ways useful for many different applications.
Materials and Methods
Protein set
The Structure Function Linkage Database (SFLD)18 is a collection of well-studied and well-curated proteins developed19 and often referred to as a “gold standard” set.12,16,20,21,54 These proteins have been manually curated and assigned to superfamilies, subgroups and families [Fig. 1(C)], based on similarity in mechanistic and specificity determinants.18 Some of these proteins have been experimentally studied; therefore, this set serves as a comparison tool for other protein classification methods. [We note that this Gold Standard set, originally published in 2005, is now represented by many more proteins. The newer data alter to some extent the original functional assignments. To address this, an updated Gold Standard set is under development (personal communication, S. Brown and P. Babbitt)].
In this work, three superfamilies manually curated by SFLD curators were evaluated: enolase, glutathione transferases (GST) and crotonase. A fourth well-studied superfamily, the peroxiredoxins (Prx), was also analyzed using this method. Prx proteins of known structure were taken from Nelson et al.45 and these are now available in the SFLD. A summary of proteins in each superfamily are provided in Table I; details about protein structures used in each subgroup and family within each superfamily are provided in Supporting Information File 4.
To accurately compare results, it is critical to create networks using the same number of proteins for each of the three edge metrics. Structure-based networks require a solved three-dimensional protein structure; thus, the set of structures available in the SFLD as of April 2013 were utilized for all network evaluations. Groups of proteins exhibiting high levels of sequence identity produced clusters that were highly similar in all three networks, cluttering network visualization and hindering analysis. Therefore, superfamily members of known structure were evaluated for redundancy. Redundancy was identified by aligning all pairs of sequences using Stretcher55 and calculating the percent identity for each pair within the superfamily. One protein, at random, was retained from each group of proteins that shared 95% full sequence identity; this representative set of non-redundant proteins for each superfamily was used in all network evaluations. The full list of representative and redundant proteins for each superfamily is provided in Supporting Information File 4.
Clustering evaluated using sequence-, structure-, and signature-based scoring methods
Networks were created for the proteins in each superfamily using ClusterMaker,56 a plugin for the Cytoscape software package57 that has implemented the Markov clustering algorithm (MCL).58 The appropriate edge metric (ASP score, TM-Align score, or BLAST score) was used for the array source with all other default parameters used. After clustering, a force directed layout based on the scoring metric was applied to arrange and visualize the networks.
Different scoring methods were used as the edge metric in each of the three networks: full-length sequence comparison using the BLAST scoring function;23 full-length structure comparison using the TM-Align scoring function;24 and active site signature comparison using the active site profile (ASP) scoring function.31 Sequence comparison edge weights were determined based on the more significant of the two reciprocal BLAST e-values between two proteins, run with default parameters. BLAST scores represent the likelihood of the sequence similarity being solely due to chance; most values are in the range [0, 1] with smaller values corresponding to higher similarity.23 Structure comparison edge weights were determined by the TM-Align score between two proteins, normalized by the average length of the two proteins. TM-Align scores represent the length-normalized structural similarity of two proteins, weighting the more similar areas stronger than the less similar areas; the scores are in the range [0, 1] with larger values corresponding to higher similarity.24 Active site similarity edge weights were determined based on the ASP scoring function, which takes into account residue identity, strong similarity, weak similarity, and gaps in the pairwise active site profile alignment and is normalized by the total length of the pairwise alignment.31 ASP scores represent the length-normalized sequential and structural similarity of the active site microenvironment of two proteins; the scores are in the range [-0.5, 1] with larger values corresponding to higher similarity.31
Active site signatures31 for each protein are determined from the structure surrounding user-identified key residues. Three key active site residues were defined for each superfamily from SFLD-identified enzymatically active residues and key residues for the remaining proteins were identified using structural overlays (Supporting Information Fig. 6) in Chimera.59 A full list of all key residues can be found in Supporting Information File 4. Active site profiling31 was performed as implemented in the Deacon Active Site Profiler (DASP).60,61 Briefly, for each protein, all residues for which any atom lies within 10 Å of the center of mass of one of the key residues are extracted and aligned N-to-C terminus to create an active site signature. Single residues and fragments of length two are eliminated from the signature. Pairwise active site signature comparison edge weights were determined using an in-house Python script that utilizes the signature similarity scoring metric previously described.31 The networks created using the BLAST, TM-Align, and ASP scoring metrics are referred to as sequence-based, structure-based, and signature-based networks, respectively.
Clusters defined by edge thresholding produce subnetworks
For each network, subnetworks or clusters were defined by the edge threshold (a “filter”) applied to the edge weights. At a given edge threshold, all edges with scores below that threshold are removed. When the threshold is applied, these missing edges produce distinct subnetworks, where the edges within the subnetwork have pairwise edge scores more significant than the threshold, and the edges that previously connected the subnetworks have been removed due to less significant scores. We explored the formation of subnetworks (or clusters) at different score thresholds, so we could compare the hierarchy of subnetwork formation in each superfamily. It is important to note that at each edge metric threshold, the MCL clustering algorithm may remove some edges that are above the threshold during the clustering process. For example, edges removed from the BLAST network during clustering are very large compared with the majority of edges that are quite small (Supporting Information Fig. 7); thus, the clustering algorithm removes the edges with the extremely large scores at the “no filter” edge threshold producing multiple subnetworks before edge threshold application.
To compare how accurately each of the three networks identified known functional groups, we counted the number of clusters that were distinct and all inclusive of a subgroup (for enolase, Prx, and GST) or family (for crotonase) at each edge threshold in each of the three networks. Subgroups or families with only one protein structure were not part of the count, and uncharacterized proteins were ignored in all clusters. The highest count for each network series was marked (Supporting Information Figs. 1–4, blue stars) and analyzed.
Signature similarity visualized using active site signature logos
Sequence logos for the protein clusters were created using WebLogo version 3.3.47 Signatures were first split into their noncontiguous fragments. To make the signature logos as accurate as possible, each signature fragment must be a consistent length for all of the proteins in a superfamily. Towards this goal, each fragment in all proteins in a superfamily was aligned based on structural overlays and both ends of the fragment were extended in each signature using the contiguous protein sequence until each fragment was a consistent length for all proteins in the superfamily. The fragments were then concatenated to form final signatures. Fragment extension and concatenation was subsequently added to DASP to more accurately group proteins based on their active site microenvironment (manuscript in prep). To create the figures, default settings from the Weblogo website (http://weblogo.berkeley.edu/) were used except for the small sample correction, which decreases the height of all of the letters in small samples; given the small sample sizes, it was important for all letters to be visible for the analysis. In the signature logos, the larger the letter, the more frequent that residue is found in that position throughout the set of active site signatures. These graphical representations allow simple comparison of the active site signatures between different clusters of proteins. Signature similarity figures were created for the enolase [Fig. 6(B)], GST [Fig. 7(A)], Prx [Supporting Information Fig. 5(A)], and crotonase [Supporting Information Fig. 5(B)] superfamilies.
Acknowledgments
Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco. J.S.F. and J.B.L. thank an anonymous reviewer for insightful comments. The authors report no conflict of interest.
Glossary
- Chl-MLE
chloromuconate cycloisomerase
- DTartD
D-tartrate dehydratase
- DipepEp
dipeptide epimerase
- GalD
galactarate dehydratase
- GlucD
glucarate dehydratase
- LFucD
L-fuconate dehydratase
- LTalGalD
l-talarate/galactarate dehydratase
- MAL
methylaspartate ammonia lyase
- ManD
mannonate dehydratase
- MLE
muconate cycloisomerase
- MLE (anti)
muconate cycloisomerase-anti
- MLE (syn)
muconate cycloisomerase-syn
- MR
mandelate racemase
- NSAR
N-succinylaminoacid racemase
- NSAR2
N-succinylaminoacid racemase 2
- OSBS
O-succinylbenzoate synthase
- RhamD
rhamnonate dehydratase
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
References
- 1.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2010;38:D46–D51. doi: 10.1093/nar/gkp1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valencia A. Automatic annotation of protein function. Curr Opin Struct Biol. 2005;15:267–274. doi: 10.1016/j.sbi.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Bork P, Bairoch A. Go hunting in sequence databases but watch out for the traps. Trends Genet. 1996;12:425–427. doi: 10.1016/0168-9525(96)60040-7. [DOI] [PubMed] [Google Scholar]
- 4.Karp PD. What we do not know about sequence analysis and sequence databases. Bioinformatics. 1998;14:753–754. doi: 10.1093/bioinformatics/14.9.753. [DOI] [PubMed] [Google Scholar]
- 5.Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol. 2009;5:e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rentzsch R, Orengo CA. Protein function prediction–the power of multiplicity. Trends Biotechnol. 2009;27:210–219. doi: 10.1016/j.tibtech.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Radivojac P, Clark WT, Oron TR, Schnoes AM, Wittkop T, Sokolov A, Graim K, Funk C, Verspoor K, Ben-Hur A, et al. A large-scale evaluation of computational protein function prediction. Nat Methods. 2013;10:221–227. doi: 10.1038/nmeth.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adai AT, Date SV, Wieland S, Marcotte EM. LGL: creating a map of protein function with an algorithm for visualizing very large biological networks. J Mol Biol. 2004;340:179–190. doi: 10.1016/j.jmb.2004.04.047. [DOI] [PubMed] [Google Scholar]
- 9.Frickey T, Lupas A. CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics. 2004;20:3702–3704. doi: 10.1093/bioinformatics/bth444. [DOI] [PubMed] [Google Scholar]
- 10.Atkinson H, Morris J, Ferrin T, Babbitt P. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS One. 2009;4:4345. doi: 10.1371/journal.pone.0004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Zagnitko O, Rodionova I, Osterman A, Godzik A. The FGGY carbohydrate kinase family: insights into the evolution of functional specificities. PLoS Comput Biol. 2011;7:e1002318. doi: 10.1371/journal.pcbi.1002318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamady M, Widmann J, Copley SD, Knight R. MotifCluster: an interactive online tool for clustering and visualizing sequences using shared motifs. Genome Biol. 2008;9:R128. doi: 10.1186/gb-2008-9-8-r128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wittkop T, Emig D, Lange S, Rahmann S, Albrecht M, Morris JH, Böcker S, Stoye J, Baumbach J. Partitioning biological data with transitivity clustering. Nat Methods. 2010;7:419–420. doi: 10.1038/nmeth0610-419. [DOI] [PubMed] [Google Scholar]
- 14.Apeltsin L, Morris J, Babbitt P, Ferrin T. Improving the quality of protein similarity network clustering algorithms using the network edge weight distribution. Bioinformatics. 2011;27:326–333. doi: 10.1093/bioinformatics/btq655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miele V, Penel S, Duret L. Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinformatics. 2011;12:116. doi: 10.1186/1471-2105-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miele V, Penel S, Daubin V, Picard F, Kahn D, Duret L. High-quality sequence clustering guided by network topology and multiple alignment likelihood. Bioinformatics. 2012;28:1078–1085. doi: 10.1093/bioinformatics/bts098. [DOI] [PubMed] [Google Scholar]
- 17.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akiva E, Brown S, Almonacid DE, Barber AE, Custer AF, Hicks MA, Huang CC, Lauck F, Mashiyama ST, Meng EC, et al. The structure–function linkage database. Nucleic Acids Res. 2014;42:D521–D530. doi: 10.1093/nar/gkt1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown SD, Gerlt JA, Seffernick JL, Babbitt PC. A gold standard set of mechanistically diverse enzyme superfamilies. Genome Biol. 2006;7:R8. doi: 10.1186/gb-2006-7-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pieper U, Chiang R, Seffernick S, Brown S, Glasner M, Kelly L, Eswar N, Sauder J, Bonanno J, Swaminathan S, et al. Target selection and annotation for the structural genomics of the amidohydrolase and enolase superfamilies. J Struct Funct Genomics. 2009;10:107–125. doi: 10.1007/s10969-008-9056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee DA, Rentzsch R, Orengo C. GeMMA: functional subfamily classification within superfamilies of predicted protein structural domains. Nucleic Acids Res. 2010;38:720–37. doi: 10.1093/nar/gkp1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerlt JA, Babbitt PC. Divergent evolution of enzymatic function: mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu Rev Biochem. 2001;70:209–46. doi: 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]
- 23.Altschul S, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Audit B, Levy ED, Gilks WR, Goldovsky L, Ouzounis CA. CORRIE: enzyme sequence annotation with confidence estimates. BMC Bioinformatics. 2007;8:S3. doi: 10.1186/1471-2105-8-S4-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marti-Renom MA, Rossi A, Al-Shahrour F, Davis FP, Pieper U, Dopazo J, Sali A. The AnnoLite and AnnoLyze programs for comparative annotation of protein structures. BMC Bioinformatics. 2007;8:S4. doi: 10.1186/1471-2105-8-S4-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verspoor K, Cohn J, Mniszewski S, Joslyn C. A categorization approach to automated ontological function annotation. Protein Sci. 2006;15:1544–1549. doi: 10.1110/ps.062184006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engin HB, Guney E, Keskin O, Oliva B, Gursoy A. Integrating structure to protein-protein interaction networks that drive metastasis to brain and lung in breast cancer. PLoS One. 2013;8:e81035. doi: 10.1371/journal.pone.0081035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin AJM, Walsh I, Domenico TD, Mičetić I, Tosatto SCE. PANADA: protein association network annotation, determination and analysis. PLoS One. 2013;8:e78383. doi: 10.1371/journal.pone.0078383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uberto R, Moomaw EW. Protein similarity networks reveal relationships among sequence, structure, and function within the cupin superfamily. PLoS One. 2013;8:e74477. doi: 10.1371/journal.pone.0074477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cammer S, Hoffman B, Speir J, Canady M, Nelson M, Knutson S, Gallina G, Baxter S, Fetrow J. Structure-based active site profiles for genome analysis and functional family subclassification. J Mol Biol. 2003;334:387–401. doi: 10.1016/j.jmb.2003.09.062. [DOI] [PubMed] [Google Scholar]
- 32.Kalyanaraman C, Imker H, Fedorov A, Fedoroc E, Glasner M, Babbitt P, Almo S, Gerlt J, Jacobson M. Discovery of a dipeptide epimerase enzymatic function guided by homology modeling and virtual screening. Structure. 2008;16:1668–1677. doi: 10.1016/j.str.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lukk T, Sakai A, Kalyanaraman C, Brown SD, Imker HJ, Song L, Fedorov AA, Fedorov EV, Toro R, Hillerich B, et al. Homology models guide discovery of diverse enzyme specificities among dipeptide epimerases in the enolase superfamily. Proc Natl Acad Sci USA. 2012;109:4122–4127. doi: 10.1073/pnas.1112081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holden HM, Benning MM, Haller T, Gerlt JA. The crotonase superfamily: divergently related enzymes that catalyze different reactions involving acyl coenzyme a thioesters. Acc Chem Res. 2001;34:145–157. doi: 10.1021/ar000053l. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z, Yin P, Lee JS, Parasuram R, Somarowthu S, Ondrechen MJ. Protein function annotation with structurally aligned local sites of activity (SALSAs) BMC Bioinformatics. 2013;14(Suppl 3):S13. doi: 10.1186/1471-2105-14-S3-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rakus J, Fedorov A, Fedorov E, Glasner M, Vick J, babbitt P, Almo S, Gerlt J. Evolution of enzymatic activities in the enolase superfamily: D-Mannonate dehydratase from Novosphingobium aromaticivorans. Biochemistry. 2007;46:12896–12908. doi: 10.1021/bi701703w. [DOI] [PubMed] [Google Scholar]
- 37.A DeBruyn, DP Martin, P Lefeuvre. New York: Molecular plant taxonomy; 2013. pp. 257–277. Humana Press. Chapter 13, Phylogenetic reconstruction methods: an overview. [Google Scholar]
- 38.Yang Z, Rannala B. Molecular phylogenetics: principles and practice. Nat Rev Genet. 2012;13:303–314. doi: 10.1038/nrg3186. [DOI] [PubMed] [Google Scholar]
- 39.Copley SD, Novak WR, Babbitt PC. Divergence of function in the thioredoxin fold suprafamily: evidence for evolution of peroxiredoxins from a thioredoxin-like ancestor. Biochemistry (Mosc) 2004;43:13981–13995. doi: 10.1021/bi048947r. [DOI] [PubMed] [Google Scholar]
- 40.Kasprzak JM, Czerwoniec A, Bujnicki JM. Molecular evolution of dihydrouridine synthases. BMC Bioinformatics. 2012;13:153. doi: 10.1186/1471-2105-13-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rojas AM, Fuentes G, Rausell A, Valencia A. The Ras protein superfamily: evolutionary tree and role of conserved amino acids. J Cell Biol. 2012;196:189–201. doi: 10.1083/jcb.201103008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fetrow JS, Siew N, Skolnick J. Structure-based functional motif identifies a potential disulfide oxidoreductase active site in the serine/threonine protein phosphatase-1 subfamily. FASEB J. 1999;13:1866–1874. doi: 10.1096/fasebj.13.13.1866. [DOI] [PubMed] [Google Scholar]
- 43.Chothia C, Gough J, Vogel C, Teichmann SA. Evolution of the protein repertoire. Science. 2003;300:1701–1703. doi: 10.1126/science.1085371. [DOI] [PubMed] [Google Scholar]
- 44.Pereira-Leal JB, Teichmann SA. Novel specificities emerge by stepwise duplication of functional modules. Genome Res. 2005;15:552–559. doi: 10.1101/gr.3102105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nelson K, Knutson S, Soito L, Klomsiri C, Poole L, Fetrow J. Analysis of the peroxiredoxin family: using active-site structure and sequence information for global classification and residue analysis. Proteins. 2011;79:947–964. doi: 10.1002/prot.22936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salsbury FR, Yuan Y, Knaggs MH, Poole LB, Fetrow JS. Structural and electrostatic asymmetry at the active site in typical and atypical peroxiredoxin dimers. J Phys Chem B. 2012;116:6832–6843. doi: 10.1021/jp212606k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrey D, Honig B. Is protein classification necessary? Toward alternative approaches to function annotation. Curr Opin Struct Biol. 2009;19:363–368. doi: 10.1016/j.sbi.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oakley AJ, Harnnoi T, Udomsinprasert R, Jirajaroenrat K, Ketterman AJ, Wilce MCJ. The crystal structures of glutathione S-transferases isozymes 1–3 and 1–4 from Anopheles dirus species B. Protein Sci. 2001;10:2176–2185. doi: 10.1110/ps.ps.21201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Achilonu I, Gildenhuys S, Fisher L, Burke J, Fanucchi S, Sewell BT, Fernandes M, Dirr HW. The role of a topologically conserved isoleucine in glutathione transferase structure, stability and function. Acta Crystallogr F Struct Biol. Cryst Commun. 2010;66:776–780. doi: 10.1107/S1744309110019135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atkinson HJ, Babbitt PC. Glutathione transferases are structural and functional outliers in the thioredoxin fold. Biochemistry. 2009;48:11108–11116. doi: 10.1021/bi901180v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mashiyama ST, Malabanan MM, Akiva E, Bhosle R, Branch MC, Hillerich B, Jagessar K, Kim J, Patskovsky Y, Seidel RD, et al. Large-scale determination of sequence, structure, and function relationships in cytosolic glutathione transferases across the biosphere. PLoS Biol. 2014;12:e1001843. doi: 10.1371/journal.pbio.1001843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Armstrong RN. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem Res Toxicol. 1997;10:2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 54.Wittkop T, Emig D, Truss A, Albrecht M, Böcker S, Baumbach J. Comprehensive cluster analysis with transitivity clustering. Nat Protoc. 2011;6:285–295. doi: 10.1038/nprot.2010.197. [DOI] [PubMed] [Google Scholar]
- 55.Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 56.Morris J, Apeltsin L, Newman A, Baumbach J, Wittkop T, Su G, Ferrin T. clusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC Bioinformatics. 2011;12:436. doi: 10.1186/1471-2105-12-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smoot ME, Ono K, Ruscheinski J, Wang P-L, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 60.Huff R. 2005. DASP: active site profiling for identification of functional sites in protein sequences and structures. Masters Thesis. North Carolina: University of North Carolina.
- 61.Huff R, Bayram E, Tan H, Knutson S, Knaffs M, Richon A, Santago P, II, Fetrow J. Chemical and structural diversity in cyclooxygenase protein active sites. Chem Biodivers. 2005;2:1533–1552. doi: 10.1002/cbdv.200590125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information