

Abstract
Caseinolytic chaperones and proteases (Clp) belong to the AAA+ protein superfamily and are part of the protein quality control machinery in cells. The eukaryotic parasite Plasmodium falciparum, the causative agent of malaria, has evolved an elaborate network of Clp proteins including two distinct ClpB ATPases. ClpB1 and ClpB2 are involved in different aspects of parasitic proteostasis. ClpB1 is present in the apicoplast, a parasite-specific and plastid-like organelle hosting various metabolic pathways necessary for parasite growth. ClpB2 localizes to the parasitophorous vacuole membrane where it drives protein export as core subunit of a parasite-derived protein secretion complex, the Plasmodium Translocon of Exported proteins (PTEX); this process is central to parasite virulence and survival in the human host. The functional associations of these two chaperones with parasite-specific metabolism and protein secretion make them prime drug targets. ClpB proteins function as unfoldases and disaggregases and share a common architecture consisting of four domains—a variable N-terminal domain that binds different protein substrates, followed by two highly conserved catalytic ATPase domains, and a C-terminal domain. Here, we report and compare the first crystal structures of the N terminal domains of ClpB1 and ClpB2 from Plasmodium and analyze their molecular surfaces. Solution scattering analysis of the N domain of ClpB2 shows that the average solution conformation is similar to the crystalline structure. These structures represent the first step towards the characterization of these two malarial chaperones and the reconstitution of the entire PTEX to aid structure-based design of novel anti-malarial drugs.
Keywords: malaria, ClpB AAA+ proteins, Plasmodium translocon of exported proteins, protein export and trafficking, chaperone, apicoplast, drug target, antimalarial design, SAXS, hybrid methods
Introduction
Malaria is a mosquito-borne parasitosis caused by the obligate intracellular protozoan Plasmodium. Efforts to eradicate this devastating disease include drug-based therapies, mosquito control, and development of a vaccine. Emergence of drug resistance to existing anti-malarial therapies among parasites constitutes a serious threat to the current disease control efforts. Plasmodium falciparum (Pfal) is responsible for its most severe and fatal form. Modern research on malaria seeks to identify and understand biological processes that are unique to the parasite. Once characterized, such processes can be targeted for drug design.
All living cells have evolved sophisticated mechanisms of protein homeostasis. Proteostasis includes the elimination of misfolded proteins or otherwise abnormal proteins, maintenance of aminoacid pools and the refolding/reactivation of salvageable proteins in cells subjected to stresses such as heat shock or starvation. Caseinolytic proteases and chaperones (Clp) constitute one large group of proteins involved in protein quality control; they belong to the superfamily of AAA+ proteins (ATPases associated with diverse cellular activities) and are found in bacteria, fungi, but also in the mitochondria of eukaryotes or the chloroplast of plants.1
They are several subgroups of Clp chaperones. ClpA, ClpC and ClpX belong to the Clp/HSP100 family of AAA+ proteins2 that also includes the molecular chaperones ClpB/HSP104.3 The hexameric chaperones ClpA, ClpC and ClpX bind and unfold specific substrate proteins before translocating the unfolded polypeptide to the protease ClpP for its final degradation.4,5 In contrast, ClpB/HSP104 proteins are essential for the survival of bacteria, fungi and plants where they act as ‘disaggregases’ assisting resolubilization and reactivation of aggregated proteins6 in response to stress. These chaperones support infectivity and host survival in a number of pathogens.7–9 Bacterial Clp proteases are bona fide targets for the screening and development of novel antibacterials.10
Sequence analyses indicate that the genome of Plasmodium falciparum encodes six Clp proteins.11 ClpP and ClpR12 are two proteases while ClpB1, ClpB2/HSP101, ClpC and ClpM are ATPases. Interestingly, despite the plethora of Clp AAA+ proteins identified in Pfal, no ClpA has been detected. Plasmodium belongs to the phylum of Apicomplexa, characterized by the presence of a peculiar organelle, the apicoplast. Apicomplexans are thought to have originated from Dinoflaggellates, a large group of photosynthetic protozoa. As such, the apicoplast is distantly related to the chloroplast of higher plants. Some 500 proteins of Plasmodium have been predicted to localize to this organelle where several prokaryotic biochemical pathways have been annotated.13 Chemical rescue experiments performed on malaria parasites lacking an apicoplast have helped define the function of this organelle and suggest that isoprenoid precursor biosynthesis is the main essential function of the apicoplast during blood stage growth.14 Other crucial biosynthetic pathways include fatty acid, heme and aminoacid synthesis. Because the apicoplast is vital to parasite survival15 and is an organelle unique to apicomplexans, it provides an enticing target for the design of novel antimalarial drugs aimed at disrupting biological pathways essential to the parasite.
In Plasmodium, all Clp proteins, except ClpB2/HSP101, are localized in the apicoplast [Fig. 1(A)]. ClpB2/HSP101 instead localizes exclusively to the encasing parasitophorous vacuole (PV) where it functions as a core subunit of a protein secretion complex, the Plasmodium Translocon of Exported proteins (PTEX) [Fig. 1(B)].16 The intracellular survival of Plasmodium within the infected red blood cells is dependent on its ability to export several hundreds of its proteins (∼7% of its proteome) that hijack and remodel the infected host cells to support its virulence and parasitic lifestyle.17–20 Parasitic proteins destined for export through the PTEX harbor a signal sequence to reach the vacuole by entering the endoplasmic reticulum (the regular secretory pathway) and a specific Plasmodium Export Element sequence (PEXEL)21–23 for their vacuolar translocation. Thus, to access the host erythrocyte cytosol, exported proteins must cross two membranes, first the parasite plasma membrane, and then the PV membrane (PVM) [Fig. 1(A,B)].
Figure 1.
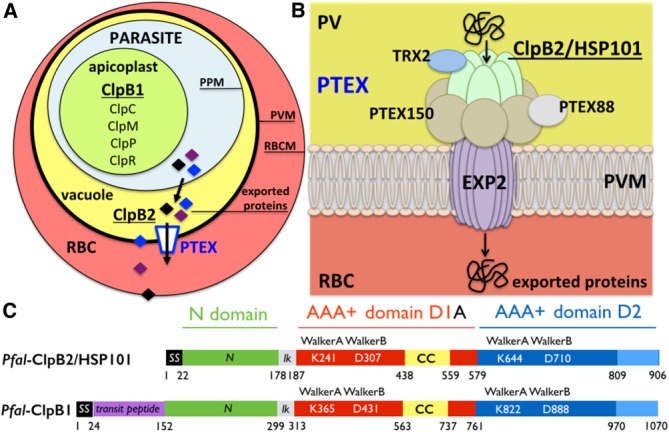
The Clp proteases and chaperones in Plasmodium and the Plasmodium translocon of exported proteins. (A) Simplified diagram showing a red blood cell (RBC) infected by a parasite encased in the parasitophorous vacuole. The nomenclature and localization of all Plasmodium Clp chaperones and proteases are show. There are two ClpB proteins, ClpB1 and ClpB2/HSP101. All Clp proteins are localized in the apicoplast except the vacuolar ClpB2/HSP101. (B) ClpB2/HSP101 is a subunit of the parasite-derived Plasmodium translocon of exported proteins (PTEX). Plasmodium proteins are translocated across the parasitophorous vacuole membrane (PVM) into the red blood cell cytoplasm through a translocation pore composed of Exported protein-2 (EXP2). Prior to their transport, exported proteins are unfolded by the hexameric AAA+ protein ClpB2/HSP101. For some cargos, unfolding also necessitates reduction of disulfide bonds by the accessory subunit thioredoxin-2 (TRX2). The other subunits, PTEX150 and PTEX88, have unknown functions. The arrows indicate the directionality of transport from the parasitophorous vacuole (yellow) towards the erythrocyte cytoplasm (red). The relative positions of the different subunits within the complex are unknown. (C) Domain organization of the two ClpB proteins present in Plasmodium falciparum: the apicoplastic ClpB1 and the vacuolar ClpB2/HSP101. The signal sequence (SS in black) targets proteins to the general secretory pathway and the transit peptide (in magenta) is a specific signal that specifies subcellular localization to the apicoplast. The N-terminal domain (in green) is linked to the two catalytic nucleotide-binding domains (NBD) D1 (in red) and D2 (in blue) by a short basic linker (lk in grey). The D1 domains of ClpB and ClpC chaperones are characterized by the presence of a coiled-coil insertion, the middle domain also named “arm” (CC in yellow). The D2 domains usually contain a C-terminal module (in light blue). Each NBD contains two signature Walker (A and B) motifs.
PTEX is a large membrane-associated complex composed of five subunits: EXP2, ClpB2/HSP101, PTEX150, TRX2 and PTEX88 [Fig. 1(B)]. A stable detergent-resistant core composed of subunits EXP2, PTEX150 and ClpB2/HSP101 has been characterized.24 The PVM-associated Exported protein-2 (EXP2)25,26 is a possible candidate for the trans-membrane protein-conducting conduit and may structurally resemble the bacterial pore-forming cytolytic α-helical toxin hemolysins.27–29 In Plasmodium, ClpB2/HSP101 actively drives the export process by harnessing the energy from ATP hydrolysis to unfold and thread the diverse cargos through the trans-membrane conduit EXP2. While ClpB2/HSP101 seems to be the primary ‘engine’ driving protein export, the malarial thioredoxin-2 (TRX2) auxiliary protein assists protein unfolding by reducing the disulfide bonds present in some cargo proteins, thus facilitating the ATP-driven export process. Protein unfolding is required for export across the PVM as it was shown that proteins must pass into the erythrocyte cytosol in an unfolded state.30 Molecular chaperones appear to play important roles in keeping parasite proteins in a ‘translocation competent’ state prior to crossing the PVM.
Attempts to generate gene complete knockouts of any of the three PTEX components EXP2, PTEX150 and ClpB2/HSP101, initially failed16 suggesting their essential functions. Mutant parasites lacking TRX2 or PTEX88 are severely impaired with considerably slower rates of development during the blood stage.31 These studies have led to the conclusions that while EXP2, HSP101, and PTEX150 play central roles during the blood infection in Plasmodium, PTEX88 and TRX2 are auxiliary yet important regulators of PTEX-mediated protein export and required for maintaining normal blood-stage growth.32 The integrity of the PTEX complex is required for efficient protein export. Ablation or inactivation of HSP101/ClpB2 in parasites results in the nearly complete block in export with substrates accumulating in the vacuole;33 furthermore this blockade affects all classes of exported proteins, whether they harbor a PEXEL motif or not.34 As a central and parasite-specific portal, PTEX appears as a weak point in the parasitic life cycle and hence an attractive drug target.
Like all class 1 Clp AAA+ proteins, Pfal- ClpB1 and ClpB2/HSP101 are modular proteins composed of a N-terminal domain involved in the binding of substrate proteins, followed by the two ATPase catalytic domains (D1 and D2) responsible for the ATP-driven unfolding of polypeptides and a short C-terminal domain [Fig. 1(C)]. They likely function as homo-hexamers where the N-domains are connected to the D1D2 ATPase catalytic core via flexible linkers and are structurally mobile, which may facilitate binding to target proteins35–37 and enhance chaperone activity.38 Switching N-domains between different HSP100 proteins alters substrate specificity accordingly, indicating that N-domains function as independent units in substrate selection.39,40 N-domains can contact substrates directly41,42 or via adaptor proteins.43 The hexameric association state and ATPase-dependent chaperone activity of the apicoplastic ClpB1 have been demonstrated in vitro; however nothing is known about its roles in vivo; it might be involved in maintaining a functional apicoplastic proteome during red blood cell infection.44
To aid design novel anti-malarials using computational tools and combinatorial screening of libraries, knowledge of the three-dimensional structure of the targeted component is a prerequisite. For ClpB1 and ClpB2, identification of the protein surfaces and structural features involved in the essential steps of cargo recognition/delivery, unfolding and/or translocation can help define and identify novel pharmacophores. PTEX appears a prime drug target and there is considerable interest in targeting the components involved in cargo unfolding such as ClpB2/HSP101 and the thioredoxin TRX2 to block protein export. The structures of malarial TRX245,46 provided valuable templates for rational structure-based drug design. Our current efforts focus on the characterization of ClpB2/HSP101 and EXP2 proteins at the core of PTEX. To this end, we describe here the first crystal structures of the N-terminal domain of ClpB1 and ClpB2/HSP101 proteins from Plasmodium and also characterize the conformation of its vacuolar ATPase N domain using solution small-angle X-ray scattering (SAXS). We analyze and compare the surface properties of the N domains from the two parasitic ClpB chaperones in terms of substrate and partner protein binding sites.
Results
Structures of the N-terminal domains of the ClpB1 and ClpB2/hsp101 ATPases from the eukaryotic pathogen plasmodium falciparum
We solved the crystal structures of the N domains from ClpB1 and ClpB2/HSP101 of Pfal at resolutions ranging from 1.7 to 2.0 Å by molecular replacement using the structures of the N domain from the ClpB proteins from Thermus thermophilus47 and Escherichia coli48 as best search models (Table I and Material and Methods). Despite relatively weak sequence identities ranging from ∼12 to 24%, the corresponding N-domains of ClpA, ClpB, ClpC and ClpV from bacteria such as Escherichia, Thermus, Bacillus and Vibrio can be superimposed with a root mean square deviation (RMSD) ranging from ∼1.5 to 2.4 Å for corresponding Cα atoms [Fig. 2(A) and Supporting Information Table S1]. The fold of the N domain is composed of eight α-helices (α1-α8) folding into a compact globular domain characteristic of all Clp chaperones belonging to the A, B and C subgroups [Fig. 2(B)]. It can be divided into two four α-helix bundles (α1-α4 and α5-α8) connected by a ∼15 residue-long loop. The excellent quality of the electron density (Supporting Information Figure S1A) enabled us to trace the entire domain of N-ClpB2/HSP101 in each of the two crystal structures reported here; the loop of N-ClpB1 showed some disorder in one of the two chains constituting the asymmetric unit.
Table I.
X-Ray Data Collection and Structure Refinement Statistics
| Data set | APS 060612 24-ID-E | APS 120912 24-ID-C | APS 10162014 24-ID-C |
|---|---|---|---|
| Protein | Pfal N ClpB2/HSP101 | Pfal N ClpB2/HSP101 | Pfal N ClpB1-sfGFP |
| Data collection statistics | PDB ID 4IOD | PDB ID 4IRF | PDB ID 4XBI |
| Wavelength | 0.97918 Å | 0.97918 Å | 0.97918 Å |
| Space group | C2221 | P22121 | P65 |
| a = 89.1 Å b = 141.88 Å c = 91.8 Å | a = 31.8 Å b = 92.1 Å c = 97.0 Å | a = 127.5 Å, c = 92.6 Å | |
| AU content | Three molecules | Two molecules | Two molecules |
| Solvent content | 58% | 45% | 52% |
| Resolution (last shell) | 44.6–1.80 Å (1.85–1.80 Å) | 66–1.65 Å (1.67–1.65 Å) | 50–2.01 Å (2.07–2.01 Å) |
| Unique reflections | 53,488 (3,809) | 35,522 (2,439) | 55,948 (3,785) |
| Completeness | 99.7% (96.7%) | 99.1% (94.9%) | 98.6% (90.4%) |
| I/σ(I) | 21.6 (3.2) | 15.0 (2.7) | 10.9 (2.7) |
| Redundancy | 9.9 (8.9) | 8.2 (8.1) | 5.0 (4.3) |
| Rsym | 7.1% (74.3%) | 6.3% (47.8%) | 7.9% (48.8%) |
| Rmeas | 7.5% (78.6%) | 6.8% (50.9%) | 8.9% (55.4%) |
| CC(1/2) | 99.9% (82.6%) | 99.8% (96.0%) | 99.6% (85.6%) |
| Refinement statistics | |||
| Resolution | 44.6–1.8 Å | 66.78–1.65 Å | 70.95–2.01 Å |
| Reflections | 53,476 | 35,144 | 55,908 |
| Work set/test set | 48,142/5,334 | 31,631/3,513 | 53,104/2,804 |
| Rfree/Rcryst | 21.2%/17.0% | 23.4%/21.4% | 20.1%/16.4% |
| Bwilson | 21.2 Å2 | 28.2 Å2 | 36.5 Å2 |
| Protein atoms, ADP | 3,524, 29.2 Å2 | 2,249, 35.6 Å2 | 5,832, 41.1 Å2 |
| Solvent atoms, ADP | 371, 34.6 Å2 | 223, 43.0 Å2 | 383, 44.9 Å2 |
| Others atoms, ADP | 3 sulfates, 53.6 Å2 | – | 5 sulfates, 79.0 Å2 |
| RMSD bonds | 0.014 Å | 0.018 Å | 0.007 Å |
| RMSD angles | 1.209° | 1.904° | 1.035° |
| Ramachandran analysis | |||
| Allowed regions | 99.6% | 99.0% | 98.2% |
| Generously allowed | 0.4% | 0.7% | 1.6% |
| Outliers | 0% | 0.3% | 0.2% |
Rsym = ∑hkl∑i |Ihkl,I − <Ihkl,i>|/∑hkl∑i |Ihkl,i| where <Ihkl,i> is the average intensity of the multiple hkl, i observations for symmetry-related reflections.
Rmeas is the redundancy independent R-factor.70
CC(1/2) percentage of correlation between intensities from random half-datasets.71
Rcryst = ∑|Fobs − Fcalc|/∑|Fobs|. Fobs and Fcalc are observed and calculated structure factors, Rfree is calculated from a set of randomly chosen reflections, and Rcryst is calculated over the remaining reflections.
ADP is the atomic displacement parameter.
Figure 2.

General architecture of the N-terminal cargo-binding domain of the Plasmodium AAA+ proteins ClpB1 and ClpB2/HSP101. (A) Structure-based sequence alignments of N domains from ClpA, ClpB and ClpC AAA+ proteins of known structures; the position of the secondary structure elements correspond to our ClpB2/HSP101 and ClpB1 N domain structures. The N domain structures from E. coli ClpA (PDB 1K6K), E. coli ClpB (PDB 1KHY), T. thermophilus ClpB (PDB 1QVR) and B. subtilis ClpC (PDB 2Y1Q) were used. (B) Crystal structures of the two ClpB N terminal domains from Plasmodium falciparum. Secondary structure elements are labeled and colored using a rainbow pattern for the N domain. (C) Two views showing the two chains (A and B) of the N domain of ClpB1 fused to superfolder GFP (in grey) present in the asymmetric unit. The magenta star highlights the different conformations adopted at the Gly-Gly linkage (in black) connecting the N domain to the sfGFP used to assist crystallization. The two-fold non-crystallographic symmetry axis that relates the two GFP chains but not the two N domains is indicated. (D) Stereo view of the two malarial ClpB N domains superposed.
We initially tried to solve the structure of N-ClpB1 by itself (Material and Methods). Bacterial expression and purification yielded homogenous protein and crystals of N-ClpB1 appeared in a single condition. However they were twinned and although a molecular replacement solution could be identified, we sought to find a way to prevent twinning. To overcome this problem, we applied carrier-driven crystallization and selected Green Fluorescent Protein (GFP)49 over Maltose Binding Protein (MBP)50 as fusion partner. We were able to crystallize a fusion protein between N-ClpB1 and a superfolder variant of GFP51 (Material and Methods). The resulting crystals were not twinned and diffracted to 2.0 Å resolution with two copies of the fusion protein in the asymmetric unit [Fig. 2(C)].
Superposition of the two copies using the GFP as reference structure reveals that the two ClpB1 adopt strikingly different orientations within the asymmetric unit [Supporting Information Fig. S2(A)]. In one copy (chain B), the C-terminal helix α8 of ClpB1 and the N-terminal α-helix of GFP align to form a single continuous helix including the G149G150 sequence linking the C-terminus of N-ClpB1 to the N-terminus of GFP. In the second copy (chain A), this linker forms a sharp break/turn resulting in a 90° angle between the two helices. This results in a local breakage of the two-fold non-crystallographic symmetry that relates the two GFP units [Fig. 2(C)]. Although the linker displays remarkable conformational plasticity, it is well resolved in the experimental electron density maps [Supporting Information Fig. S1(B)]. Most importantly, the two ClpB1 N domain copies are superimposable, thus showing that fusion to the GFP using a very short linker did not distort the structure of the N-terminal domain [Supporting Information Figure S2(B)]. These are the first structures of N domains from eukaryotic ClpBs and in particular of a fragment of the vacuolar ATPase ClpB2/HSP101, an essential core subunit of the PTEX.
Surface properties of the two ClpB N domain proteins in plasmodium
Within the N domains, the two four-helix bundles are related to each other by a pseudo two-fold symmetry axis [Fig. 3(A)]. This topology results in the most salient feature of these structures: a solvent-accessible grove that is delineated by helices α1/α2 and their pseudo-symmetry related counterparts α5/α6. This surface is composed of hydrophobic residues [Fig. 3(B)] and can be referred to as a “hydrophobic patch.” The ∼15 residue-long loops connecting the two halves are slightly basic in contrast with the so-called “acidic loop” present in all N domains of ClpA ATPases;52 it is well ordered and adopts the same conformation in the two different crystal forms of N-ClpB2/HSP101 we obtained (Material and Methods).
Figure 3.

Pseudo two-fold internal symmetry creates a hydrophobic patch at the surface of the N terminal domains of the two ClpB chaperones from Plasmodium. (A) Two four-α-helical bundles related by a pseudo two-fold axis perpendicular to the plane of the figure (red symbol) constitute the N domain (left). The first bundle (α1– α4) is colored in green; the second (α5–α8) is colored in blue. This arrangement, shown here for N-ClpB2 creates a cleft. The two bundles are shown superposed (right). (B) Solvent accessible surface representation highlighting the hydrophobic nature of the cleft present in N-ClpB2 (left) and N-ClpB1 (right). Residues delineating these putative binding sites are labeled. The same orientation is used in A and B.
In Plasmodium ClpB2/HSP101, the corresponding surface is wider and flatter and seems more suited to the promiscuous binding of unfolded and/or hydrophobic polypeptides. In ClpA, the same hydrophobic patch was shown to bind to peptides with low affinity and loose specificity.52 This particular surface also overlaps partially with the cargo-specific binding site revealed in the structure of the ClpV-VipB peptide53 (Supporting Information Fig. S3). However, the ClpV protein N domain contains a short extra N-terminal helix appended to the eight α-helical bundle common to all N domains of ClpA, ClpB and ClpC HSP100 AAA+ proteins; this architecture results in an hydrophobic groove that specifically accommodates the α-helical VipB peptide (Supporting Information Fig. S3).
The specificity of individual AAA+ proteins is achieved by their direct or adaptor-mediated association with their proteinaceous substrates. ClpA and ClpC ATPases have been shown to recognize some of their substrates through adaptor proteins that dock to their N-terminal domains and deliver their cargo for subsequent unfolding by the D1 and D2 catalytic domains. Only a few AAA+ Clp proteins have been crystallized with their adaptor or their specific cargo proteins. These include the N-ClpA/ClpS adaptor complex from Escherichia,54,55 the ClpC/MecA adaptor complex56 from Bacillus (Fig. 4) and the N-ClpV/VipB cargo peptide complex from Vibrio.53 ClpA, ClpB, ClpC, and ClpV share structurally similar N-terminal domains. Although structurally unrelated, the bacterial adaptors ClpS and MecA primarily use a single α-helix to interact with a common binding site at the surface of the N domains of ClpA and ClpC. Strikingly, in both cases a glutamate residue from the adaptor (E64 in ClpS and E184 in MecA) interacts with this pocket of the Clp protein. This “hot-spot” for adaptor binding is delineated by the connecting loop and helices α2, α4, α5, and α7 [Fig. 4(A)]. We compared the electrostatic features of the N domains from Clp proteins representative of the three subgroups (i.e., ClpA/B/C): the bacterial ClpA and ClpC and our two malarial ClpB structures. For each of these proteins representing three subclasses of Clp proteins, a shallow pocket is present. However, the electrostatic potential of malarial ClpB2/HSP101 shows a remarkable charge distribution asymmetry on this specific face of the protein [Fig. 4(B)] that sets it apart from its homologues.
Figure 4.

Modalities of interaction between N domains from different AAA+ proteins of the Clp subfamily and their know adaptors. (A) The structures of the E. coli ClpS/ClpA N domain (PDB 1LZW) and the B. subtilis MecA/ClpC N domain (PDB 2Y1R) complexes are displayed next to our Plasmodium ClpB1 and ClpB2/HSP101 N domain structures. In both complexes, the structurally unrelated adaptors use a conserved glutamate residue to interact with the N-domain of their cognate Clp ATPase. (B) Electrostatic potential properties as calculated using APBS72 for the binding interfaces of ClpA and ClpC with their respective adaptors; the structurally equivalent surface in the ClpB1 and ClpB2/HSP101 N domains are shown in the same orientation. Red, blue and white surfaces correspond to negative, positive and neutral surface potential, respectively. The cross indicates the binding pockets accommodating residues E64 (in ClpS bound to ClpA) and E184 (in MecA bound to ClpC) contributed by the two adaptor proteins. The ‘hot-spot’ for adaptor binding is marked with an × symbol.
Unlike ClpA and ClpC, ClpB proteins do not seem to require adaptors to bind their substrates. The two ClpB chaperones from Plasmodium are localized in two distinct cellular compartments and thus interact with different subsets of substrates, in different subcellular contexts. ClpB1 most likely functions as a ‘house-keeping’ chaperone in the apicoplast. On the other hand, the vacuolar ClpB2/HSP101 is a quite specialized chaperone involved in the specific export across the vacuolar membrane of a subset of malarial proteins; the molecular mechanisms governing cargo selection and triage are not presently understood and it is likely that some other PTEX subunit, namely the core subunit PTEX150 that tightly associates with ClpB2/HSP101 and EXP2, might play this role of delivering the diverse cargos to the chaperone for their translocation.
Conformation of the N-terminal domain of the plasmodium ClpB2/hsp101 ATPase in solution
ClpA/B/C proteins function as homo-hexamers. Purified N-terminal domains of ClpB1 and ClpB2, together with the N-ClpB1-GFP fusion protein [Fig. 5(A)] were analyzed by size exclusion chromatography (SEC). The three proteins behaved as monomers [Fig. 5(B)]. We further characterized the association state and conformation of the N domain of ClpB2/HSP101 in solution using SAXS57 (Material and Methods). Kratky plots indicate that the protein is folded in solution and is overall compact [Fig. 5(C)]. We derived the experimental values of radius of gyration (RG) values by Guinier analysis of scattered intensities at very small angles [Table II and Fig. 5(D)]. Experimental scattering curves were fitted against theoretical curves calculated using our crystal structures [Fig. 5(E)]. The pair distance distribution function (P(r)) estimated by Fourier inversion of the experimental intensities was compared to those calculated from our crystal structures model and used to estimate Dmax (the longest intra-molecular distance) and RG values independent of the Guinier analysis [Table II and Fig. 5(F)]. The experimental RG value of 16.3 Å measured for N-ClpB2/HSP101 is similar to the value calculated from the X-ray structure. The excellent agreement between theoretical and experimental scattering curves (as quantified by a discrepancy factor of χ2 = 1.008) and the overlap of the associated P(r) distributions indicate that the average conformation sampled by the N domain of ClpB2/HSP101 in solution is similar to our crystal structures and the monomeric state observed by SEC. It also suggests that the N-terminal domain of ClpB2/HSP101 is a rather rigid structure where the loop adopts the conformation observed in the crystal structures and does not sample a wide range of conformations.
Figure 5.

Homogeneity of the N domains of malarial ClpB1 and ClpB2 proteins purified from E. coli and solution analysis by SAXS of the Plasmodium ClpB2/HSP101 N domain. (A) Coomassie-stained SDS-PAGE analysis of N-ClpB1 (17.2 kDa), N-ClpB2 (16.8 kDa) and the N-ClpB1sfGFP fusion (43.2 kDa) proteins. (B) Size exclusion chromatography analysis of the three purified proteins used for structure determination on a Superdex S75 HR 10/30 column. The three proteins elute as monomers. Elution volumes for three molecular mass standards are indicated with arrows. (C) Kratky plot showing that Pfal-ClpB2/HSP101 N domain is folded in solution. (D) Guinier analysis. Guinier plot shown with the corresponding experimental value for the radius of gyration. The angular range (expressed in QRG values) used for the regression is indicated. Guinier's law is applied in its most conservative form within a QRG range not exceeding 1.3. (E) Scattering curve fit. The experimental curve (in color) is fitted against the curves calculated from the X-ray structures (in black) with CRYSOL, assuming a monomeric state of the N-domain in solution. The discrepancy factor for the fit is indicated. (F) Pair distance distribution functions. Experimental curve (in color) and curve calculated from the X-ray structures (in black) with GNOM. Dmax and RG experimental values inferred from the analysis are indicated.
Table II.
Small Angle X-ray Scattering Analysis
| X-ray structures |
SAXS | SAXS |
||||
|---|---|---|---|---|---|---|
| Models |
Guinier analysis | Fourier analysis |
||||
| Protein | Sequence Massa | RG | Dmax | RG | RG | Dmax |
| N ClpB2b | 16,782 Da | 16.3 Å | 53 Å | 16.9 ± 0.1Å | 16.9 ± 0.1Å | 55 ± 3 Å |
Theoretical masses calculated for the monomer of the protein constructs described in this study.
The theoretical RG and Dmax values correspond to the monomer of the X-ray structure reported in this study.
Conclusion
This study describes the first high-resolution crystal structures of the N-terminal domain from the two ClpB chaperones from the eukaryotic pathogen causing malaria, the parasite Plasmodium falciparum. The two Plasmodium ClpB chaperones are prime drug targets. Because of their ubiquitous distribution and the conserved architecture of their catalytic core, AAA+ proteins are challenging targets for the design of selective inhibitors. Nevertheless, such small molecules inhibitors have been described. A di-benzyl-quinazoline-diamine was the first reported reversible inhibitor for the p97/VCP ATPase58 offering a promising new therapeutic avenue in the treatment of cancer. More recently, small compounds named ciliobrevins were characterized as specific inhibitors of the motor cytoplasmic dynein.59 Disrupting cargo recognition, either through direct binding or via adaptors, might prove another viable strategy. Using these crystal structures as templates, future analyses of their protein surfaces might reveal potential binding sites that can be targeted for inhibitor design using fragment-based and computational screening of chemical libraries. The characterization of the other Plasmodium Clp ATPases and their yet to be discovered effectors or adaptors will also identify novel targets for anti-malarial design. For ClpB2/HSP101, this also represents a step towards the determination of the structure of the full-length ATPase through a hybrid approach combining SAXS, electron microscopy and X-ray crystallography. Upon reconstitution of the entire PTEX, our structures will facilitate future modeling, biochemical and, biophysical studies aimed at deciphering the mechanisms of cargo recognition, unfolding, and delivery by ClpB2/HSP101 to the predicted protein-translocating channel in the vacuolar membrane of Plasmodium.
Material and Methods
Protein expression and purification
A codon optimized synthetic gene (DNA2.0 Inc.) encoding full-length Plasmodium falciparum ClpB2/HSP101 (Pfal-ClpB2/HSP101: PF3D7_1116800) was used as starting material to construct several expression vectors. Two expression constructs encompassing residues A27-L188 (long-N101) or Q32-T177 (short-N101) were cloned into a pJexp401 vector (DNA2.0 Inc.) (for long-N101) or a pCDF vector (Novagen) (for short-N101) to express the corresponding proteins as fusions harboring a cleavable deca-histidine tag at their C-terminus. Both constructs lacked residues M1 through C26 corresponding to the signal sequence of Pfal-ClpB2/HSP101.
A codon optimized synthetic gene (Invitrogen Life Technologies) encoding residues G152-G298 corresponding to the N-terminal domain of Plasmodium falciparum ClpB1 (Pfal-ClpB1: PF3D7_0816600) was used as starting material to construct two expression vectors. Residues M1-I151 correspond to the signal sequence and the apicoplast targeting transit peptide. A first construct encoded the N-domain of ClpB1 cloned in the pET28b vector (Novagen) to be expressed as an N-terminal hexa-histidine-tagged cleavable fusion protein. A second construct encoding the N-ClpB1 gene fused to the superfolder GFP gene with a single G residue linker connecting residues G253 of ClpB1 and S2 of sfGFP; the N-ClpB1-sfGFP fusion protein contained a cleavable C-terminal octa-histidine tag.
All proteins were expressed using C43(DE3) E. coli cells grown in LB media at 37°C until they reached OD600 = 0.6 and protein expression was induced with 0.8 mM IPTG. Cells were harvested, washed in 150 mM KCl and 20 mM Tris pH = 7.8, and stored at −80°C until processing. Thawed cell pellets were resuspended in lysis buffer (500 mM NaCl, 20 mM Tris pH = 7.8, 15% glycerol) supplemented with 2.8 mM β-mercaptoethanol (β-ME), 0.2 mM of phenylmethylsulfonyl fluoride (PMSF), and 1 tablet of EDTA-free protease inhibitor cocktail (Roche). All subsequent steps were carried out at 4°C or on ice. Cells were disrupted by three passes through a C-3 Emulsiflex (Avestin) pressurized at 15,000 psi. The lysate was clarified by centrifugation at 25,000 g for 1 hr, after which the resulting supernatant corresponding to the total soluble extract was applied onto a gravity-flow column (BioRad) packed with 5-10 mL of Cobalt-NTA IMAC resin (Qiagen). Non-specifically bound bacterial proteins were subsequently washed from the column using Cobalt-A wash buffer (12.5 mM imidazole, 500 mM NaCl, 20 mM Tris pH = 7.8, 10% glycerol, 2.8 mM β-ME and 0.2 mM PMSF). The protein was eluted from the column with Cobalt-B buffer (125 mM imidazole, 500 mM NaCl, 20 mM Tris pH = 7.8, 10% glycerol, 2.8 mM β-ME, and 0.2 mM PMSF). The IMAC eluate was concentrated to 0.5-1 mL using a 10 kDa cutoff centricon, and desalted using a PD-10 desalting column (BioRad) equilibrated in 100 mM NaCl, 20 mM Tris pH = 7.8, 2% glycerol, 2.8 mM β-ME and 0.2 mM PMSF, to remove imidazole. Following desalting, the protein was purified by ion exchange chromatography (IEX) on a CaptoS (ClpB2) or a CaptoQ (for ClpB1-sfGFP) HiTrap column (GE Healthcare). The sample was then treated with thrombin for 24 hr at 4°C (0.25 units enzyme/mg of protein) to remove the histidine purification tag. Following thrombin treatment, the sample was further purified on a mixture of Nickel-NTA IMAC (Qiagen) and Benzamidine Sepharose (GE Healthcare) resins to simultaneously subtract unprocessed tagged-proteins, tags, and thrombin. The final purification step consisted in a size exclusion chromatography (SEC) on a Superdex 200 HR16/60 or a Superdex 75 HR10/30 gel-filtration column (GE Healthcare) equilibrated in 150 mM NaCl, 20 mM Tris pH = 7.8, 2% glycerol, and 7 mM β-ME to condition samples for crystallization trials.
Crystallization
Crystals of the Pfal-N-ClpB1-sfGFP fusion protein grew at room temperature in 2.0M Ammonium sulfate and 100 mM Hepes at pH = 7.0 at a protein concentration of 13–17 mg/mL and belong to hexagonal space group P65 with unit cells parameters a = 127.5 Å, c = 92.6 Å and a solvent content of 52% corresponding to two molecules of fusion protein in the asymmetric unit. The N-terminal domain of Pfal-ClpB2/HSP101 was concentrated at 30 mg/mL and yielded multiple crystallization conditions at 4°C. A first crystal form grew at 4°C in 20% PEG 3350, 10% glycerol and 50 mM CAPSO buffer at pH = 9.4 and belong to orthorhombic space group P22121 with unit cell parameters a = 31.8 Å b = 92.1 Å and c = 97.0 Å for two copies in the asymmetric unit and a solvent content of 45%. A second form of crystals belonging to orthorhombic space group C2221 was obtained in 2.2M ammonium sulfate with unit cell parameters a = 89.1 Å b = 141.9 Å and c = 91.8 Å for three copies in the asymmetric unit and a solvent content of 58%.
Structure determination and refinement
All diffraction data were collected on beamlines 24-ID-C or 24-ID-E at the Advanced Photon Source in the Argonne National Laboratory using crystals cryoprotected in mother liquor supplemented with 20–25% glycerol. Data were indexed, scaled and reduced using XDS.60 For N-ClpB2, the C2221 1.8 Å resolution structure was solved by molecular replacement using the structures of E. coli CplB (PDB ID 1KHY) and T. thermophilus ClpB (PDB ID 1QVR) N terminal domains as search probes. Molecular replacement using Phaser61 combined with partial automatic rebuilding in place with Phenix62 yielded a preliminary model that was manually improved in Coot.63 ADP refinement was carried in Phenix using TLS restraints combined with individual isotropic ADP refinement. This structure was then used to determine the P22121 1.65 Å resolution structure by molecular replacement using Phaser and refined with Phenix using TLS restraints combined with individual isotropic ADP refinement. For N-ClpB1-sfGFP, the structure was solved at 2.0 Å resolution by molecular replacement in Phaser using the structures of the N domains from E. coli, T. thermophilus ClpBs and our P. falciparum ClpB2 N domain structure (PDB 4IRF and 4IOD, this work) and the superfolder GFP structure (PDB 2B3P) as search probes. The resulting maps enabled partial automatic rebuilding of the model with further rebuilding in Coot. Refinement was carried out in Phenix using a strategy combining simulated annealing and individual ADP refinement. At the last stage of refinement, TLS refinement was performed where each chain was split into two TLS groups corresponding to the ClpB1 N domain and the GFP. For each structure, all molecules observed in the asymmetric unit were refined independently without applying non-crystallographic symmetry restraints.
Small angle X-ray scattering
Solution scattering data were collected at the SIBYLS high-throughput SAXS beamline 12.3.164 of the Advanced Light Source at the Lawrence Berkeley National Laboratory. Proteins and complexes were first prepared as described in the above section. Samples were further purified by gel filtration (Superdex 75 HR 10/30) equilibrated in the SAXS experimental buffer 200 mM NaCl, 20 mM Tris pH = 8.0, 2% glycerol, 0.5 mM EDTA and 5 mM fresh DTT with concentrations ranging from 1 to 25 mg/mL as estimated by UV absorption spectroscopy using the extinction coefficient values calculated from the amino-acid compositions. All intensity curves were collected with a sample-to-detector distance of 1.5m and an X-ray wavelength of λ = 1 Å and each contained about 500 data points. This corresponds to a Q ranging from 0.001 to 0.32 Å−1, with Q the scattering vector defined as Q = 4πsinθ/λ where 2θ and λ are the scattering angle and X-ray wavelength, respectively. For each sample several exposures were sequentially recorded (0.5, 1, 2, and 4 sec). Corresponding to each protein sample, data were collected on a buffer sample under identical experimental conditions. A beamline-specific software was used for radial averaging of images and for subtracting buffer signal from protein signal.65 Each scattering profile was visually inspected to check for aggregation, repulsive or attractive interactions, X-ray induced damage, detector saturation or anomalies in buffer subtraction. Following data reduction, analysis of experimental scattering data was performed using Scatter (http://bl1231.als.lbl.gov/saxs_protocols/index.php and http://www.bioisis.net/) to determine the radius of gyration (RG) by Guinier analysis, P(r) and Dmax. Kratky plots [Q2.I(Q) vs. Q] were used to assess the particle flexibility and folded nature.66 Scattering curves corresponding to the X-ray structures reported in this study and fitted against the experimental scattering curves were calculated using CRYSOL67 or FoXS68 with default parameters; the discrepancy factor χ2 was used to estimate the quality of the fits between experimental and calculated scattering curves. Pair distance distribution functions (P(r)) were derived by Fourier transform using GNOM69 to estimate Dmax, the longest distance occurring within the particle, and RG, independently of Guinier analysis.
Acknowledgments
The authors are grateful to Kevin Dyer from the Advanced Light Source SYBILS beamline 12.3.1 for his help with SAXS data collection. They acknowledge the DOE Integrated Diffraction Analysis (IDAT) grant DE-AC02-05CH11231 that supports the development and implementation of the SIBYLS beamline at the Advanced Light Source of the Lawrence Berkeley National Laboratory. They thank M. Capel, K. Rajashankar, N. Sukumar, J. Schuermann, I. Kourinov, and F. Murphy at NECAT beamlines 24-ID at the Advanced Photon Source at the Argonne National Laboratory
Glossary
- Pfal
Plasmodium falciparum
- AAA+
ATPase associated with diverse activities
- Clp
caseinolytic protease
- PTEX
Plasmodium translocon of exported proteins
- PV
parasitophorous vacuole
- PVM
parasitophorous vacuole membrane
- GFP
green fluorescent protein
- SAXS
small angle X-ray scattering
- RMSD
root-mean-square deviation
- ADP
atomic displacement parameters
- SEC
size exclusion chromatography
- Ec
Escherichia coli
- Tth
Thermus thermophilus
- Bsu
Bacillus subtilis
- Vch
Vibrio cholera
Author Contributions
Performed the experiments: APAY, AK, DC, and PFE. Designed the experiments: APAY, AK, DC, and PFE. Contributed reagents/materials/analysis: APAY, AK, DC, and PFE. Wrote the paper: APAY, AK, and PFE. Contributed equally to this work: APAY and AK.
Accession Numbers
Atomic coordinates and structure factors have been deposited at the RCSB Protein Data Bank (http://www.rcsb.org/pdb) under accession numbers 4IOD and 4IRF for the ClpB2/HSP101 N domain and 4XBI for the ClpB1 N domain GFP fusion.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- 1.Nishimura K, van Wijk KJ. Organization, function and substrates of the essential Clp protease system in plastids. Biochim Biophys Acta. 2014;1847:915–930. doi: 10.1016/j.bbabio.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 2.Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 3.Doyle SM, Wickner S. Hsp104 and ClpB: protein disaggregating machines. Trend Biochem Sci. 2009;34:40–48. doi: 10.1016/j.tibs.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 4.Baker TA, Sauer RT. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta. 2012;1823:15–28. doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu AY, Houry WA. ClpP: a distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 2007;581:3749–3757. doi: 10.1016/j.febslet.2007.04.076. [DOI] [PubMed] [Google Scholar]
- 6.Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 7.Krobitsch S, Clos J. A novel role for 100 kD heat shock proteins in the parasite Leishmania donovani. Cell Stress Chaperones. 1999;4:191–198. doi: 10.1379/1466-1268(1999)004<0191:anrfkh>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capestany CA, Tribble GD, Maeda K, Demuth DR, Lamont RJ. Role of the Clp system in stress tolerance, biofilm formation, and intracellular invasion in Porphyromonas gingivalis. J Bacteriol. 2008;190:1436–1446. doi: 10.1128/JB.01632-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meibom KL, Dubail I, Dupuis M, Barel M, Lenco J, Stulik J, Golovliov I, Sjostedt A, Charbit A. The heat-shock protein ClpB of Francisella tularensis is involved in stress tolerance and is required for multiplication in target organs of infected mice. Mol Microbiol. 2008;67:1384–1401. doi: 10.1111/j.1365-2958.2008.06139.x. [DOI] [PubMed] [Google Scholar]
- 10.Brotz-Oesterhelt H, Sass P. Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int J Med Microbiol. 2014;304:23–30. doi: 10.1016/j.ijmm.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 11.El Bakkouri M, Pow A, Mulichak A, Cheung KL, Artz JD, Amani M, Fell S, de Koning-Ward TF, Goodman CD, McFadden GI, Ortega J, Hui R, Houry WA. The Clp chaperones and proteases of the human malaria parasite Plasmodium falciparum. J Mol Biol. 2010;404:456–477. doi: 10.1016/j.jmb.2010.09.051. [DOI] [PubMed] [Google Scholar]
- 12.El Bakkouri M, Rathore S, Calmettes C, Wernimont AK, Liu K, Sinha D, Asad M, Jung P, Hui R, Mohmmed A, Houry WA. Structural insights into the inactive subunit of the apicoplast-localized caseinolytic protease complex of Plasmodium falciparum. J Biol Chem. 2013;288:1022–1031. doi: 10.1074/jbc.M112.416560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ralph SA, van Dooren GG, Waller RF, Crawford MJ, Fraunholz MJ, Foth BJ, Tonkin CJ, Roos DS, McFadden GI. Tropical infectious diseases: metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat Rev Microbiol. 2004;2:203–216. doi: 10.1038/nrmicro843. [DOI] [PubMed] [Google Scholar]
- 14.Yeh E, DeRisi JL. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011;9:e1001138. doi: 10.1371/journal.pbio.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodman CD, Su V, McFadden GI. The effects of anti-bacterials on the malaria parasite Plasmodium falciparum. Mol Biochem Parasitol. 2007;152:181–191. doi: 10.1016/j.molbiopara.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 16.de Koning-Ward TF, Gilson PR, Boddey JA, Rug M, Smith BJ, Papenfuss AT, Sanders PR, Lundie RJ, Maier AG, Cowman AF, Crabb BS. A newly discovered protein export machine in malaria parasites. Nature. 2009;459:945–949. doi: 10.1038/nature08104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crabb BS, Cooke BM, Reeder JC, Waller RF, Caruana SR, Davern KM, Wickham ME, Brown GV, Coppel RL, Cowman AF. Targeted gene disruption shows that knobs enable malaria-infected red cells to cytoadhere under physiological shear stress. Cell. 1997;89:287–296. doi: 10.1016/s0092-8674(00)80207-x. [DOI] [PubMed] [Google Scholar]
- 18.Waterkeyn JG, Wickham ME, Davern KM, Cooke BM, Coppel RL, Reeder JC, Culvenor JG, Waller RF, Cowman AF. Targeted mutagenesis of Plasmodium falciparum erythrocyte membrane protein 3 (PfEMP3) disrupts cytoadherence of malaria-infected red blood cells. EMBO J. 2000;19:2813–2823. doi: 10.1093/emboj/19.12.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Ooij C, Tamez P, Bhattacharjee S, Hiller NL, Harrison T, Liolios K, Kooij T, Ramesar J, Balu B, Adams J, Waters AP, Janse CJ, Haldar K. The malaria secretome: from algorithms to essential function in blood stage infection. PLoS Pathog. 2008;4:e1000084. doi: 10.1371/journal.ppat.1000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maier AG, Rug M, O'Neill MT, Brown M, Chakravorty S, Szestak T, Chesson J, Wu Y, Hughes K, Coppel RL, Newbold C, Beeson JG, Craig A, Crabb BS, Cowman AF. Exported proteins required for virulence and rigidity of Plasmodium falciparum-infected human erythrocytes. Cell. 2008;134:48–61. doi: 10.1016/j.cell.2008.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marti M, Good RT, Rug M, Knuepfer E, Cowman AF. Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science. 2004;306:1930–1933. doi: 10.1126/science.1102452. [DOI] [PubMed] [Google Scholar]
- 22.Hiller NL, Bhattacharjee S, van Ooij C, Liolios K, Harrison T, Lopez-Estrano C, Haldar K. A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science. 2004;306:1934–1937. doi: 10.1126/science.1102737. [DOI] [PubMed] [Google Scholar]
- 23.Gruring C, Heiber A, Kruse F, Flemming S, Franci G, Colombo SF, Fasana E, Schoeler H, Borgese N, Stunnenberg HG, Przyborski JM, Gilberger TW, Spielmann T. Uncovering common principles in protein export of malaria parasites. Cell Host Microbe. 2012;12:717–729. doi: 10.1016/j.chom.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 24.Bullen HE, Charnaud SC, Kalanon M, Riglar DT, Dekiwadia C, Kangwanrangsan N, Torii M, Tsuboi T, Baum J, Ralph SA, Cowman AF, de Koning-Ward TF, Crabb BS, Gilson PR. Biosynthesis, localisation and macromolecular arrangement of the Plasmodium falciparum translocon of exported proteins (PTEX) J Biol Chem. 2012;287:7871–7884. doi: 10.1074/jbc.M111.328591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischer K, Marti T, Rick B, Johnson D, Benting J, Baumeister S, Helmbrecht C, Lanzer M, Lingelbach K. Characterization and cloning of the gene encoding the vacuolar membrane protein EXP-2 from Plasmodium falciparum. Mol Biochem Parasit. 1998;92:47–57. doi: 10.1016/s0166-6851(97)00224-7. [DOI] [PubMed] [Google Scholar]
- 26.Riglar DT, Rogers KL, Hanssen E, Turnbull L, Bullen HE, Charnaud SC, Przyborski J, Gilson PR, Whitchurch CB, Crabb BS, Baum J, Cowman AF. Spatial association with PTEX complexes defines regions for effector export into Plasmodium falciparum-infected erythrocytes. Nat Commun. 2013;4:1415. doi: 10.1038/ncomms2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace AJ, Stillman TJ, Atkins A, Jamieson SJ, Bullough PA, Green J, Artymiuk PJ. E-coli hemolysin E (HlyE, ClyA, SheA): X-ray crystal structure of the toxin and observation of membrane pores by electron microscopy. Cell. 2000;100:265–276. doi: 10.1016/s0092-8674(00)81564-0. [DOI] [PubMed] [Google Scholar]
- 28.Eifler N, Vetsch M, Gregorini M, Ringler P, Chami M, Philippsen A, Fritz A, Muller SA, Glockshuber R, Engel A, Grauschopf U. Cytotoxin ClyA from Escherichia coli assembles to a 13-meric pore independent of its redox-state. EMBO J. 2006;25:2652–2661. doi: 10.1038/sj.emboj.7601130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mueller M, Grauschopf U, Maier T, Glockshuber R, Ban N. The structure of a cytolytic alpha-helical toxin pore reveals its assembly mechanism. Nature. 2009;459:726–730. doi: 10.1038/nature08026. [DOI] [PubMed] [Google Scholar]
- 30.Gehde N, Hinrichs C, Montilla I, Charpian S, Lingelbach K, Przyborski JM. Protein unfolding is an essential requirement for transport across the parasitophorous vacuolar membrane of Plasmodium falciparum. Mol Microbiol. 2009;71:613–628. doi: 10.1111/j.1365-2958.2008.06552.x. [DOI] [PubMed] [Google Scholar]
- 31.Matthews K, Kalanon M, Chisholm SA, Sturm A, Goodman CD, Dixon MW, Sanders PR, Nebl T, Fraser F, Haase S, McFadden GI, Gilson PR, Crabb BS, de Koning-Ward TF. The Plasmodium translocon of exported proteins (PTEX) component thioredoxin-2 is important for maintaining normal blood-stage growth. Mol Microbiol. 2013;89:1167–1186. doi: 10.1111/mmi.12334. [DOI] [PubMed] [Google Scholar]
- 32.Matz JM, Matuschewski K, Kooij TW. Two putative protein export regulators promote Plasmodium blood stage development in vivo. Mol Biochem Parasitol. 2013;191:44–52. doi: 10.1016/j.molbiopara.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 33.Beck JR, Muralidharan V, Oksman A, Goldberg DE. PTEX component HSP101 mediates export of diverse malaria effectors into host erythrocytes. Nature. 2014;511:592–595. doi: 10.1038/nature13574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elsworth B, Matthews K, Nie CQ, Kalanon M, Charnaud SC, Sanders PR, Chisholm SA, Counihan NA, Shaw PJ, Pino P, Chan JA, Azevedo MF, Rogerson SJ, Beeson JG, Crabb BS, Gilson PR, de Koning-Ward TF. PTEX is an essential nexus for protein export in malaria parasites. Nature. 2014;511:587–591. doi: 10.1038/nature13555. [DOI] [PubMed] [Google Scholar]
- 35.Lee S, Choi JM, Tsai FT. Visualizing the ATPase cycle in a protein disaggregating machine: structural basis for substrate binding by ClpB. Mol Cell. 2007;25:261–271. doi: 10.1016/j.molcel.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Effantin G, Ishikawa T, De Donatis GM, Maurizi MR, Steven AC. Local and global mobility in the ClpA AAA+ chaperone detected by cryo-electron microscopy: functional connotations. Structure. 2010;18:553–562. doi: 10.1016/j.str.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang T, Ploetz EA, Nagy M, Doyle SM, Wickner S, Smith PE, Zolkiewski M. Flexible connection of the N-terminal domain in ClpB modulates substrate binding and the aggregate reactivation efficiency. Proteins. 2012;80:2758–2768. doi: 10.1002/prot.24159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chow IT, Barnett ME, Zolkiewski M, Baneyx F. The N-terminal domain of Escherichia coli ClpB enhances chaperone function. FEBS Lett. 2005;579:4242–4248. doi: 10.1016/j.febslet.2005.06.055. [DOI] [PubMed] [Google Scholar]
- 39.Kirstein J, Schlothauer T, Dougan DA, Lilie H, Tischendorf G, Mogk A, Bukau B, Turgay K. Adaptor protein controlled oligomerization activates the AAA+ protein ClpC. EMBO J. 2006;25:1481–1491. doi: 10.1038/sj.emboj.7601042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Djuranovic S, Hartmann MD, Habeck M, Ursinus A, Zwickl P, Martin J, Lupas AN, Zeth K. Structure and activity of the N-terminal substrate recognition domains in proteasomal ATPases. Mol Cell. 2009;34:580–590. doi: 10.1016/j.molcel.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 41.Wojtyra UA, Thibault G, Tuite A, Houry WA. The N-terminal zinc binding domain of ClpX is a dimerization domain that modulates the chaperone function. J Biol Chem. 2003;278:48981–48990. doi: 10.1074/jbc.M307825200. [DOI] [PubMed] [Google Scholar]
- 42.Bonemann G, Pietrosiuk A, Diemand A, Zentgraf H, Mogk A. Remodelling of VipA/VipB tubules by ClpV-mediated threading is crucial for type VI protein secretion. EMBO J. 2009;28:315–325. doi: 10.1038/emboj.2008.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maurizi MR, Xia D. Protein binding and disruption by Clp/Hsp100 chaperones. Structure. 2004;12:175–183. doi: 10.1016/j.str.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 44.Ngansop F, Li H, Zolkiewska A, Zolkiewski M. Biochemical characterization of the apicoplast-targeted AAA+ ATPase ClpB from Plasmodium falciparum. Biochem Biophys Res Commun. 2013;439:191–195. doi: 10.1016/j.bbrc.2013.08.064. [DOI] [PubMed] [Google Scholar]
- 45.Sharma A, Dixit S. Structural insights into thioredoxin-2: a component of malaria parasite protein secretion machinery. Sci Rep. 2011;1:179. doi: 10.1038/srep00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng M, Cascio D, Egea PF. Crystal structure and solution characterization of the thioredoxin-2 from Plasmodium falciparum, a constituent of an essential parasitic protein export complex. Biochem Biophys Res Commun. 2015;456:403–409. doi: 10.1016/j.bbrc.2014.11.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee S, Sowa ME, Watanabe YH, Sigler PB, Chiu W, Yoshida M, Tsai FT. The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell. 2003;115:229–240. doi: 10.1016/s0092-8674(03)00807-9. [DOI] [PubMed] [Google Scholar]
- 48.Li J, Sha B. Crystal structure of the E. coli Hsp100 ClpB N-terminal domain. Structure. 2003;11:323–328. doi: 10.1016/s0969-2126(03)00030-3. [DOI] [PubMed] [Google Scholar]
- 49.Suzuki N, Hiraki M, Yamada Y, Matsugaki N, Igarashi N, Kato R, Dikic I, Drew D, Iwata S, Wakatsuki S, Kawasaki M. Crystallization of small proteins assisted by green fluorescent protein. Acta Cryst. 2010;D66:1059–1066. doi: 10.1107/S0907444910032944. [DOI] [PubMed] [Google Scholar]
- 50.Moon AF, Mueller GA, Zhong X, Pedersen LC. A synergistic approach to protein crystallization: combination of a fixed-arm carrier with surface entropy reduction. Protein Sci. 2010;19:901–913. doi: 10.1002/pro.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 52.Xia D, Esser L, Singh SK, Guo F, Maurizi MR. Crystallographic investigation of peptide binding sites in the N-domain of the ClpA chaperone. J Struct Biol. 2004;146:166–179. doi: 10.1016/j.jsb.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 53.Pietrosiuk A, Lenherr ED, Falk S, Bonemann G, Kopp J, Zentgraf H, Sinning I, Mogk A. Molecular basis for the unique role of the AAA+ chaperone ClpV in type VI protein secretion. J Biol Chem. 2011;286:30010–30021. doi: 10.1074/jbc.M111.253377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zeth K, Ravelli RB, Paal K, Cusack S, Bukau B, Dougan DA. Structural analysis of the adaptor protein ClpS in complex with the N-terminal domain of ClpA. Nat Struct Biol. 2002;9:906–911. doi: 10.1038/nsb869. [DOI] [PubMed] [Google Scholar]
- 55.Guo F, Esser L, Singh SK, Maurizi MR, Xia D. Crystal structure of the heterodimeric complex of the adaptor, ClpS, with the N-domain of the AAA+ chaperone, ClpA. J Biol Chem. 2002;277:46753–46762. doi: 10.1074/jbc.M208104200. [DOI] [PubMed] [Google Scholar]
- 56.Wang F, Mei Z, Qi Y, Yan C, Hu Q, Wang J, Shi Y. Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature. 2011;471:331–335. doi: 10.1038/nature09780. [DOI] [PubMed] [Google Scholar]
- 57.Blanchet CE, Svergun DI. Small-angle X-ray scattering on biological macromolecules and nanocomposites in solution. Ann Ref Phys Chem. 2013;64:37–54. doi: 10.1146/annurev-physchem-040412-110132. [DOI] [PubMed] [Google Scholar]
- 58.Chou TF, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, Patricelli MP, Hodder P, Rosen H, Deshaies RJ. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A. 2011;108:4834–4839. doi: 10.1073/pnas.1015312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Firestone AJ, Weinger JS, Maldonado M, Barlan K, Langston LD, O'Donnell M, Gelfand VI, Kapoor TM, Chen JK. Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature. 2012;484:125–129. doi: 10.1038/nature10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kabsch W. Xds. Acta Cryst. 2010;D66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 64.Classen S, Hura GL, Holton JM, Rambo RP, Rodic I, McGuire PJ, Dyer K, Hammel M, Meigs G, Frankel KA, Tainer JA. Implementation and performance of SIBYLS: a dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the advanced light source. J Appl Cryst. 2013;46:1–13. doi: 10.1107/S0021889812048698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, Tsutakawa SE, 2nd, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nature Meth. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rambo RP, Tainer JA. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 2011;95:559–571. doi: 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Svergun DI, Barberato C, Koch MH. CRYSOL: a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Cryst. 1995;28:768–773. [Google Scholar]
- 68.Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys J. 2013;105:962–974. doi: 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Svergun DI. Determination of the regularization parameter in indirect transform methods using perceptual criteria. J Appl Cryst. 1992;25:495–503. [Google Scholar]
- 70.Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- 71.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information