

Abstract
Cdc48 (also known as p97 or VCP) is an essential and highly abundant, double-ring AAA+ ATPase, which is ubiquitous in archaea and eukaryotes. In archaea, Cdc48 ring hexamers play a direct role in quality control by unfolding and translocating protein substrates into the degradation chamber of the 20S proteasome. Whether Cdc48 and 20S cooperate directly in protein degradation in eukaryotic cells is unclear. Two regions of Cdc48 are important for 20S binding, the pore-2 loop at the bottom of the D2 AAA+ ring and a C-terminal tripeptide. Here, we identify an aspartic acid in the pore-2 loop as an important element in 20S recognition. Importantly, mutation of this aspartate in human Cdc48 has been linked to familial amyotrophic lateral sclerosis (ALS). In archaeal or human Cdc48 variants, we find that mutation of this pore-2 residue impairs 20S binding and proteolytic communication but does not affect the stability of the hexamer or rates of ATP hydrolysis and protein unfolding. These results suggest that human Cdc48 interacts functionally with the 20S proteasome.
Keywords: AAA+ machine, ATP-dependent degradation, protein unfolding, proteasome
Introduction
Protein quality control in all cells depends upon protein remodeling machines of the AAA+ (ATPases associated with a variety of cellular activities) superfamily. Frequently, these enzymes work with self-compartmentalized peptidases to carry out protein degradation. In these cases, a AAA+ ring hexamer binds a specific protein substrate, unfolds it, and then translocates the polypeptide through an axial channel and into the degradation chamber of the peptidase.1 In eukaryotes, for example, the 20S proteasome interacts with the Rpt1-6 unfolding ring in the 19S regulatory particle to form the 26S proteasome. In archaea, the 20S proteasome can work with two different AAA+ enzymes, PAN (an Rpt1-6 homolog) or Cdc48, which is often called p97 or VCP.2 Cdc48 is also present in eukaryotic cells, where it plays roles in cell-cycle regulation, reassembly of nuclear and Golgi membranes, and protein degradation mediated by autophagy or the ubiquitin-proteasome-system.2–4 Although eukaryotic Cdc48 binds to the 20S proteasome in vitro,5 it is not known if this interaction is biologically important.
Each Cdc48 subunit contains an N-domain and two AAA+ modules, which form distinct D1 and D2 rings in the hexamer [Fig. 1(A)].6,8 Binding of archaeal Cdc48 to the 20S proteasome aligns the Cdc48 axial channel with the 20S pore, allowing unfolded substrates to be spooled into the 20S chamber for degradation.6 Two sets of Cdc48 residues are important for 20S binding [Fig. 1(B)]. First, a peptide motif consisting of a hydrophobic residue, tyrosine, and any residue (HbYX) at the C-terminus of each Cdc48 subunit docks into clefts on 20S.2 Similar tripeptide-docking interactions stabilize 20S complexes with PAN and the Rpt1-6 ring.9 Second, interactions mediated by residues in the pore-2 loop at the bottom of the D2 ring appear to stabilize Cdc48-20S complexes.5 Intriguingly, human exome sequencing identified Cdc48 mutations in this loop, which are linked to familial amyotrophic lateral sclerosis (ALS), an ultimately fatal disease that affects protein homeostasis and quality control.10,11 Here, we characterize the effects of changing residues in this pore loop, especially an aspartic acid, on the biochemical activities of Cdc48. A conservative Asp→Asn substitution impairs binding to and proteolytic communication with the 20S proteasome but has little effect on other Cdc48 functions. The same substitution as well as an Asp→His mutation are ALS-linked mutations. Hence, impairment of formation of a Cdc48•20S complex in human cells may contribute to disease development.
Figure 1.
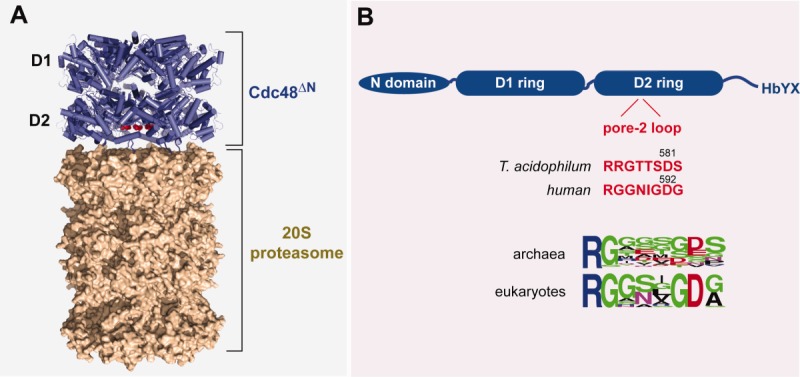
Cdc48 and the 20S proteasome. A: Model for the T. acidophilum Cdc48ΔN•20S proteasome based on the 3CF1 and 1PMA crystal structures and a low-resolution structure determined by electron microscopy.6 The red spheres mark the position of Asp592 in human Cdc48, corresponding to Asp581 in T. acidophilum Cdc48. B: In T. acidophilum and humans, the Cdc48 enzyme consists of a N-terminal domain, D1 and D2 AAA+ domains that mediate ATP hydrolysis and machine function, and a C-terminal tail with a terminal HbYX motif that stabilizes binding to the 20S proteasome. The positions and sequences of the pore-2 loop in the D2 ring of T. acidophilum and human Cdc48 are shown in red. Sequence conservation within this loop in archaeal and eukaryotic Cdc48 orthologs is shown in WebLogo representation.7
Results
Identification of a key 20S-binding residue in the pore-2 loop
Studies of Cdc48 from the archaeon Thermoplasma acidophilum showed that deletion of four residues (580–583) from the pore-2 loop of the D2 ring reduced apparent 20S affinity ∼10-fold.5 To determine the effects of substitutions for individual residues in this region, we constructed single R575Q, R576Q, G577A, T579A, S580A, D581N, and S582A mutations [Fig. 2(A)] in a variant of T. acidophilum Cdc48 from which the N-terminal domain and the C-terminal 20 amino acids, including the HbYX motif, were deleted (taCdc48ΔN/ΔC20). We were unable to construct the T578A mutant. Deletion of the N domain strengthens 20S binding and increases degradation activity, whereas deletion of the C-terminal 20 residues makes 20S binding completely dependent upon the pore-2 loop of the D2 ring.2,5
Figure 2.
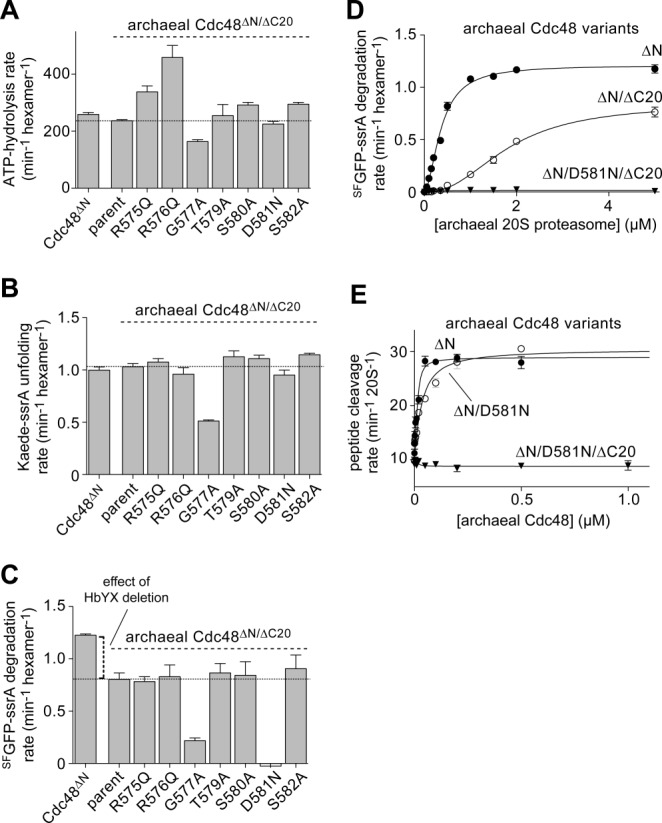
Effects of pore-2 loop mutations in T. acidophilum Cdc48 variants lacking the N domain and HbYX tails. A: Rates of hydrolysis of ATP (5 mM) by T. acidophilum Cdc48 variants (0.3 µM) were measured at 45°C with ATP regeneration. B: Rates of Kaede-ssrA (10 µM) unfolding by taCdc48 variants (0.3 µM) were measured at 45°C with 5 mM ATP and ATP regeneration. C: Rates of SFGFP-ssrA (10 µM) degradation by Cdc48 variants (0.3 µM) and the T. acidophilum 20S proteasome (5 µM) were measured at 45°C with 5 mM ATP and ATP regeneration. D: Rates of SFGFP-ssrA (10 µM) degradation were measured as a function of increasing ta20S proteasome concentration in the presence of taCdc48 variants (50 nM) with 5 mM ATP and ATP regeneration. Lines are fits to the Hill equation. E: Rates of nonapeptide (10 µM) cleavage by the ta20S proteasome (10 nM) were assayed in the presence of increasing concentrations of taCdc48 variants with 2 mM ATP at 45°C. The lines are fits to a quadratic equation for near-stoichiometric binding. In all panels, values are plotted as averages (n ≥ 3) ± SEM and the parental enzyme is taCdc48ΔN/ΔC20.
The parental taCdc48ΔN/ΔC20 enzyme and the T579A, S580A, D581N, and S582A variants catalyzed ATP hydrolysis at similar rates, the R575Q and R576Q mutants had increased hydrolysis activity, and the G577A mutant displayed a modest decrease in ATP turnover [Fig. 2(A)]. Next we assayed the rate at which the parent and mutant enzymes unfolded split Kaede-ssrA, a model protein substrate that loses fluorescence upon denaturation.12 All of the pore-loop variants except G577A had activities similar to the parent enzyme in this assay [Fig. 2(B)], establishing that these mutations do not alter the intrinsic protein-unfolding activity of archaeal Cdc48. Most of these mutants also supported maximal rates of 20S degradation of an ssrA-tagged variant of super-folder green fluorescent protein (SFGFP-ssrA) similar to the parental enzyme [Fig. 2(C); Table I]. The G577A variant showed a substantial Vmax defect in 20S degradation of SFGFP-ssrA, presumably reflecting the reduced ATP-hydrolysis and unfolding activities of this mutant. Notably, however, the D581N Cdc48 variant supported no detectable degradation over a wide range of 20S concentrations [Fig. 2(C,D)]. The D581N and R576Q mutants also failed to simulate degradation of a nonapeptide by 20S, whereas the other mutants exhibited the same maximal peptide cleavage rate as the parental enzyme (Table I). Why does the R576Q mutant support protein degradation but not peptide cleavage? The simplest possibility is that this variant binds 20S but does not open the axial gating pore unless active translocation of a protein substrate forces opening of the pore. By contrast, the D581N mutation appears to affect binding or functional communication with the 20S proteasome.
Table I.
Parameters for 20S binding and SFGFP-ssrA degradation
| Nonapeptide-cleavage stimulation |
SFGFP-ssrA degradation |
|||
|---|---|---|---|---|
| taCdc48ΔN Variant | Vmax (min−1) | Kapp (nM) | Vmax (min−1) | Kapp (μM) |
| Parent | 28 | 4.2 | 1.20 | 0.36 |
| ΔC20 | 13 | 45 | 0.84 | 1.8 |
| R575Q/ΔC20 | 12 | 60 | 0.79 | 1.8 |
| R576Q/ΔC20 | No stimulation | 0.80 | 1.2 | |
| G577A/ΔC20 | 13 | 27 | 0.19 | 2.2 |
| T579A/ΔC20 | 12 | 24 | 0.80 | 1.0 |
| S580A/ΔC20 | 13 | 43 | 0.86 | 1.6 |
| D581N/ΔC20 | No stimulation | No degradation | ||
| S582A/ΔC20 | 13 | 61 | 0.88 | 2.2 |
The interaction of the T. acidophilum 20S proteasome with T. acidophilum taCdc48ΔN and variants was assayed by titration experiments similar to those shown in Figs. 2D and 2E. For peptide cleavage, data were fit to a quadratic equation for tight binding. For protein degradation, data were fit to the Hill equation. Kapp for the peptide-cleavage experiments is the concentration of the taCdc48ΔN variant required for half-maximal stimulation. Kapp for SFGFP-ssrA proteolysis is the concentration of the ta20S proteasome required for half-maximal degradation. The ta20S proteasome alone cleaves the nonapeptide substrate at a rate of 9.5 min−1 but does not degrade SFGFP-ssrA. Values are averages (n ≥ 3) with SEM errors of 5–10%.
Partial rescue of d581 mutations by a functional HbYX motif
The experiments described above were performed using archaeal Cdc48 variants lacking the C-terminal HbYX motif, which stabilizes docking with the 20S proteasome. To determine if Asp581 in the pore-2 loop of the D2 ring is important for 20S binding/communication when the HbYX tails are present, we constructed and assayed the activities of the Cdc48ΔN/D581N and Cdc48ΔN/D581A variants. Compared to the parent enzyme, both of these variants hydrolyzed ATP at similar rates [Fig. 3(A)] and had similar Kaede-ssrA unfolding activities [Fig. 3(B)]. In combination with 20S, both Cdc48ΔN/D581N and Cdc48ΔN/D581A also supported degradation of SFGFP-ssrA but at 50% or less of the parental rate [Fig. 3(C)]. In titration experiments, Cdc48ΔN/D581N also supported degradation of SFGFP-ssrA at roughly half of the parental rate even when 20S was saturating [Fig. 3(D)], indicating that the mutant protease has a mild defect in engagement, unfolding, and/or translocation of this protein substrate. The apparent binding constant (Kapp) in these assays increased from 0.36 µM for the parental enzyme to 1.2 µM for the Cdc48ΔN/D581N mutant. We also titrated increasing concentrations of Cdc48ΔN and Cdc48ΔN/D581N against a fixed concentration of 20S and assayed cleavage of a nonapeptide that is degraded very slowly by 20S alone [Fig. 2(E)]. Both variants stimulated the peptidase activity of 20S to similar extents, but approximately 10-fold higher concentrations of Cdc48ΔN/D581N were required to achieve half-maximal stimulation. The 20S-binding defect of the Cdc48ΔN/D581N variant was similar in magnitude to the defect observed previously for Cdc48ΔN/Δ580–583,5 suggesting that Asp581 is the key 20S-binding residue within the pore-2 loop.
Figure 3.
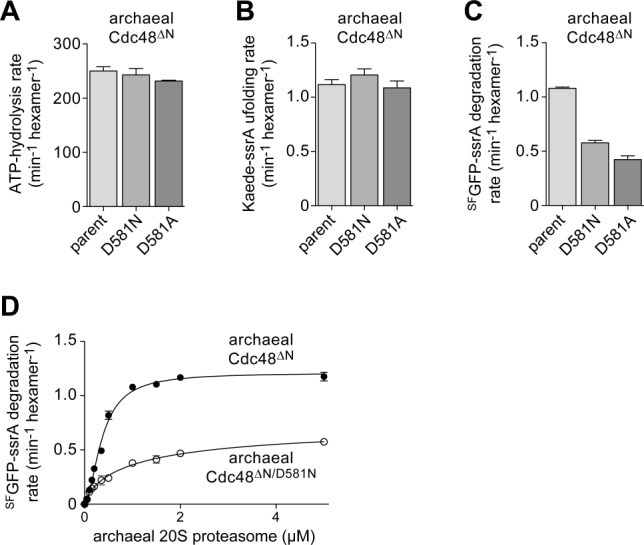
Archaeal D581 mutants show impaired 20S-proteasome binding and proteolytic communication in the presence of the HbYX tails. A: Rates of hydrolysis of ATP (5 mM) by taCdc48ΔN variants (0.3 µM) were measured at 45°C. B: Rates of unfolding of Kaede-ssrA (10 µM) by taCdc48ΔN variants (0.3 µM) with 5 mM ATP and ATP regeneration. C: Rates of SFGFP-ssrA (10 µM) degradation by taCdc48ΔN variants (0.3 µM) and the ta20S proteasome (5 µM) at 45°C with 5 mM ATP and ATP regeneration. D: Rates of SFGFP-ssrA (10 µM) degradation by taCdc48ΔN variants (50 nM) and increasing concentrations of the ta20S proteasome with 5 mM ATP and ATP regeneration. Lines are fits to the Hill equation. In all panels, values are plotted as averages (n ≥ 3) ± SEM.
An ALS mutation in human cdc48 impairs 20S communication
Asp581 in T. acidophilum Cdc48 corresponds to Asp592 in human Cdc48 [Fig. 1(B)], which is mutated to Asn or His in patients with familial ALS.10,11 Substrates degraded by human Cdc48•20S complexes have not been identified. Thus, to determine if the D592N disease allele impairs binding and proteolytic communication with human 20S, we used a human variant (hsCdc48ΔN/YY) engineered to recognize ssrA-tagged substrates as a consequence of an LA→YY mutation in the pore-1 loop of the D1 ring.5,13 In this hsCdc48ΔN/YY background, the D592N mutation had little effect on the rate of ATP hydrolysis or unfolding of Kaede-ssrA but resulted in an ∼50% decrease in the rate of degradation of SFGFP-ssrA [Fig. 4(A–C)]. Because of limited quantities of human 20S, the degradation experiment was performed using 0.5 µM human 20S and 1 µM hsCdc48ΔN/YY. Hence, reduced degradation activity could result from partial Cdc48-20S binding or from mild defects in the activity of the proteolytic complex. In the hsCdc48ΔN/YY/ΔC3 background, which is also missing the HbYX motif, the D592N mutation modestly reduced the rate of basal ATP hydrolysis [Fig. 4(A)], was only slightly defective for Kaede-ssrA unfolding [Fig. 4(B)], but almost completely eliminated 20S degradation of SFGFP-ssrA [Fig. 4(C)]. Thus, a major effect of the human D592N Cdc48 mutation is alteration of functional interactions with the human 20S proteasome.
Figure 4.
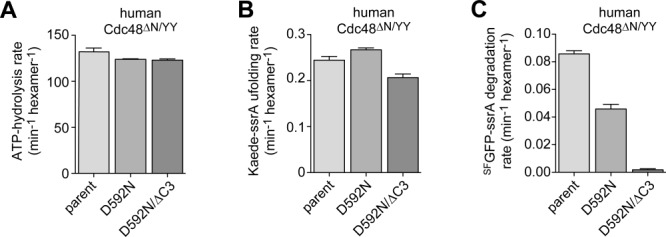
Effects of the ALS-linked D592N mutation on the enzymatic activities of human Cd48ΔN/YY. A: Rates of hydrolysis of ATP (5 mM) by hsCdc48ΔN/YY variants (1 µM) were measured at 37°C. B: Rates of unfolding of Kaede-ssrA (10 µM) by hsCdc48ΔN/YY variants (1 µM) were measured at 37°C in the presence of 5 mM ATP and ATP regeneration. C: Rates of SFGFP-ssrA (10 µM) degradation by hsCdc48ΔN/YY variants (1 µM) and the human 20S proteasome (0.5 µM) were measured at 37°C with 5 mM ATP and ATP regeneration. In all panels, values are plotted as averages (n ≥ 3) ± SEM.
Discussion
Mutations in the gene encoding human Cdc48 are linked to sporadic and familial forms of the neurodegenerative disorder ALS, which is characterized by accumulation of ubiquitinated protein aggregates.10,11 Most of these ALS mutations are located in the interface between the D1 AAA+ ring of Cdc48 and its N-terminal domain, which binds ubiquitin and adaptor proteins and normally packs against D1 in a coplanar fashion.6,14–16 Some ALS mutations seem to increase conformational flexibility of the N domain, thereby affecting binding of specific Cdc48 cofactors.17–20 Hence, it has been proposed that these mutations do not cause a general loss of Cdc48 function but rather affect specialized biological roles, consistent with the finding that transgenic mice carrying these alleles initially develop normally but develop disease as they age.21
The discovery of ALS-linked D592N and D592H mutations in the D2 pore-2 loop of human Cdc48 was intriguing,10,11 as previous studies showed that a deletion in this loop impaired archaeal Cdc48 binding to the 20S proteasome.5 Thus, we hypothesized that pore-2 loop ALS mutations might destabilize a human Cdc48•20S proteasome, perhaps leading to accumulation of ubiquitinated protein aggregates. Our biochemical experiments provide some support for this hypothesis, as we observe that the human D592N mutation weakens 20S binding and causes a defect in degradation activity but results in no significant alteration in ATPase or 20S-independent unfolding activity. In archaeal Cdc48, the corresponding mutation (D581N) also impairs 20S binding and reduces steady-state proteolytic activity of the complex ∼50%, as does the D581A mutation. These mild proteolytic defects could arise from slower productive substrate engagement, from the mutant Cdc48•20S complex being a poorer unfoldase than mutant Cdc48 alone, and/or from a reduced rate of substrate translocation. The aspartic acid affected by these mutations is highly conserved in eukaryotic Cdc48 [Fig. 1(B)] and is the key 20S-interaction residue in the pore-2 loop of T. acidophilum Cdc48. Although the mechanism by which the aspartic acid in the pore-2 loop stabilizes 20S binding is not known, we speculate that this negatively charged side chain may form a salt bridge with the positively charged α-amino group of the segment of the 20S alpha subunit that mediates access to the degradation chamber. By this model, the D592H mutation might have more severe consequences than the D592N mutation, as a consequence of steric or electrostatic clashes with 20S residues. Interestingly, a conserved aspartate in the pore-2 loops of the Rpt1-6 subunits of the 19S regulatory particle also contributes to 20S binding and proteolytic communication in the 26S proteasome.22
The role of eukaryotic Cdc48 in degradation of ubiquitinated proteins is uncertain. It is widely assumed that Cdc48 unfolds or extracts target proteins from membranes, allowing their subsequent shuttling to the 26S proteasome for degradation.23–25 However, other studies hint at a more direct role for eukaryotic Cdc48 in degradation, as archaeal Cdc48 interacts with the 20S proteasome to form a robust AAA+ degradation machine and eukaryotic Cdc48 retains 20S-binding determinants.2,5 As the ALS-linked human D592N mutation weakens 20S binding/communication, we suggest that disease could result from impaired degradation of specific substrates by Cdc48•20S or from increased activity of the 26S proteasome, caused by reduced competition for 20S binding by the mutant Cdc48 protein.
Materials and Methods
Cloning, expression, and purification
Mutations were created by PCR-based mutagenesis and verified by DNA sequencing. The expression and purification strategies for the taCdc48 and ta20S variants, human Cdc48 and E. coli ClpXΔN-His6 have been described.2,5,26 SFGFP-ssrA was expressed in E. coli BL21 (DE3) RIL cells at 25°C for 4 h after adding 1 mM IPTG at an OD600 of 1 and was purified by Ni2+-NTA affinity and ion-exchange chromatography as described.27 Kaede-ssrA was expressed, purified, and photo-cleaved as reported.12
The human 20S proteasome was isolated from whole-blood erythrocytes by a modification of an established protocol.28 Briefly, cells were lysed with a Dounce homogenizer in lysis buffer (20 mM HEPES- KOH, pH 7.5, 80 mM KOAc, 5 mM MgOAc2, and 0.1% (v/v) Triton-X100), and cellular debris was removed by centrifugation at 30,000 × g for 30 min. The supernatant was bound to DEAE Sepharose Fast Flow resin (GE Healthcare), washed with 20 mM HEPES-KOH (pH 7.5), 80 mM KOAc, and 5 mM MgOAc2, and 20S was eluted with an otherwise identical buffer containing 500 mM KOAc. The sample was then loaded onto a Superose 6 10/30 column (GE Healthcare) equilibrated in 50 mM Tris-HCl (pH 7.5), 1 M NaCl, 5 mM MgCl2, 1 mM DTT, and 1 mM EDTA. Fractions containing the 20S peptidase were combined and dialyzed against 20 mM HEPES (pH 7.5), 100 mM NaCl, 5 mM MgCl2, loaded onto a MonoQ (GE Healthcare) column and eluted by applying a linear gradation to 1 M NaCl. The human 20S peptidase was further purified by gel filtration on a S200 16/60 (GE Healthcare) column equilibrated in 50 mM HEPES-KOH (pH 7.5), 100 mM NaCl, 1 mM EDTA, and 1 mM DTT and was flash frozen in liquid nitrogen and stored at −80°C in single aliquots that were used once after thawing.
Biochemical assays
ATP hydrolysis by taCdc48 variants was measured by using an NADH-coupled colorimetric assay in the presence of 5 mM ATP and an ATP-regeneration system (10 U ml−1 pyruvate kinase, 10 U ml−1 lactate dehydrogenase, 1 mM NADH, and 7.5 mM phosphoenolpyruvate). Reactions were performed with Cdc48 variants (0.3 µM hexamer) in 30 µL of taPD buffer (50 mM HEPES-KOH, pH 7.5, 100 mM KCl, and 20 mM MgCl2) at 45°C. Reaction components were preincubated at 45°C for 10 min before starting the reactions by adding ATP. For human Cdc48 variants (1 µM), ATP hydrolysis was measured in hsPD buffer (50 mM HEPES-KOH, pH 7.5, 100 mM KCl, 20 mM MgCl2, and 3% (v/v) glycerol) at 37°C. Protein unfolding, measured by changes in photo-cleaved Kaede-ssrA fluorescence (excitation, 540 nm; emission, 580 nm), was assayed at 45°C in taPD buffer for archaeal Cdc48 variants and at 37°C in hsPD buffer for human Cdc48 variants in the presence of 5 mM ATP and an ATP-regeneration system (20 U mL−1 pyruvate kinase; 15 mM phosphoenolpyruvate). Degradation of SFGFP-ssrA was measured by loss of native fluorescence (excitation 467 nm; emission, 511 nm) using archaeal 20S and Cdc48 variants at 45°C in taPD buffer or human 20S and Cdc48 variants at 37°C the presence of 5 mM ATP and an ATP-regeneration system (20 U mL−1 pyruvate kinase; 15 mM phosphoenolpyruvate). To account for slow protein degradation of unfolded substrates by the free 20S proteasome, the rate of 20S degradation of SFGFP-ssrA after unfolding by E. coli ClpXΔN-His6 was subtracted from degradation rates determined in experiments with Cdc48 variants. The assay for nonapeptide degradation has been described previously.5
Acknowledgments
The authors thank Charlotte Lorentz for help in the initial steps in the project. They thank Ute E. Burkhardt (Dana Farber Cancer Institute) for providing human erythrocytes. The authors confirm no conflict of interest.
References
- 1.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 2.Barthelme D, Sauer RT. Identification of the Cdc48*20S proteasome as an ancient AAA+ proteolytic machine. Science. 2012;337:843–846. doi: 10.1126/science.1224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baek GH, Cheng H, Choe V, Bao X, Shao J, Luo S, Rao H. Cdc48: a Swiss army knife of cell biology. J Amino Acids. 2013;2013:183421. doi: 10.1155/2013/183421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14:117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- 5.Barthelme D, Sauer RT. Bipartite determinants mediate an evolutionarily conserved interaction between Cdc48 and the 20S peptidase. Proc Natl Acad Sci USA. 2013;110:3327–3332. doi: 10.1073/pnas.1300408110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barthelme D, Chen JZ, Grabenstatter J, Baker TA, Sauer RT. Architecture and assembly of the archaeal Cdc48*20S proteasome. Proc Natl Acad Sci USA. 2014;111:E1687–E1694. doi: 10.1073/pnas.1404823111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeLaBarre B, Brunger AT. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol. 2003;10:856–863. doi: 10.1038/nsb972. [DOI] [PubMed] [Google Scholar]
- 9.Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL. Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's alpha ring opens the gate for substrate entry. Mol Cell. 2007;27:731–744. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abramzon Y, Johnson JO, Scholz SW, Taylor JP, Brunetti M, Calvo A, Mandrioli J, Benatar M, Mora G, Restagno G, Chio A, Traynor BJ. Valosin-containing protein (VCP) mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33:2231 e2231–2231 e2236. doi: 10.1016/j.neurobiolaging.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez-Lage M, Falcone D, Hernandez DG, Arepalli S, Chong S, Schymick JC, Rothstein J, Landi F, Wang YD, Calvo A, Mora G, Sabatelli M, Monsurro MR, Battistini S, Salvi F, Spataro R, Sola P, Borghero G, Consortium I, Galassi G, Scholz SW, Taylor JP, Restagno G, Chio A, Traynor BJ. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glynn SE, Nager AR, Baker TA, Sauer RT. Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat Struct Mol Biol. 2012;19:616–622. doi: 10.1038/nsmb.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothballer A, Tzvetkov N, Zwickl P. Mutations in p97/VCP induce unfolding activity. FEBS Lett. 2007;581:1197–1201. doi: 10.1016/j.febslet.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 14.DeLaBarre B, Brunger AT. Nucleotide dependent motion and mechanism of action of p97/VCP. J Mol Biol. 2005;347:437–452. doi: 10.1016/j.jmb.2005.01.060. [DOI] [PubMed] [Google Scholar]
- 15.Rouiller I, DeLaBarre B, May AP, Weis WI, Brunger AT, Milligan RA, Wilson-Kubalek EM. Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat Struct Biol. 2002;9:950–957. doi: 10.1038/nsb872. [DOI] [PubMed] [Google Scholar]
- 16.Tang WK, Li D, Li CC, Esser L, Dai R, Guo L, Xia D. A novel ATP-dependent conformation in p97 N-D1 fragment revealed by crystal structures of disease-related mutants. EMBO J. 2010;29:2217–2229. doi: 10.1038/emboj.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Saiz V, Buchberger A. Imbalances in p97 co-factor interactions in human proteinopathy. EMBO Rep. 2010;11:479–485. doi: 10.1038/embor.2010.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ju JS, Miller SE, Hanson PI, Weihl CC. Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J Biol Chem. 2008;283:30289–30299. doi: 10.1074/jbc.M805517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ritz D, Vuk M, Kirchner P, Bug M, Schutz S, Hayer A, Bremer S, Lusk C, Baloh RH, Lee H, Glatter T, Gstaiger M, Aebersold R, Weihl CC, Meyer H. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat Cell Biol. 2011;13:1116–1123. doi: 10.1038/ncb2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weihl CC, Pestronk A, Kimonis VE. Valosin-containing protein disease: inclusion body myopathy with Paget's disease of the bone and fronto-temporal dementia. Neuromuscul Disord. 2009;19:308–315. doi: 10.1016/j.nmd.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Badadani M, Nalbandian A, Watts GD, Vesa J, Kitazawa M, Su H, Tanaja J, Dec E, Wallace DC, Mukherjee J, Caiozzo V, Warman M, Kimonis VE. VCP associated inclusion body myopathy and paget disease of bone knock-in mouse model exhibits tissue pathology typical of human disease. PLoS One. 2010;5:e13183. doi: 10.1371/journal.pone.0013183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beckwith R, Estrin E, Worden EJ, Martin A. Reconstitution of the 26S proteasome reveals functional asymmetries in its AAA+ unfoldase. Nat Struct Mol Biol. 2013;20:1164–1172. doi: 10.1038/nsmb.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braun S, Matuschewski K, Rape M, Thoms S, Jentsch S. Role of the ubiquitin-selective CDC48(UFD1/NPL4) chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J. 2002;21:615–621. doi: 10.1093/emboj/21.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, Jentsch S. Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell. 2001;107:667–677. doi: 10.1016/s0092-8674(01)00595-5. [DOI] [PubMed] [Google Scholar]
- 25.Christianson JC, Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol. 2014;21:325–335. doi: 10.1038/nsmb.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim YI, Burton RE, Burton BM, Sauer RT, Baker TA. Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol Cell. 2000;5:639–648. doi: 10.1016/s1097-2765(00)80243-9. [DOI] [PubMed] [Google Scholar]
- 27.Nager AR, Baker TA, Sauer RT. Stepwise unfolding of a beta barrel protein by the AAA+ ClpXP protease. J Mol Biol. 2011;413:4–16. doi: 10.1016/j.jmb.2011.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groettrup M, Ruppert T, Kuehn L, Seeger M, Standera S, Koszinowski U, Kloetzel PM. The interferon-gamma-inducible 11 S regulator (PA28) and the LMP2/LMP7 subunits govern the peptide production by the 20 S proteasome in vitro. J Biol Chem. 1995;270:23808–23815. doi: 10.1074/jbc.270.40.23808. [DOI] [PubMed] [Google Scholar]