

Abstract
The anti-diabetic drug metformin (1,1-dimethylbiguanide hydrochloride) reduces both the incidence and mortality of several types of cancer. Metformin has been shown to selectively kill cancer stem cells, and triple-negative breast cancer (TNBC) cell lines are more sensitive to the effects of metformin as compared to luminal breast cancer. However, the mechanism underlying the enhanced susceptibility of TNBC to metformin has not been elucidated. Expression profiling of metformin-treated TNBC lines revealed fatty acid synthase (FASN) as one of the genes most significantly downregulated following 24 h of treatment, and a decrease in FASN protein was also observed. Since FASN is critical for de novo fatty acid synthesis and is important for the survival of TNBC, we hypothesized that FASN downregulation facilitates metformin-induced apoptosis. Profiling studies also exposed a rapid metformin-induced increase in miR-193 family members, and miR-193b directly targets the FASN 3′UTR. Addition of exogenous miR-193b mimic to untreated TNBC cells decreased FASN protein expression and increased apoptosis of TNBC cells, while spontaneously immortalized, non-transformed breast epithelial cells remained unaffected. Conversely, antagonizing miR-193 activity impaired the ability of metformin to decrease FASN and cause cell death. Further, the metformin-stimulated increase in miR-193 resulted in reduced mammosphere formation by TNBC lines. These studies provide mechanistic insight into metformin-induced killing of TNBC.
Electronic supplementary material
The online version of this article (doi:10.1007/s12672-014-0188-8) contains supplementary material, which is available to authorized users.
Keywords: Metformin, Metformin Treatment, TNBC Cell, Mammosphere Formation, TNBC Cell Line
Introduction
Fatty acids are fundamental to energy production and storage, cellular structure, and as intermediates in the biosynthesis of hormones and other biologically important molecules. Metabolic dysfunction in human cancers has been well-recognized for a century following discovery by Otto Warburg, who postulated that anaerobic glycolysis was a major driver of tumorigenesis in human cancers [1]. Tumor cells frequently display alterations in lipid metabolism characterized by reliance on de novo fatty acid biosynthesis [2, 3] regardless of the availability of pre-formed extracellular lipids derived from the diet or adipose stores in the body [4, 5]. Fatty acid synthase (FASN), an enzyme required for de novo fatty acid synthesis, catalyzes the formation of long-chain fatty acids from acetyl-CoA and malonyl-CoA in an NADPH-dependent manner. FASN expression is increased in a wide range of tumors as compared to normal tissues and correlates with increased tumor grade and severity of clinical outcome (reviewed in [6]). The term “lipogenic switch” refers to overexpression and hyperactivity of acetyl-CoA carboxylase (ACACA), which generates malonyl-CoA. Silencing of ACACA or FASN kills breast cancer cells while non-transformed human breast epithelial cells are unaffected [7], confirming the importance of lipogenesis in cancer cell survival [8]. These observations suggested that de novo fatty acid synthesis provides a survival advantage to tumor cells and the possibility that FASN could serve as a target for anti-cancer therapeutics [6, 9–12]. Indeed, the inhibition of FASN by the obesity drug orlistat or analogues of cerulenin blocked proliferation and induced apoptosis of tumor cells in culture [13–15] and reduced growth of xenograft tumors in mice [16, 17], but did not affect normal differentiated cells in vitro [18]. More recently, various novel inhibitors of FASN have also demonstrated anti-cancer activity [12, 19–21].
It is now well-recognized that the anti-diabetic drug metformin reduces the incidence and mortality of breast cancer and other cancer types [22, 23]. The ability of metformin to improve disease-free and overall survival of breast cancer patients is currently being evaluated in phase III prospective randomized clinical trials [24–26]. Metformin blocked the ability of a high fat diet to stimulate growth of transplanted colon cancer cells, decreased diet-induced increases in serum insulin and phosphorylation of AKT, and was associated with reduced expression of FASN in colon cancer cells [27]. Metformin action in this model was suggested to be mediated by metformin-induced activation of AMP-dependent kinase (AMPK) and phosphorylation of AMPK substrates such as ACACA. More recently, the direct targeting of AMPK by metformin was shown to decrease the ACACA/FASN-driven lipogenic switch critical to self-renewal and pluripotency during the induction of pluripotent stem (iPS) cells from somatic cells [28, 29]. Recent studies by Thor and colleagues suggested both direct and indirect effects of metformin on breast cancer cells in vitro and in vivo [30–33] and implicated additional signaling molecules such as Stat3 as potential targets [34].
Triple-negative breast cancer (TNBC) is a subtype of breast cancer with the lowest 5-year survival rate, for which there is currently no effective targeted therapy. Intriguingly, metformin reduced proliferation and increased death in TNBC cells [30, 32–34]. We previously demonstrated that in comparison to luminal A breast cancer cell lines, TNBC lines are more sensitive to metformin with a tenfold lower IC50 [33]. Further, the response of TNBC cells to metformin is modulated by the concentration of glucose present in the culture media, with the super-physiological concentrations present in most media (15–17 mM) dampening the response of TNBC cell lines to metformin [33]. Metformin is also known to be particularly potent against breast cancer stem cells (CSCs) [35, 36], and TNBC cell lines contain a higher number of stem-like tumor-initiating cells [37, 38]. In order to gain insight into the initiating events by which metformin kills TNBC cells, we conducted global expression profiling of messenger RNAs (mRNAs) and micro RNAs (miRNAs) at early timepoints following metformin treatment of TNBC cells. Here, we report that metformin causes a dramatic reduction in FASN mRNA and protein that is facilitated by metformin-mediated upregulation of a miRNA that targets the FASN 3′UTR.
Methods
Cell Culture and Treatments
Human breast cancer-derived cell lines or alternate cell lines MCF7 and T47D (luminal A), MDA-MB-231, MDA-MB-468, BT-549, HCC70 (triple negative), MCF10A, and HEC50 were obtained from the American Type Culture Collection (Manassas, VA) or University of Colorado Cancer Center (UCCC) Tissue Culture Shared Resource. The identity of all cell lines was authenticated by analysis of short-tandem repeats by the UCCC DNA Sequencing and Analysis Shared Resource. All cell lines were confirmed to be free of mycoplasma contamination (using a MycoAlert detection kit, (Lonza Walkersville Inc, Walkersville, MD). All lines were maintained in DMEM; Nutrient Mix-F-12 (D-MEM/F-12 1:1; (Life Technologies/Invitrogen, Grand Island, NY), supplemented with 5–10 % fetal bovine serum (FBS; Sigma Aldrich Co., St. Louis, MO, or Thermo Fisher Scientific, Waltham, MA) with 2.50 mM l-glutamine and antibiotic/antimycotic solution containing 10,000 U/mL penicillin and 10,000 μg/mL streptomycin and 25 μg/mL amphotericin B (Hyclone™, Logan, UT). Media for BT-549 were additionally supplemented with 100 ng/mL insulin. Cells were maintained at 37 °C in a humidified atmosphere containing 95 % air and 5 % CO2. Cells were plated in 10 % FBS, then starved with glucose-free medium (Invitrogen) DMEM/F12 media with 0–5 % FBS for 24 h, then replenished with pure glucose supplemented at 5 or 17 mM/L glucose (Sigma Aldrich Co.). Metformin (1,1-dimethylbiguanide hydrochloride) (MP Biomedicals LLC, Solon OH) was dissolved in sterile water and used at the indicated concentration for each specific study. Cerulenin (Cat No. C2389) and C75 (Cat. No C5490) (Sigma) were dissolved in DMSO and used at indicated concentrations.
Microarrays
mRNA
MDA-MB-468 cells were seeded in DMEM medium containing 0.5 % FBS with either 5 or 17 mM glucose, with or without 10 mM metformin for 24 h. RNA was harvested using Trizol (Life Technologies) as previously described [33]. The integrity of the RNA was assessed and high quality RNA (RNA integrity number >7.0) was used in accordance with Affymetrix GeneChip© Whole Transcript (WT) sense target labeling assay protocol (Affymetrix, Santa Clara, CA). rRNA reduction, first round double strand-cDNA synthesis, ss-cDNA fragment, and labeling were performed as described by the Affymetrix GeneChip© WT sense target labeling assay manual. Affymetrix Human Exon 1.0 ST arrays were hybridized overnight in accordance to manual guidelines and performed in the University of Colorado Cancer Center Microarray Core Facility. Signal intensity estimate and P value for each of the samples were processed to determine the fold change in each cell line between control and metformin-treated samples. Analysis was performed using Partek (St. Louis, MO) and/or Genespring software (Agilent, Santa Clara, CA). Gene expression was filtered on statistical significance (Student’s t test, P < =0.05, Benjamini Hochberg correction) and genes with fold changes over control ≥2.0 or ≤ −2.0 were accepted for further analysis.
miRNA
MDA-MB-468, HCC70, T47D, and MCF7 cells were grown in DMEM/F12 medium containing 5 % FBS with either 5 or 10 mM glucose for 24 h, then treated with or without 10 mM metformin for 6 h (n = 3 replicates per group), and harvested in Trizol. RNA quality was assessed as above, and hybridizations to Affymetrix miRNA 2.0 arrays were performed by the core facility listed above and subsequent expression and statistical analysis performed as above.
Transfections
Lipofectamine 2000 (Life Technologies/Invitrogen) was incubated with miR-193b mimic or scrambled negative control (Life Technologies/Ambion) in serum-free media for 20 min prior to addition to cells at a final concentration of 50 nM. Cells were incubated at 37 °C for 4 h before the addition of FBS to a final concentration of 10 %. Protein was harvested 48 h post transfection.
Stable Cell Lines
Lentiviral vectors containing the miR-193b antagonist miR-193b-Zip, or the scrambled control miR-Scr-Zip (System Biosciences, Mountain View, CA), were prepared by the transfection of 293FT cells and virus harvested at 48 and 72 h. Filtered virus was added to MDA-MB-468, MDA-MB-231, and BT-549 cells in the presence of 6 μg/ml of polybrene, and stable cells were selected using 0.50 μg/ml (MDA-MB-468) or 1 μg/ml of puromycin (MDA-MB-231 or BT549) (Sigma Aldrich Co.).
Western Blot
Whole cell protein extracts were prepared using RIPA lysis buffer, 30–50 μg of total protein was size-separated on SDS-PAGE and transferred to PVDF. Blots were probed with antibodies to FASN (Antibody #3189), phospho-ATP-citrate lyase (Ser455; P-ACLY, Antibody #4331), ATP-citrate lyase (ACLY; Antibody #43320), phospho-acetyl-CoA carboxylase (Ser79; P-ACC, Antibody #3661), acetyl-CoA carboxylase (ACC; Antibody #3676), phospho-AMPKα (Thr172; P-AMPK, Antibody #2531), AMPKα (AMPK; Antibody #2532), PARP (Antibody #9542), or IGF-I receptor β (IGF1R; Antibody #3018) which were all obtained from Cell Signaling Technology, Inc. (Beverly MA). Antibodies to β-actin (AC-75), glyceraldehyde 3-phosphate dehydrogenase [GAPDH (clone GAPDH-71.1)], and alpha tubulin (clone B-5-1-2) and secondary antibody rabbit anti-mouse horseradish peroxidase (HRP) were obtained from Sigma Aldrich Co. PSTAIR antibody was obtained from Abcam (Cambridge, MA). Goat anti-rabbit HRP was obtained from MP Biomedicals. Goat anti-mouse or rabbit immunoglobulin labeled with Alexa Fluor® 680 was obtained from Molecular Probes, (Eugene, OR). For quantitation, blots were scanned using an Odyssey Infrared Imager (LI-COR Biosciences, Lincoln, NE), and FASN signal normalized to GAPDH expression.
miRNA qRT-PCR
MDA-MB-468 cells were treated with 10 mM metformin for indicated time points (0, 6, 12, or 24 h), and RNA was harvested using Trizol. For mature miRNAs, the Taqman miRNA Reverse Transcription kit (Applied Biosystems Inc., (ABI) Foster City, CA) was used to generate complementary DNA (cDNA) from 50 ng of total RNA, using miRNA-specific primers for miR-193a-3p, miR-193b, and U6 (for normalization). MiRNA-specific reverse transcription primers and probes were used for qRT-PCR. For the pri-miRNAs, 1 μg of total RNA was treated with DNase1 (Life Technologies/Invitrogen) for 15 min at room temperature. RNA was reverse-transcribed into cDNA in a reaction containing reaction buffer, 10 mM dNTPs, RNase inhibitor, random hexamers (250 ng), and 50 U of MuLV-RT (ABI). The reaction proceeded at 22 °C for 10 min, then at 37 °C for 1 h. The miR-193b pri-taqman assay (Hs03303897_pri) and miR-193a pri-taqman assay (Hs03303307_pri) containing probes specific for these miRNAs were obtained from ABI and used for qPCR. All qPCR was performed on the iQ5 cycler (Biorad, Hercules, CA) using Taqman Universal PCR Master Mix (ABI #4324018). The relative miRNA levels were calculated using the comparative Ct method (ΔΔCt). Briefly, the Ct (cycle threshold) values for U6 or 18S were subtracted from Ct values of the miRNA to achieve the ΔCt value. The 2−ΔCt was calculated and then divided by a control sample to achieve the relative miRNA levels (ΔΔCt). Reported values are the means and standard errors of three biological replicates.
Luciferase Assays
To determine if miR-193b directly targets a bioinformatically predicted site in the FASN 3’UTR, HEC50 cells (15,000 cells per well) were plated into a 96-well plate and either transfected with 50 nM of a negative control mimic, mimic for miR-193b, antagonist for miR-193b (α193, Thermo Scientific/Dharmacon, Pittsburg, PA) or a combination of the mimic and antagonist. After 24 h, the complete FASN 3′UTR cloned downstream of luciferase firefly reporter plasmid pMIR-REPORT (Origene, Rockville, MD) (0.196 μg) and a Renilla luciferase normalization plasmid pRL-SV40 (0.004 μg) were introduced using Lipofectamine 2000. Cells were harvested 48 h later for analysis using the Dual Luciferase Reporter assay system (Promega, Madison, WI).
Real-Time Quantitative Reverse Transcriptase-PCR (qRT-PCR) for FASN
MDA-MB-468, MD-MB-231, and BT-549 cells were seeded in medium containing 5 mM glucose for 24 h then treated with 5 or 10 mM metformin for 0–72 h, and RNA was harvested using Buffer RLT RNeasy Mini kit (Qiagen, Germantown, MD). For qRT-PCR analysis of FASN, cDNA was created using Verso cDNA Synthesis Kit (Cat #AB-1453/A, Thermo Scientific, Houston, TX) and 1 μg of total RNA. Pre-designed gene-specific primer and probe sets were obtained from SA Biosciences (Valencia, CA), for human FASN gene (Cat No#330001 PPH01012B) NM_004104.4; and beta-actin (ACTB) (Cat No#330001 PPH00073 G) NM_001101.3. qRT-PCR synthesis of FASN was performed using DyNAmo Flash SYBR Green qPCR kit (Cat No#F-415 L, ABI) according to the manufacturer’s protocol on an ABI 7500 Fast Real-Time PCR System. The relative mRNA levels were calculated using the comparative Ct method (ΔΔCt). Briefly, the Ct (cycle threshold) values for the BACTIN were subtracted from Ct values of the FASN mRNA to achieve the ΔCt value. The 2−ΔCt was calculated and then divided by a control sample to achieve the relative mRNA levels (ΔΔCt). Reported values are the means and standard errors of three biological replicates.
Apoptosis Assays
Vibrant Apoptosis Kit with YO-PRO®-1 and PI (Molecular Probes) was used in accordance with the manufacturer’s protocol as described previously [39]. In brief, MDA-MB-231 and BT-549 cells were plated in 10 % FBS DMEM/F-12 medium, then starved with glucose-free DMEM/F12 media (Invitrogen) with 0–5 % FBS for 24 h, then replenished with 5 mM glucose (Sigma Aldrich Co.) in the presence or absence of 5 or 10 mM metformin for 72 h. Cells were harvested after the treatment period, washed with chilled PBS, and counted for cell density of ~1 × 106 cells/mL in PBS in 1-mL assay. Cells were then stained using Vibrant apoptosis assay, as per manufacturer’s guidelines. Gated percentage of cells in region A2 is representative of dead cells and is indicated with percentage of dead cells relative to the entire population of gated cells, excluding debris. Total and dead cells were counted, and the percentage of dead cells was calculated.
HOECHST 33342 Assay. MDA-MB-231 and BT-549 cells were treated as outlined in Membrane Permeability/Dead Cell Apoptosis kit and stained with Hoechst 33342 NucBlue® Live Read Probes Reagent (Molecular Probes) as previously described.
Live/Dead® Viability/Cytotoxicity kit. MDA-MB-231 and BT-549 cells grown on sterile coverslips and treated as outlined in Membrane Permeability/Dead Cell Apoptosis kit then washed with PBS followed by fixation using 70 % methanol for 30 min and stained with live/dead viability/cytotoxicity kit for mammalian cells (Molecular Probes) as per manufacturer’s guidelines and described in [40]. Experiments are representative of three independent experiments ± SE.
Mammosphere Assay
Transduced BT-549 or MDA-MB-231 cells expressing either miR-Scr-Zip or miR-193b-Zip or parental cell lines (MDA-MB-468, BT-549, MDA-MB-231) were cultured in suspension in Mammocult™ complete medium (Stem Cell Technologies Inc, Vancouver, BC, Canada) at 1,000 cells/mL. The propagation of spheres in vitro was conducted with gentle centrifugation and disassociation into single cells to generate mammospheres, a procedure similar to that described previously [41]. The day following plating, cultures were treated with increasing concentrations of metformin (0–10 mM), cerulenin (0–10 μM), or C75 (0–15 μM) for 7–10 days. Primary mammospheres were collected and disassociated with 0.25 % trypsin into single cells, counted, and re-seeded at 1,000 cells/mL in Mammocult™ complete medium to generate secondary passage (2P) mammospheres. On the day following plating the 2P cultures, we re-treated cells with indicated concentrations of metformin, cerulenin, or C75 for another 7–10 days. 2P mammospheres were counted and imaged using a Nikon phase microscope at ×10 power. Mammospheres greater than 75 μm in diameter were counted.
Flow Cytometric Analysis
Single-cell suspension from 2P mammosphere cultures were obtained as described above. Cells were washed once with phosphate-buffered saline (PBS) and then harvested with 0.05 % trypsin/0.025 % EDTA. Detached cells were washed with PBS containing 5 % BSA and resuspended with wash buffer at 106 cells/100 μl. Combinations of fluorochrome-conjugated monoclonal antibodies obtained from BD Biosciences (San Diego, CA) against human CD44 (FITC; cat. #555478) and CD24 (PE; cat.#555428) or their respective isotype controls were added to the cell suspension at concentrations recommended by the manufacturer and incubated at 4 °C in the dark for 30–40 min. The labeled cells were then processed by Gallios (Beckman Coulter, Danvers, MA) flow cytometry processor at the University of Colorado Denver Flow Cytometry Shared Resource. Aldehyde dehydrogenase (ALDH) positive cells were quantitated using the ALDEFLUOR kit (Stem Cell Technologies) described [42].
Results
Metformin Decreases Expression of FASN and Many Other Genes Encoding Proteins Involved in Fatty Acid and Cholesterol Biosynthesis
Gene expression profiling revealed that the expression of FASN decreased 8.2-fold in MDA-MB-468 cells treated with 10 mM metformin for 24 h. Interestingly, seven of the top ten genes decreased by metformin (Table 1) are involved in fatty acid and cholesterol biosynthesis. The change in FASN expression was the second highest, with the greatest change being an 8.9-fold decrease in squalene epoxidase (SQLE). SQLE catalyzes the first oxygenation step in sterol biosynthesis and is a rate-limiting enzyme in this biosynthetic pathway. The third most highly decreased gene was HMGCS1 (decreased by eightfold), which encodes an enzyme involved in cholesterol biosynthesis that condenses acetyl-CoA with acetoacetyl-CoA to form HMG-CoA. LSS, which encodes lanosterol synthase (2,3-oxidosqualene-lanosterol cyclase) that catalyzes the first step in the biosynthesis of cholesterol, steroid hormones, and vitamin D, was downregulated by 6.7-fold. INSIG1 (insulin-induced gene 1 protein), downregulated by 6.1-fold, is an insulin-induced gene encoding an endoplasmic reticulum membrane protein that serves a critical role in regulating the transcription of genes involved in fatty acid and cholesterol biosynthesis by binding to the sterol-sensing domains of SREBP cleavage-activating protein (SCAP). ACAT2 (acetyl-coenzyme A acetyltransferase 2) (5.5-fold downregulated by metformin) encodes an enzyme involved in lipid metabolism and MVD (5.3-fold downregulated), which encodes the mevalonate pyrophosphate decarboxylase, catalyzes the conversion of mevalonate pyrophosphate into isopentenyl pyrophosphate in the early steps in cholesterol biosynthesis. Additionally, we list the top fatty acid biosynthetic, lipid metabolic, and lipid biosynthetic processes altered by metformin in MDA-MB-468 cells (Table 2). In summary, the top genes decreased by metformin are involved in cholesterol biosynthesis and lipid metabolism (Table 3). In this study, we specifically focus upon the mechanisms regulating the changes in FASN expression since it was strongly decreased by metformin and is a critical enzyme required for de novo fatty acid synthesis.
Table 1.
Top 15 Genes downregulated by 10 mM metformin treatment for 24 h in MDA-MB-468 cells at 5 mM glucose
| Gene name | Description | Fold change |
|---|---|---|
| SQLE | Squalene epoxidase | −8.9 |
| FASNa | Fatty acid synthase | −8.2 |
| HMGCS1 | 3-Hydroxy-3-methylglutaryl-CoA synthase 1 | −8.0 |
| TF | Transferrin | −6.7 |
| LSSa | Lanosterol synthase | −6.7 |
| INSIG1 | Insulin-induced gene 1 | −6.1 |
| SKA2 | Spindle and kinetochore-associated complex subunit 2 | −6.0 |
| HIST1H3A | Histone cluster 1, H3a | −5.7 |
| HIST1H1A | Histone cluster 1, H1a | −5.6 |
| ACAT2 | Acetyl-CoA acetyltransferase 2 | −5.5 |
| MVD | Mevalonate (diphospho)decarboxylase | −5.3 |
| HMGCR | 3-Hydroxy-3-methylglutaryl-CoA reductase | −5.3 |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 | −5.3 |
| DHCR24 | 24-Dehydrocholesterol reductase | −5.0 |
| IL22RA2 | Interleukin 22 receptor, alpha 2 | −5.0 |
aThese genes were also significantly downregulated in MDA-MB-231 treated with 10 mM metformin at 5 mM glucose
Table 2.
Top fatty acid biosynthetic and lipid metabolic/biosynthesis processes altered by metformin in MDA-MB-468 cells
| Gene name | Description | Fold change |
|---|---|---|
| FASNa | Fatty acid synthase | −8.2 |
| HMGCS1 | 3-Hydroxy-3-methylglutaryl-CoA synthase 1 | −8.0 |
| ACAT2 | Acetyl-CoA acetyltransferase 2 | −5.5 |
| SCD | Stearoyl-CoA desaturase | −4.31 |
| ACLY | ATP-citrate lyase | −4.26 |
| ACSL1 | Acyl-CoA synthetase long-chain family member 4 | −2.69 |
| PC | Pyruvate carboxylase | −2.40 |
| ACSL4 | Acyl-CoA synthetase long-chain family member 4 | −2.18 |
aThese genes were also significantly down regulated in MDA-MB-231 treated with 10 mM metformin at 5 mM glucose
Table 3.
115F Genes down regulated by metformin and altered by miR-193
| Gene symbol | RefSeq | Fold‐change (g5 met vs. g5) | p value (g5 met vs. g5) | Prediction algorithm | Description |
|---|---|---|---|---|---|
| AFAP1‐AS | NR_026892 | −2.89766 | 1.61E‐06 | MiRanda | AFAP1 antisense RNA (non‐protein coding) |
| ARPC4 | NM_005718 | −2.06384 | 5.77E‐05 | MiRanda | Actin-related protein 2/3 complex, subunit 4, 20 kDa |
| CCDC71 | NM_022903 | −2.01874 | 0.0025781 | MiRanda | Coiled‐coil domain containing 71 |
| CCM2 | NM_031443 | −2.18803 | 1.50E‐06 | MiRanda | Cerebral cavernous malformation 2 |
| CLSTN1 | NM_001009566 | −2.1818 | 2.83E‐06 | Target Scan | Calsyntenin 1 |
| CNOT6 | NM_015455 | −2.25598 | 4.71E‐07 | Target Scan | CCR4‐NOT transcription complex, subunit 6 |
| CREB3L4 | NM_130898 | −3.35141 | 2.39E‐05 | MiRanda | cAMP responsive element binding protein 3‐like 4 |
| FASN | NM_004104 | −8.19682 | 1.75E‐08 | MiRanda | Fatty acid synthase |
| FEN1 | NM_004111 | −2.42731 | 7.58E‐08 | MiRanda | Flap structure‐specific endonuclease 1 |
| HN1L | NM_144570 | −2.40156 | 1.10E‐07 | Leivonen et al. 2011 | Hematological and neurological expressed 1‐like |
| HNRNPUL2 | NM_001079559 | −2.55145 | 7.07E‐09 | Target Scan | Heterogeneous nuclear ribonucleoprotein U‐like |
| IDI1 | NM_004508 | −3.94434 | 1.34E‐10 | Target Scan | Isopentenyl‐diphosphate delta isomerase 1 |
| IGSF3 | NM_001542 | −2.20126 | 9.90E‐06 | Leivonen et al. 2011 | Immunoglobulin superfamily, member 3 |
| IVD | NM_002225 | −3.21389 | 5.95E‐06 | MiRanda | Isovaleryl‐CoA dehydrogenase |
| KIT | NM_000222 | −3.11623 | 7.14E‐06 | MiRanda | Kit Hardy‐Zuckerman 4 feline sarcoma viral oncogene homologue |
| KRT19 | NM_002276 | −2.18542 | 5.94E‐07 | Leivonen et al. 2011 | Keratin 19 |
| LETM1 | NM_012318 | −2.16657 | 1.13E‐05 | Leivonen et al. 2011 | Leucine zipper‐EF‐hand-containing transmembrane protein 1 |
| MAP3K11 | NM_002419 | −2.50877 | 5.11E‐06 | MiRanda | Mitogen‐activated protein kinase kinase kinase 11 |
| MRPL28 | NM_006428 | −2.76247 | 7.05E‐06 | MiRanda | Mitochondrial ribosomal protein L28 |
| MUC1 | NM_001018016 | −3.7317 | 1.29E‐06 | MiRanda | Mucin 1, cell surface associated |
| NCAPD2 | NM_014865 | −2.0363 | 5.16E‐07 | Leivonen et al. 2011 | Non‐SMC condensin I complex, subunit D2 |
| NSDHL | NM_015922 | −2.25004 | 1.65E‐06 | Leivonen et al. 2011 | NAD(P)-dependent steroid dehydrogenase‐like |
| ODC1 | NM_002539 | −2.25215 | 6.14E‐07 | MiRanda | Ornithine decarboxylase 1 |
| QPRT | NM_014298 | −2.19681 | 2.52E‐05 | MiRanda | Quinolinate phosphoribosyltransferase |
| RBM14 | NM_006328 | −3.15718 | 5.21E‐07 | MiRanda | RNA binding motif protein 14 |
| RFT1 | NM_052859 | −2.03807 | 0.0002474 | Target Scan | RFT1 homologue (S. cerevisiae) |
| SCNN1A | NM_001038 | −2.86166 | 1.56E‐06 | MiRanda | sodium channel, non-voltage‐gated 1 alpha |
| SDF2L1 | NM_022044 | −2.11541 | 9.90E‐05 | MiRanda | Stromal cell‐derived factor 2‐like 1 |
| SH3TC2 | NM_024577 | −2.21405 | 0.0001689 | MiRanda | SH3 domain and tetratricopeptide repeats 2 |
| SLC23A2 | NM_005116 | −2.12693 | 0.00058 | Target Scan | Solute carrier family 23 (nucleobase transporters), member 2 |
| SLC39A7 | NM_006979 | −2.54712 | 2.56E‐06 | MiRanda | Solute carrier family 39 (zinc transporter), member 7 |
| ST6GALNAC2 | NM_006456 | −2.15331 | 7.38E‐05 | Leivonen et al. 2011 | ST6 (alpha‐N‐acetyl‐neuraminyl‐2,3‐beta‐galactosyl‐1,3)‐N‐ac |
| SURF4 | NM_033161 | −2.21445 | 2.40E‐08 | Leivonen et al. 2011 | Surfeit 4 |
| TM7SF2 | NM_003273 | −3.63654 | 1.69E‐08 | Leivonen et al. 2011 | Transmembrane 7 superfamily member 2 |
| TMED2 | NM_006815 | −2.05481 | 5.36E‐06 | MiRanda | Transmembrane emp24 domain trafficking protein 2 |
| TXLNA | NM_175852 | −2.08624 | 5.46E‐06 | Leivonen et al. 2011 | Taxilin alpha |
| WDR82 | NM_025222 | −2.57578 | 1.34E‐05 | Target scan | WD repeat domain 82 |
To validate results obtained by gene expression profiling, qRT-PCR for FASN mRNA was performed, and results indicated that the metformin-induced decrease in FASN mRNA is detectable as early as 4 h following metformin treatment of MDA-MB-468 cells and that inhibition of FASN expression was maintained over a 72-h period (Fig. 1a). Consistent data were observed in two additional TNBC cell lines, BT-549 and MDA-MB-231 (Fig. 1a). The decrease in RNA levels was reflected at the protein level, where metformin treatment caused a decrease in FASN protein at 24 and 48 h (Fig. 1b). The decrease in FASN protein level was quantified in the lower panels shown in Fig. 1b and is significant at 48 h.
Fig. 1.
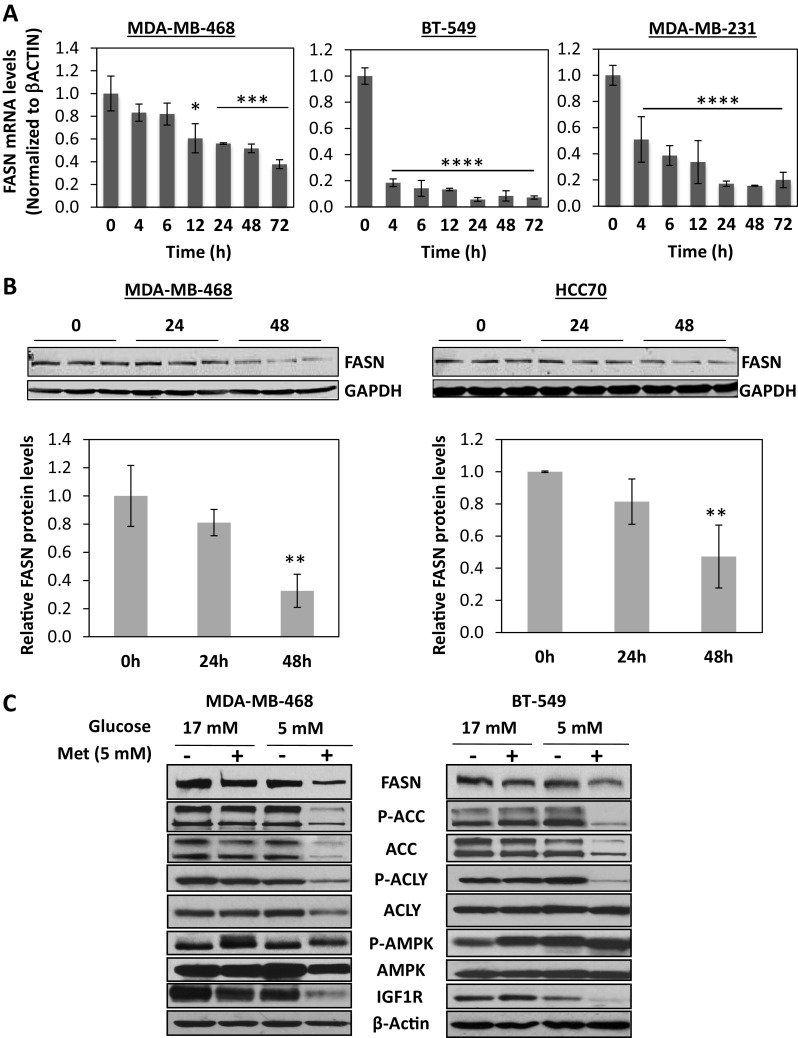
Metformin decreases FASN mRNA and protein in TNBC cell lines. a MDA-MB-468, BT-549, and MDA-MB-231 cells were treated with either 5 mM (BT-549 and MDA-MB-468) or 10 mM (MDA-MB-231) metformin for 0–72 h in 5 mM glucose, and qRT-pCR for FASN was performed. Shown are the averages of triplicate values, normalized to β-actin expression. *P < 0.01, ***P < 0.001, ****P < 0.0001, ANOVA with Tukey’s multiple comparisons test. b TN cell lines (HCC70 and MDA-MB-468) were plated in media containing 10 % FBS and changed to media with 5 % FBS, 5 mM glucose the next day. After 24 h, 10 mM metformin was added and cells harvested 24 and 48 h later. Immunoblots were probed for FASN protein. Blots were scanned, quantitated using an Odyssey infrared imager, and normalized to GAPDH (bottom). **P < 0.01, ANOVA with Tukey’s multiple comparisons test. c MDA-MB-468 cells were glucose starved for 24 h, then replenished with media containing 5 or 17 mM glucose for 24 h and treated with or without 5 mM metformin for 24 h. Whole-cell protein extracts were prepared and the level of FASN, P-ACC, ACC, P-ACLY, ACLY, P-AMPK, AMPK, IGFIR, and β-actin determined by immunoblot analysis. Experiments are representative of three individual experiments
We previously demonstrated that TNBC lines are more sensitive to metformin with an IC50 tenfold lower than that observed in luminal A cell lines and that the super-physiological concentrations of glucose present in most media (15–17 mM) dampen the response of TNBC cell lines to metformin [33]. Therefore, we decided to examine how varying glucose concentrations affected the ability of metformin to downregulate FASN protein. MDA-MB-468 cells were glucose starved for 24 h prior to supplementation with medium containing either 17 or 5 mM glucose for 24 h. Cells were then treated with or without metformin for 24 h, and the amount of FASN protein was quantitated by immunoblot analysis, along with other components of the fatty acid synthesis pathway and signaling proteins of interest. Metformin-induced decrease in FASN protein was detectable in cells cultured in 5 mM glucose, but not in the presence of 17 mM glucose. ACC was also decreased in metformin-treated cells in 5 mM, but not at 17 mM glucose (Fig. 1c). In contrast, total ACLY was not altered in BT-549 cells by metformin treatment at either glucose concentrations, but is altered in MDA-MB-468 cells. Phosphorylation of both ACC and ACLY was decreased by metformin when glucose was present at 5 mM but not at 17 mM (Fig. 1c). The amount of AMP-dependent protein kinase (AMPK), which is known to be activated by metformin [43], was increased by metformin in the presence of either 5 or 17 mM glucose (Fig. 1c). The amount of the insulin-like growth factor 1 receptor (IGF1R) was also significantly decreased by metformin treatment in a glucose concentration-dependent manner (Fig. 1c).
Metformin Upregulates miR-193a-3p and miR-193b in TNBC Cells
miRNA expression profiling of breast cancer cells treated with metformin for 6 h demonstrated that members of the miR-193 family were significantly increased in TNBC cell lines (Fig. 2a). The increase in miR-193a-3p and miR-193b following 12 and 24 h of metformin treatment was confirmed using qRT-PCR (Fig. 2b, c, left panels). Further, the primary miRNAs that give rise to miR-193a-3p and miR-193b (pri-mir-193a and pri-mir-193b) also increased significantly as early as 6 h post metformin treatment (Fig. 2b, c, right panels), suggesting that the upregulation of these miRNAs was mediated at the transcriptional level.
Fig. 2.

Metformin upregulates miR-193 family members in TNBC cells. Cells were plated in media containing 10 % FBS and changed to media with 5 % FBS, 5 or 10 mM glucose the next day. After 24 h, 10 mM metformin was added to plates (n = 3) for 6 h prior to harvesting RNA. miRNA expression profiling was performed on Affymetrix miRNA 2.0 chips. a Table showing fold change with P ≤ 0.05 for miR-193 family members in TN cell lines. Taqman qRT-PCR for mature miR-193a-3p (b, left) and the primary transcript (b, right) and mature miR-193b (c, left) and primary transcript (c, right) for MDA-MB-468-treated cells. For miRNA transcript quantification, *P < 0.05, **P < 0.01, ANOVA with Tukey’s multiple comparisons test. For primiRNA quantification, **P < 0.01, ***P < 0.001, unpaired t test
miR-193b Targets FASN and Kills Breast Cancer Cells, But Not Normal Cells
Endogenous levels of mature miR-193b as quantified by qRT-PCR were higher in luminal A cell lines compared to TNBC cell lines (Fig. 3a), and this corresponded with our previous findings [44]. Treatment of BT-549 cells with a miR-193b mimic caused a dramatic decrease in FASN protein levels (Fig. 3b) in a dose-dependent manner (Fig. 3c). The decrease in FASN protein coincided with a marked increase in PARP cleavage, indicating the induction of apoptosis (Fig. 3b, c). In contrast, the transfection of miR-193 mimic into normal immortalized mammary epithelial cell line MCF10A had no effect upon PARP cleavage (Fig. 3d).
Fig. 3.
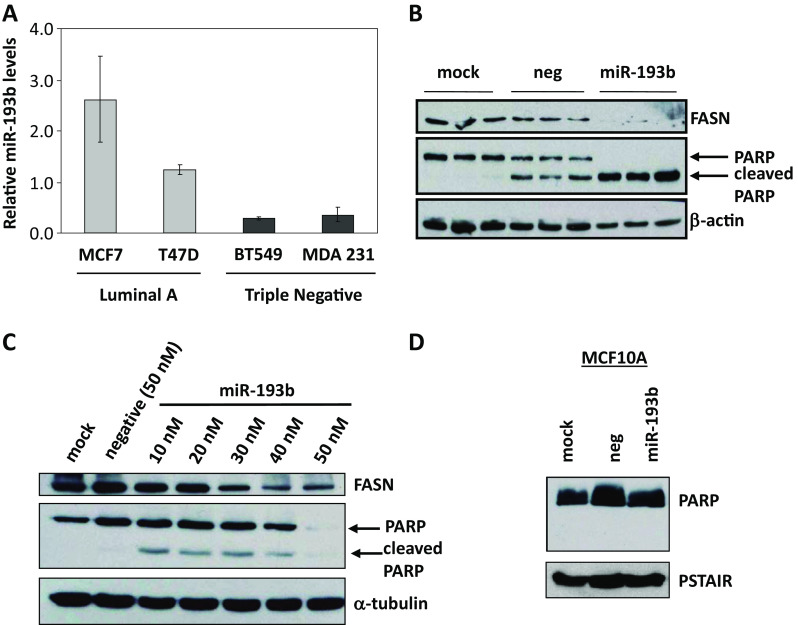
MiR-193b targets FASN and induces apoptosis of TNBC cells, but not normal mammary epithelial cells. a Mature miR-193b levels are assessed in luminal A and TN breast cancer cells by qRT-PCR. b Transfection of miR-193b mimic into BT-549 cells for 72 h decreases FASN protein and increases apoptosis as detected by PARP cleavage. c Transfection of a miR-193b mimic into BT-549 cells for 72 h causes a dose-dependent decrease in FASN and induction of apoptosis. d No apoptosis is observed when miR-193b is transfected into MCF10A (normal immortalized) cells
Bioinformatic prediction analysis revealed a putative miR-193 target sequence in the FASN 3′UTR located between nucleotides 514 and 520 (Fig. 4a). To show that miR-193 directly targets the FASN transcript, we performed a luciferase reporter assay in which the region of the 3′UTR of FASN containing the putative miR-193b binding site was placed downstream of the luciferase reporter gene. Cells were either mock-transfected or transfected with a negative control miRNA mimic, a miR-193b mimic (193), a miR-193b antagomiR (α193), or a combination of the mimic and antagomiR (193 + α193). These cells were subsequently transfected with a transfection control Renilla luciferase vector in addition to either the empty vector (EV) or FASN 3′UTR. A dual luciferase assay was performed 48 h later. Luciferase activity from the empty vector was not affected by any of the mimics, antagomiR, or the combination of both (Fig. 4b). However, the expression of luciferase from the FASN 3′ UTR construct was significantly decreased in the presence of the miR-193b mimic, demonstrating direct targeting of the FASN 3′ UTR by miR193 (Fig. 4b). When miR-193b activity was blocked with antagomiR, luciferase activity was restored to the same level observed in the negative control, demonstrating that the effect is specific to miR-193b.
Fig. 4.

MiR-193b directly targets FASN, and the inhibition of miR193b reduces metformin-mediated reduction of FASN. a MiR-193b is predicted to target a sequence in the 3′UTR of FASN. b Cells were transfected with transfection reagent alone (mock), negative control mimic, miR-193b mimic, a 193b antagomiR (α193), or combination antagomiR and mimic. Twenty-four hours later, reporter vector containing the FASN 3′UTR downstream of the luciferase gene or an empty vector (EV) and renilla luciferase vector (as a loading control) were transfected. Dual luciferase reporter assay was performed 24 h later. Shown are relative luciferase units (luciferase divided by the renilla values) normalized to the mock condition. For FASN 3′UTR compared to EV, *P < 0.05, two-way ANOVA with Dunnett’s multiple comparisons test. c MDA-MB-468 (left) and BT-549 (right) cells stably expressing a miR-193b antagonist (miR-193b-Zip) or a scrambled control (miR-Scr-Zip) were seeded in 5 mM glucose prior to 5 mM metformin treatment for 24 h. Immunoblot analysis on whole cell lysates was performed for FASN, P-ACC, ACC, P-ACLY, ACLY, AMPK, and IGFIR, with β-actin shown as a loading control. Data are representative of three independent experiments
Inhibition of miR-193b Reduces the Ability of Metformin to Decrease FASN Protein and Kill TNBC Cells
Based upon the above observations, we postulated that the upregulation of miR-193 family members might mediate the downregulation of FASN by metformin. To gain functional insight into the role of miR-193 in metformin action, stable cell lines were engineered that express a miR-193b antagomir (miR-193b-Zip) to inhibit the activity of miR-193b or a non-targeting control (miR-Scr-Zip) in MDA-MB-468 and BT-549 cells. These stable cell lines were treated with 5 or 10 mM metformin for 24 h. In the miR-Scr-Zip cells, metformin treatment caused a dramatic downregulation of FASN, ACC, and IGF1R and decreased phosphorylation of ACC and ACLY (Fig. 4c). In cells expressing the miR-193b-Zip, in which miR-193b activity was repressed, the metformin-induced downregulation of FASN was dampened (Fig. 4c). This result indicated that the suppression of miR-193b activity overcomes the ability of metformin to upregulate this miRNA and thereby affects the degree to which metformin decreases the expression of proteins in the fatty acid synthesis pathway as well as IGF1R in TNBC. Similar results were shown in an additional TNBC line, MDA-MB-231 (Supplemental Fig. 1).
We next examined the proliferation and induction of apoptosis in this same panel of stable cell lines to determine if blocking miR-193 affected the ability of metformin to kill TNBC cells. Metformin treatment reduced the proliferation of miR-Scr-Zip cells, but had no effect on proliferation in the miR-193-Zip cells (Supplemental Fig. 2). Treatment of BT-549 cells expressing miR-Scr-Zip with metformin induced apoptosis as detected by YO-PRO1/PI staining with the Hoechst and LIVE/DEAD Viability/Cytotoxicity kit (Fig. 5a). Metformin-treated miR-Scr-Zip cells significantly increased the percentage of dead cells, as indicated by Yo-Pro1 staining by 13.8 % (or 1.4 fold difference), whereas miR-193b-Zip cells show no change in percentage of dead cells (Fig. 5a). Metformin treatment increased the percentage of Hoechst staining in miR-Scr-Zip cells, whereas the inhibition of 193b did not (Fig. 5b). Lastly, metformin-induced apoptosis, assessed by the uptake of ethidiumD1 (EthD1) staining, was dramatically lower in the miR-193b-Zip cells compared to miR-Scr-Zip control cells which had high levels of EthD1 marking apoptotic cells and low calcein-AM staining (marking live cells). In other words, in the presence of the miR-193b-Zip, which inhibits miR-193b activity, metformin was unable to induce the marked cell death (detected using the LIVE/DEAD Viability kit) observed in the miR-Scr-Zip expressing cells (Fig. 5c). Similar results were obtained in MDA-MB-231 cells (Supplemental Fig. 3).
Fig. 5.

Inhibition of miR-193b impairs the ability of metformin to kill TNBC lines. BT-549 cells stably expressing miR-193b-Zip and miR-Scr-Zip were starved of glucose for 24 h prior to replenishing with medium containing 5 mM glucose for an additional 24 h. Cells were treated with 5 mM metformin for an additional 48 h prior to harvesting for apoptosis assays. a Cells were stained with YO-PRO1/PI stain and processed by flow cytometry. Data shown is representative of triplicate samples. Cells shown in region (A2) are indicative of dead cells, and percentage is representative of total cells harvested. Percent of apoptotic cells are quantified (right), normalized to miR-Scr control cells, error bars are SEM. **P < 0.01, two-way ANOVA with Dunnett’s multiple comparisons test. b Cells were stained with HOECHST dye, and apoptotic cells are indicated with white arrows. Percent of apoptotic cells are quantified (right), normalized to miR-Scr-Zip control cells. Error bars are SEM. ****P < 0.0001, two-way ANOVA with Dunnett’s multiple comparisons test. c Cells were processed for immunofluorescent analysis with LIVE/DEAD Viability/Cytotoxicity kit. Membrane-permeant calcein AM is cleaved by esterases in live cells resulting in bright green fluorescence, and membrane-impermeant to ethidium homodimer-1 labels nucleic acids of membrane-compromised cells resulting in a bright red color
Attenuation of miR-193b Abrogates Metformin-Mediated Inhibition of Mammosphere Formation
It is now well-accepted that a population of breast cancer stem cells (CSCs) exist in the heterogenous cell population that exists in most TNBC cell lines including BT-549 cells. These CSCs have a robust self-renewal and tumor-initiating capacity, and these cells are largely resistant to conventional chemotherapy. In recent years, metformin has been shown to suppress self-renewal of CSCs in vitro using mammosphere assays as well as using in vivo assays, although the direct mechanism of this inhibitory action is poorly understood [35, 36]. A study by Pandey and colleagues found that resveratrol suppressed the growth of CSCs by inhibiting FASN activity [45]. Specifically, resveratrol downregulated FASN protein levels and significantly reduced cell viability and mammosphere formation. We hypothesized that since metformin induced the upregulation of miR-193b, which decreased FASN expression and protein levels, that metformin would also cause a reduction in mammosphere formation by inducing apoptosis in CSCs. To test this hypothesis, we examined the ability of BT-549 cells expressing miR-193b-Zip to form mammospheres. Metformin reduced mammosphere formation in miR-Scr-Zip controls; however, the metformin effect on mammosphere formation was significantly reduced with miR-193b-Zip construct (Fig. 6a, b, Supplemental Figs. 4–6). Comparison between miR-Scr-Zip vs. miR-193b-Zip treated with 5 mM metformin show that miR-193b-Zip attenuates metformin-mediated inhibition of mammospheres (45.17 ± 0.9, n = 6 (miR-193b-Zip + 5 met) vs. 18.3 ± 1.8, n = 6 (miR-Scr-Zip + 5 met) p < 0.0001). Furthermore, when second passage (2P) mammospheres were stained to quantitate the presence of CD24low/− CD44high/+ cells by flow cytometry, we found that antagonizing miR-193b reduced metformin-mediated inhibition of CD24low/− CD44high/+ expression (Fig. 6c). Relative to miR-Scr-Zip control cells, metformin causes a reduction in the CD24−/low/CD44+/high population from 93.56 ± 0.3 % (n = 3) to 83.01 ± 0.6 % (n = 3), P = <0.0001. In other words, metformin decreased CD24−/low/CD44+/high CSC population by 10.55 %. In contrast, in cells transfected with miR-193b-Zip construct (inhibiting the activity of miR-193b), metformin did not show a statistically significant change when compared to vehicle ((93.16 ± 0.32 % (n = 3) vs. 94.53 ± 1.24 % (n = 3), P = 0.125, NS). These data provide the first evidence that the attenuation of miR-193b activity reversed the metformin-mediated reduction in CD44 positivity and mammosphere formation, suggesting that metformin-induced upregulation of miR-193b at least partially mediated the ability of metformin to reduce FASN protein expression and to reduce mammosphere formation. Cells from the 2P mammospheres were also examined for expression of ALDEFLUOR, and we found that metformin reduced ALDH expression relative to DAB-treated controls from 8.4 ± 0.38 % (n = 3) (miR-Scr-Zip vehicle control) ALDHpos population to 2.1 ± 0.86 % (n = 3) (miR-Scr-Zip 5 mM metformin) (P = 0.0026) ALDHpos population. In contrast, metformin treatment of cells expressing miR-193b-Zip cells did not significantly change the percentage of ALDHpos cells (7.7 ± 0.21 % (n = 3) (miR-193b-Zip control) to 6.9 ± 0.26 % (n = 3) (miR-193b-Zip 5 mM metformin), P = 0.76, NS) (Fig. 6d). Similar results were shown using MDA-MB-231 transfected cells with miR-Scr-Zip and miR-193b-Zip (Supplemental Figs. 4–6).
Fig. 6.
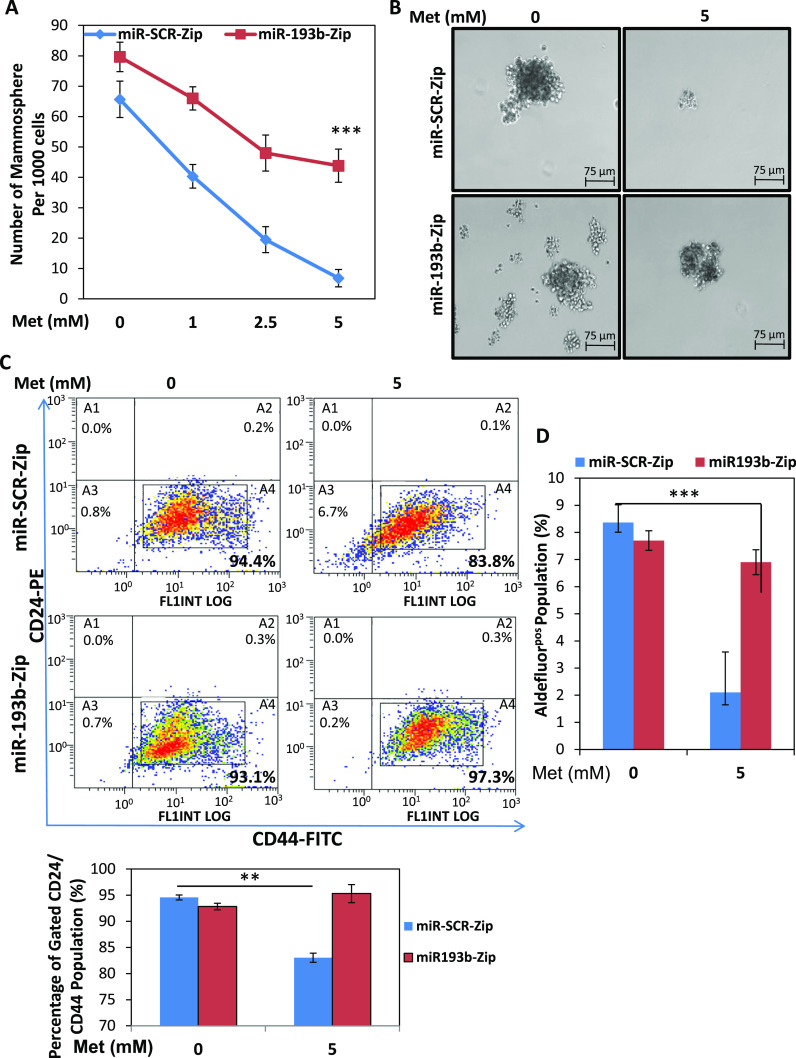
Inhibition of miR-193b reduces metformin-mediated inhibition of mammosphere formation. a BT-549 cells stably expressing miR-193b-Zip and miR-Scr-Zip were seeded in non-adherent conditions in Mammocult medium for 24 h prior to dose–response treatment (0–5 mM) of metformin. Second passage mammospheres (2P) counted from six biological replicates (n = 6), ***P < 0.0001 relative to untreated controls. b Images of the mammospheres were taken using Nikon phase-contrast microscope with either 5 mM metformin or vehicle control treatment. c (top) Generated mammospheres were then collected and stained with CD24/CD44 then processed by flow cytometry. Flow cytometric profiles are shown for 5 mM metformin or vehicle control treated cells. (bottom) Bar graph is representative of triplicate repetitions of experiment for gated population of cells expressing CD24−/low and CD44+/high. d Mammospheres were processed for Aldefluor expression using ALDEFLUOR assay. Flow cytometric profile of Aldefluorpos cells were quantified by calculating the percentage of fluorescent cells compared with a DEAB staining reaction. Bar is representative of triplicates +/− SE. Data are representative of three independent experiments. Error bars are SEM. **P < 0.01, ***P < 0.001, two-way ANOVA with Dunnett’s multiple comparisons test
To test whether the direct inhibition of FASN reduces CSCs in TNBC cell lines, we used two direct chemical modulators of FASN that antagonize FASN activity. Cerulenin forms an epoxy group that directly reacts with the ketoacyl synthesis domain of FASN [46, 47]. In addition, we used another FASN inhibitor, C75. We used these potent FASN inhibitors to test the ability of the direct inhibition of FASN to attenuate mammosphere formation as compared to metformin (Supplementary Fig. 7). Our results show that metformin displayed similar reduction in mammosphere number similar to cerulenin and C75 FASN inhibitors (Supplementary Fig. 7 a,b,c). Compare to vehicle control, all compounds significantly reduced mammosphere formation as observed by Nikon image of mammospheres formed (Supplementary Fig. 7d).
Discussion
Epidemiological evidence from the Nurse’s Health Study and the Women’s Health Study provided evidence that women taking metformin for the control of type 2 diabetes had a lower incidence of breast cancer and that women on metformin that did develop breast cancer had better outcomes than those not on metformin [48–50]. These observations have led to a dramatic increase in the number of studies investigating the effect of metformin upon breast cancer as well as the initiation of clinical trials involving metformin [25, 51]. TNBC has a worse prognosis than other breast cancer subtypes and lacks any effective targeted therapy. Intriguingly, metformin preferentially kills TNBC cell lines as compared to the luminal subtype [30, 32, 34]. This suggests that TNBC cells have unique features that render them more sensitive to metformin action. Interestingly, CSCs have increased expression and reliance on lipogenic enzymes, including FASN [28, 29, 52, 53]. The ACACA/FASN-driven lipogenic switch couples the Warburg Effect to anabolic metabolism and enables conversion of somatic cells to stem cells [28]. Since TNBC have a much higher proportion of CSCs [38], the ability of metformin to target FASN could explain why TNBC are more sensitive to metformin [32, 33], if CSCs are more reliant on FASN.
Although perturbations in the metabolism of tumor cells, including aberrant expression of FASN, may be an early event in carcinogenesis [6, 54], FASN is tightly hormonally regulated by estrogen and progesterone receptors [55–64] in luminal estrogen receptor positive breast cancers cells. In several different types of cancer, aggressive features such as migration/invasion, metastasis, and chemoresistance are dependent on FASN and lipogenesis [65–67]. These features are prominent in TNBC, and consequently targeting FASN with an agent such as metformin may impact TNBC more than other subtypes of breast cancers.
The molecular signaling pathways activated by metformin treatment have been largely elucidated in liver and muscle cells [68–70]. Metformin causes an activation of AMPK signaling, with downstream effects on mTOR, the ribosomal protein S6 kinase, and eIF4EBP1 [36]. As a result, one of the main effects of metformin treatment is an overall decrease in protein synthesis [71]. The ability of super-physiologic concentrations of glucose (glucose greater than 5 mM) to block metformin action likely reflects the activation of molecules such as AKT and mTOR by a glucose-dependent mechanism, which is not blocked by metformin-mediated activation of AMPK. Chronic energy excess, as defined by an increased level of glucose, can alter metformin efficacy as described previously [33]. Here, we assessed the effects of metformin on TNBC cell lines in supraphysiological (17 mM) versus normal glucose (5 mM) and found that high glucose diminishes the metformin-mediated abrogation of FASN and phosphorylation of other components of the fatty acid synthesis pathway.
We cannot rule out additional effects related to greater flux of carbon through metabolic pathways that contribute to metformin resistance. There is evidence that the function of drugs that activate AMPK, including metformin, is dependent on their ability to repress key lipogenic enzymes [72]. Metformin causes a decrease in lipogenesis by also targeting ACC and the transcription factor SREBP-1 in normal cells [73]. Activation of AMPK through means other than metformin also causes a time and dose-dependent decrease in FASN expression in TNBC cells [74]. Our results demonstrate that FASN is significantly reduced in TNBC cells by 10 mM metformin. Interestingly, seven of the top ten genes decreased by metformin are components of the fatty acid and cholesterol biosynthesis pathways.
Relatively few studies have examined the consequences of metformin treatment on miRNA expression in cancer cells [75–77]. Here, we report that the expression of the miR-193 family is rapidly increased by metformin treatment and facilitates metformin-induced downregulation of FASN by directly targeting the FASN 3′UTR. Furthermore, the downregulation of FASN is integral to the ability of metformin to induce apoptosis and decrease mammosphere formation in TNBC. Our data are the first to show that direct inhibition of FASN activity can attenuate CSCs in TNBC cell lines. Our results indicate that direct inhibition of FASN with either cerulenin or C75 can reduce CSCs and metformin is similarly capable of reducing mammosphere formation through direct suppression of lipogenesis by reducing FASN levels. Since there are toxicities associated with FASN inhibitors, for instance C75 caused drastic weight loss and decreased food intake in vivo [78], using metformin may be a better strategy to reduce FASN and consequent mammosphere formation with limited in vivo toxicity.
Metformin was recently found to positively regulate expression of Dicer [76], the enzyme responsible for the final maturation step in miRNA biogenesis. We previously demonstrated that TNBC express Dicer at much lower levels than luminal breast cancers [44]. Several miRNAs have been identified as targeting FASN either directly or indirectly [79–82]. Our data demonstrate that metformin-induced upregulation of miR-193b and targeting of FASN by this miRNA facilitates the ability of metformin to decrease FASN protein. Our data demonstrate that the metformin-dependent increase in the expression of miR-193 is involved in its ability to downregulate FASN, increase apoptosis, and reduce mammosphere formation in TNBC. Downregulation of FASN is clearly an important component of metformin action, although our gene expression profiling data also suggest that metformin reduces many other genes in the fatty acid and cholesterol synthesis pathways (illustrated in Supplemental Fig. 8). This study establishes that FASN is a critical target of metformin in TNBC cells and that downregulation of FASN is, at least in part, facilitated by miR-193b.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
(PDF 1.57 mb)
Acknowledgments
This work was covered in part by the S. G. Komen Foundation for the Cure Grant K100575 to ADT, SMA, JKR, NIH P01 PAR-10-245 to SMA, and the AMC Women’s Cancer Fund/Salah Foundation (JKR). We also acknowledge the following University of Colorado Cancer Center Shared Resource facilities supported by NIH/NCI P30CA046934: the DNA Sequencing and Analysis Shared Resource, the Microarray Shared Resource, the Flow Cytometry Shared Resource, and the Protein Production, Monoclonal antibody and Tissue Culture Shared Resource.
Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Reema S. Wahdan-Alaswad and Dawn R. Cochrane contributed equally to the manuscript.
References
- 1.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269–270. [PubMed] [Google Scholar]
- 2.Hilvo M, et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 2011;71(9):3236–3245. doi: 10.1158/0008-5472.CAN-10-3894. [DOI] [PubMed] [Google Scholar]
- 3.Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953;13(1):27–29. [PubMed] [Google Scholar]
- 4.Ookhtens M, et al. Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am J Physiol. 1984;247(1 Pt 2):R146–R153. doi: 10.1152/ajpregu.1984.247.1.R146. [DOI] [PubMed] [Google Scholar]
- 5.Sabine JR, Abraham S, Chaikoff IL. Control of lipid metabolism in hepatomas: insensitivity of rate of fatty acid and cholesterol synthesis by mouse hepatoma BW7756 to fasting and to feedback control. Cancer Res. 1967;27(4):793–799. [PubMed] [Google Scholar]
- 6.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 7.Chajes V, et al. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Res. 2006;66(10):5287–5294. doi: 10.1158/0008-5472.CAN-05-1489. [DOI] [PubMed] [Google Scholar]
- 8.Alo PL, et al. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer. 1996;77(3):474–482. doi: 10.1002/(SICI)1097-0142(19960201)77:3<474::AID-CNCR8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 9.Menendez JA, Lupu R. Oncogenic properties of the endogenous fatty acid metabolism: molecular pathology of fatty acid synthase in cancer cells. Curr Opin Clin Nutr Metab Care. 2006;9(4):346–357. doi: 10.1097/01.mco.0000232893.21050.15. [DOI] [PubMed] [Google Scholar]
- 10.Menendez JA, Lupu R, Colomer R. Inhibition of tumor-associated fatty acid synthase hyperactivity induces synergistic chemosensitization of HER-2/neu-overexpressing human breast cancer cells to docetaxel (taxotere) Breast Cancer Res Treat. 2004;84(2):183–195. doi: 10.1023/B:BREA.0000018409.59448.60. [DOI] [PubMed] [Google Scholar]
- 11.Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. 2006;9(4):358–365. doi: 10.1097/01.mco.0000232894.28674.30. [DOI] [PubMed] [Google Scholar]
- 12.Turrado C, et al. New synthetic inhibitors of fatty acid synthase with anticancer activity. J Med Chem. 2012;55(11):5013–5023. doi: 10.1021/jm2016045. [DOI] [PubMed] [Google Scholar]
- 13.Knowles LM, et al. Inhibition of fatty-acid synthase induces caspase-8-mediated tumor cell apoptosis by up-regulating DDIT4. J Biol Chem. 2008;283(46):31378–31384. doi: 10.1074/jbc.M803384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pizer ES, et al. Fatty acid synthase (FAS): a target for cytotoxic antimetabolites in HL60 promyelocytic leukemia cells. Cancer Res. 1996;56(4):745–751. [PubMed] [Google Scholar]
- 15.Samudio I, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120(1):142–156. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chuang HY, Chang YF, Hwang JJ. Antitumor effect of orlistat, a fatty acid synthase inhibitor, is via activation of caspase-3 on human colorectal carcinoma-bearing animal. Biomed Pharmacother. 2011;65(4):286–292. doi: 10.1016/j.biopha.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 17.Kridel SJ, et al. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004;64(6):2070–2075. doi: 10.1158/0008-5472.CAN-03-3645. [DOI] [PubMed] [Google Scholar]
- 18.Deepa PR, et al. Therapeutic and toxicologic evaluation of anti-lipogenic agents in cancer cells compared with non-neoplastic cells. Basic Clin Pharmacol Toxicol. 2012;110(6):494–503. doi: 10.1111/j.1742-7843.2011.00844.x. [DOI] [PubMed] [Google Scholar]
- 19.McGowan MM, et al. A proof of principle clinical trial to determine whether conjugated linoleic acid modulates the lipogenic pathway in human breast cancer tissue. Breast Cancer Res Treat. 2013;138(1):175–183. doi: 10.1007/s10549-013-2446-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puig T, et al. A novel inhibitor of fatty acid synthase shows activity against HER2 + breast cancer xenografts and is active in anti-HER2 drug-resistant cell lines. Breast Cancer Res. 2011;13(6):R131. doi: 10.1186/bcr3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puig T, et al. Novel inhibitors of fatty acid synthase with anticancer activity. Clin Cancer Res. 2009;15(24):7608–7615. doi: 10.1158/1078-0432.CCR-09-0856. [DOI] [PubMed] [Google Scholar]
- 22.Romero IL, et al. Relationship of type II diabetes and metformin use to ovarian cancer progression, survival, and chemosensitivity. Obstet Gynecol. 2012;119(1):61–67. doi: 10.1097/AOG.0b013e3182393ab3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gandini S, et al. Metformin and breast cancer risk. J Clin Oncol. 2013;31(7):973–974. doi: 10.1200/JCO.2012.46.3596. [DOI] [PubMed] [Google Scholar]
- 24.Dowling RJ, et al. Metformin in cancer: translational challenges. J Mol Endocrinol. 2012;48(3):R31–R43. doi: 10.1530/JME-12-0007. [DOI] [PubMed] [Google Scholar]
- 25.Goodwin PJ, Ligibel JA, Stambolic V. Metformin in breast cancer: time for action. J Clin Oncol. 2009;27(20):3271–3273. doi: 10.1200/JCO.2009.22.1630. [DOI] [PubMed] [Google Scholar]
- 26.Niraula S, et al. Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res Treat. 2012;135(3):821–830. doi: 10.1007/s10549-012-2223-1. [DOI] [PubMed] [Google Scholar]
- 27.Algire C, et al. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010;17(2):351–360. doi: 10.1677/ERC-09-0252. [DOI] [PubMed] [Google Scholar]
- 28.Vazquez Martin A, et al. The mitochondrial H(+)-ATP synthase and the lipogenic switch: new core components of metabolic reprogramming in induced pluripotent stem (iPS) cells. Cell Cycle. 2013;12(2):207–218. doi: 10.4161/cc.23352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vazquez‐Martin A, et al. Metformin limits the tumourigenicity of iPS cells without affecting their pluripotency. Sci Rep. 2012;2:964. doi: 10.1038/srep00964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alimova IN, et al. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8((6):909–915. doi: 10.4161/cc.8.6.7933. [DOI] [PubMed] [Google Scholar]
- 31.Giles ED, et al. Obesity and overfeeding affecting both tumor and systemic metabolism activates the progesterone receptor to contribute to postmenopausal breast cancer. Cancer Res. 2012;72(24):6490–6501. doi: 10.1158/0008-5472.CAN-12-1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu B, et al. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8(13):2031–2040. doi: 10.4161/cc.8.13.8814. [DOI] [PubMed] [Google Scholar]
- 33.Wahdan‐Alaswad R, et al. Glucose promotes breast cancer aggression and reduces metformin efficacy. Cell Cycle. 2013;12(24):3759–3769. doi: 10.4161/cc.26641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng XS, et al. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. 2012;11(2):367–376. doi: 10.4161/cc.11.2.18813. [DOI] [PubMed] [Google Scholar]
- 35.Hirsch HA, et al. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69(19):7507–7511. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zakikhani M, et al. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66(21):10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 37.Creighton CJ, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106(33):13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Honeth G, et al. The CD44+/CD24− phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008;10(3):R53. doi: 10.1186/bcr2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siddiqui RA, et al. Anticancer properties of propofol-docosahexaenoate and propofol-eicosapentaenoate on breast cancer cells. Breast Cancer Res. 2005;7(5):R645–R654. doi: 10.1186/bcr1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanlioglu AD, et al. Surface TRAIL decoy receptor-4 expression is correlated with trail resistance in MCF7 breast cancer cells. BMC Cancer. 2005;5:54. doi: 10.1186/1471-2407-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dontu G, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17(10):1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ginestier C, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhuang Y, Miskimins WK. Cell cycle arrest in metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1. J Mol Signal. 2008;3:18. doi: 10.1186/1750-2187-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cochrane DR, et al. MicroRNAs link estrogen receptor alpha status and dicer levels in breast cancer. Horm Cancer. 2010;1(6):306–319. doi: 10.1007/s12672-010-0043-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pandey PR, et al. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res Treat. 2011;130(2):387–398. doi: 10.1007/s10549-010-1300-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Funabashi H, et al. Binding site of cerulenin in fatty acid synthetase. J Biochem. 1989;105(5):751–755. doi: 10.1093/oxfordjournals.jbchem.a122739. [DOI] [PubMed] [Google Scholar]
- 47.Pizer ES, et al. Inhibition of fatty acid synthesis induces programmed cell death in human breast cancer cells. Cancer Res. 1996;56(12):2745–2747. [PubMed] [Google Scholar]
- 48.Chlebowski RT. Obesity and breast cancer outcome: adding to the evidence. J Clin Oncol. 2012;30(2):126–128. doi: 10.1200/JCO.2011.39.7877. [DOI] [PubMed] [Google Scholar]
- 49.Chlebowski RT, et al. Diabetes, metformin, and breast cancer in postmenopausal women. J Clin Oncol. 2012;30(23):2844–2852. doi: 10.1200/JCO.2011.39.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knowler WC, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiralerspong S, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27(20):3297–3302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Knobloch M, et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature. 2013;493(7431):226–230. doi: 10.1038/nature11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, et al. PPARgamma maintains ERBB2-positive breast cancer stem cells. Oncogene. 2013;32(49):5512–5521. doi: 10.1038/onc.2013.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Milgraum LZ, et al. Enzymes of the fatty acid synthesis pathway are highly expressed in situ breast carcinoma. Clin Cancer Res. 1997;3(11):2115–2120. [PubMed] [Google Scholar]
- 55.Esslimani Sahla M, et al. Increased expression of fatty acid synthase and progesterone receptor in early steps of human mammary carcinogenesis. Int J Cancer. 2007;120(2):224–229. doi: 10.1002/ijc.22202. [DOI] [PubMed] [Google Scholar]
- 56.Chalbos D, et al. Progestin‐induced fatty acid synthetase in human mammary tumors: from molecular to clinical studies. J Steroid Biochem Mol Biol. 1992;43(1‐3):223–228. doi: 10.1016/0960-0760(92)90211-Z. [DOI] [PubMed] [Google Scholar]
- 57.Joyeux C, Chalbos D, Rochefort H. Effects of progestins and menstrual cycle on fatty acid synthetase and progesterone receptor in human mammary glands. J Clin Endocrinol Metab. 1990;70(5):1438–1444. doi: 10.1210/jcem-70-5-1438. [DOI] [PubMed] [Google Scholar]
- 58.Chalbos D, et al. Expression of the progestin‐induced fatty acid synthetase in benign mastopathies and breast cancer as measured by RNA in situ hybridization. J Natl Cancer Inst. 1990;82(7):602–606. doi: 10.1093/jnci/82.7.602. [DOI] [PubMed] [Google Scholar]
- 59.Chalbos D, et al. Regulation of fatty acid synthetase by progesterone in normal and tumoral human mammary glands. Rev Esp Fisiol. 1990;46(1):43–46. [PubMed] [Google Scholar]
- 60.Chambon M, et al. Progestins and androgens stimulate lipid accumulation in T47D breast cancer cells via their own receptors. J Steroid Biochem. 1989;33(5):915–922. doi: 10.1016/0022-4731(89)90240-9. [DOI] [PubMed] [Google Scholar]
- 61.Joyeux C, Rochefort H, Chalbos D. Progestin increases gene transcription and messenger ribonucleic acid stability of fatty acid synthetase in breast cancer cells. Mol Endocrinol. 1989;3(4):681–686. doi: 10.1210/mend-3-4-681. [DOI] [PubMed] [Google Scholar]
- 62.Chalbos D, et al. Fatty acid synthetase and its mRNA are induced by progestins in breast cancer cells. J Biol Chem. 1987;262(21):9923–9926. [PubMed] [Google Scholar]
- 63.Martel PM, et al. S14 protein in breast cancer cells: direct evidence of regulation by SREBP‐1c, superinduction with progestin, and effects on cell growth. Exp Cell Res. 2006;312(3):278–288. doi: 10.1016/j.yexcr.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 64.Menendez JA, Lupu R, Colomer R. Obesity, fatty acid synthase, and cancer: serendipity or forgotten causal linkage? Mol Genet Metab. 2005;84(3):293–295. doi: 10.1016/j.ymgme.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 65.Menendez JA, et al. Novel signaling molecules implicated in tumor‐associated fatty acid synthase‐dependent breast cancer cell proliferation and survival: role of exogenous dietary fatty acids, p53‐p21WAF1/CIP1, ERK1/2 MAPK, p27KIP1, BRCA1, and NF‐kappaB. Int J Oncol. 2004;24(3):591–608. [PubMed] [Google Scholar]
- 66.Seguin F, et al. The fatty acid synthase inhibitor orlistat reduces experimental metastases and angiogenesis in B16‐F10 melanomas. Br J Cancer. 2012;107(6):977–987. doi: 10.1038/bjc.2012.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zaytseva YY, et al. Inhibition of fatty acid synthase attenuates CD44‐associated signaling and reduces metastasis in colorectal cancer. Cancer Res. 2012;72(6):1504–1517. doi: 10.1158/0008-5472.CAN-11-4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fryer LG, Parbu‐Patel A, Carling D. The Anti‐diabetic drugs rosiglitazone and metformin stimulate AMP‐activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277(28):25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- 69.Musi N, et al. Metformin increases AMP‐activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51(7):2074–2081. doi: 10.2337/diabetes.51.7.2074. [DOI] [PubMed] [Google Scholar]
- 70.Zhou G, et al. Role of AMP‐activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dowling RJ, et al. Metformin inhibits mammalian target of rapamycin‐dependent translation initiation in breast cancer cells. Cancer Res. 2007;67(22):10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 72.Guo D, et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII‐expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci U S A. 2009;106(31):12932–12937. doi: 10.1073/pnas.0906606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao X, et al. Regulation of lipogenesis by cyclin‐dependent kinase 8‐mediated control of SREBP‐1. J Clin Invest. 2012;122(7):2417–2427. doi: 10.1172/JCI61462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee KH, et al. Targeting energy metabolic and oncogenic signaling pathways in triple‐negative breast cancer by a novel adenosine monophosphate‐activated protein kinase (AMPK) activator. J Biol Chem. 2011;286(45):39247–39258. doi: 10.1074/jbc.M111.264598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bao B, et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev Res (Phila) 2012;5(3):355–364. doi: 10.1158/1940-6207.CAPR-11-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blandino G, et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c‐MYC. Nat Commun. 2012;3:865. doi: 10.1038/ncomms1859. [DOI] [PubMed] [Google Scholar]
- 77.Oliveras‐Ferraros C, et al. Micro(mi)RNA expression profile of breast cancer epithelial cells treated with the anti‐diabetic drug metformin: induction of the tumor suppressor miRNA let‐7a and suppression of the TGFbeta‐induced oncomiR miRNA‐181a. Cell Cycle. 2011;10(7):1144–1151. doi: 10.4161/cc.10.7.15210. [DOI] [PubMed] [Google Scholar]
- 78.Loftus TM, et al. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science. 2000;288(5475):2379–2381. doi: 10.1126/science.288.5475.2379. [DOI] [PubMed] [Google Scholar]
- 79.Mao JH, et al. microRNA‐195 suppresses osteosarcoma cell invasion and migration in vitro by targeting FASN. Oncol Lett. 2012;4(5):1125–1129. doi: 10.3892/ol.2012.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Park JH, et al. Murine hepatic miRNAs expression and regulation of gene expression in diet‐induced obese mice. Mol Cells. 2011;31(1):33–38. doi: 10.1007/s10059-011-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shirasaki T, et al. MicroRNA‐27a regulates lipid metabolism and inhibits hepatitis C virus replication in human hepatoma cells. J Virol. 2013;87(9):5270–5286. doi: 10.1128/JVI.03022-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhong D, et al. MicroRNA‐613 represses lipogenesis in HepG2 cells by downregulating LXRalpha. Lipids Health Dis. 2013;12:32. doi: 10.1186/1476-511X-12-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1.57 mb)