

Abstract
Hydrogen peroxide (H2O2) is involved in various signal transduction pathways and cell fate decisions. The mechanism of the so called “redox signaling” includes the H2O2-mediated reversible oxidation of redox sensitive cysteine residues in enzymes and transcription factors thereby altering their activities. Depending on its intracellular concentration and localization, H2O2 exhibits either pro- or anti-apoptotic activities. In comparison to normal cells, cancer cells are characterized by an increased H2O2 production rate and an impaired redox balance thereby affecting the microenvironment as well as the anti-tumoral immune response. This article reviews the current knowledge about the intracellular production of H2O2 along with redox signaling pathways mediating either the growth or apoptosis of tumor cells. In addition it will be discussed how the targeting of H2O2-linked sources and/or signaling components involved in tumor progression and survival might lead to novel therapeutic targets.
Introduction
Hydrogen peroxide (H2O2) is next to the superoxide anion and hydroxyl radical a key member of the class of reactive oxygen species (ROS), which are in particular generated via the respiratory chain cascade but also as byproducts of the cellular metabolism including protein folding. In contrast to the superoxide anion and hydroxyl radical, the less reactive H2O2 is involved in many physiological processes such as hypoxic signal transduction, cell differentiation and proliferation but also plays a role in mediating immune responses. However, it exerts its effects depending on the cellular context, its local concentration as well as its exposure time [1, 2]. Thus H2O2 is no more considered as an unwanted rather toxic byproduct, but plays an important role in the control of vital cellular processes.
Tumor cells are characterized by an enhanced metabolic activity resulting in changes of the cellular redox state that has to handle the production of high levels of ROS [3]. In many cancer cells persistently upregulated H2O2-dependent signaling pathways are involved in cell differentiation, growth and survival, yet high levels of H2O2 can also induce cell cycle arrest or apoptosis in cells. Due to this dual functionality of H2O2 robust cellular anti-oxidative systems are thought to be essential for maintaining the cellular redox homeostasis. Several defense systems against oxidative stress have been shown to be upregulated in cancer cells via the transcription factor nuclear factor-erythroid 2 p45-related factor 2 (Nrf2) [4]. These include the thioredoxin/thioredoxin reductase (Trx/TrxR) system, peroxiredoxins (Prxs) and several glutathione S-transferases (GSTs), which are involved in mediating the cellular redox homeostasis, but still allow redox modifications of specific redox-sensitive proteins thereby triggering redox signaling events. In this review we will address how (i) cellular H2O2 is produced and how it regulates certain signaling pathways, (ii) tumor cells cope with enhanced H2O2 levels to escape from oxidative stress, (iii) potential redox-sensors might be correlated with tumorigenesis, and how (iv) H2O2-modulated processes/pathways might be used as therapeutic targets.
Sources of H2O2
Reactive oxygen species (ROS) represent a class of oxygen-containing chemical compounds that are defined by their reactivity towards biological targets, including lipids, proteins and DNA [3]. The most prominent member of this class is the superoxide anion (O2−), largely produced by either the mitochondrial electron transport chain, in particular its complexes I, II and III, or by NAD(P)H oxidases (NOXs). The O2− is rapidly converted to H2O2 by distinct superoxide dismutases (SODs) (Fig. 1) or to hydroxyl radicals (OH●) [5]. While O2− released into the mitochondrial matrix is directly converted by SOD2 into the less reactive H2O2, O2− released by the complex III into the mitochondrial intermembrane space can be exported via voltage-dependent anion channels (VDAC) into the cytosol followed by a SOD1-mediated conversion into H2O2 [6, 7]. In addition, cellular membrane-associated NOXs transferring electrons from NAD(P)H across cell membranes to molecular oxygen (O2) are producers of superoxide anions. Via NOX2 O2− can be transported into the extracellular space, where it can be either converted to H2O2 by SOD3 or re-imported via chloride channels [8]. Given that SODs are characterized as highly efficient enzymes the intracellular (cytosol - SOD1, mitochondria - SOD2) and extracellular (SOD3) balance is shifted towards the formation of H2O2, which diffuses relatively free or is receptor-mediated transported across biological membranes [9] thereby acting as an intra- and intercellular signaling molecule (Fig. 1). In contrast, the hydroxyl radical (OH●) is considered as the most reactive ROS species. Due to its high reactivity towards lipids, proteins and DNA, it has a short half-life thereby limiting its diffusion but causing damage largely at its site of production.
Fig. 1.
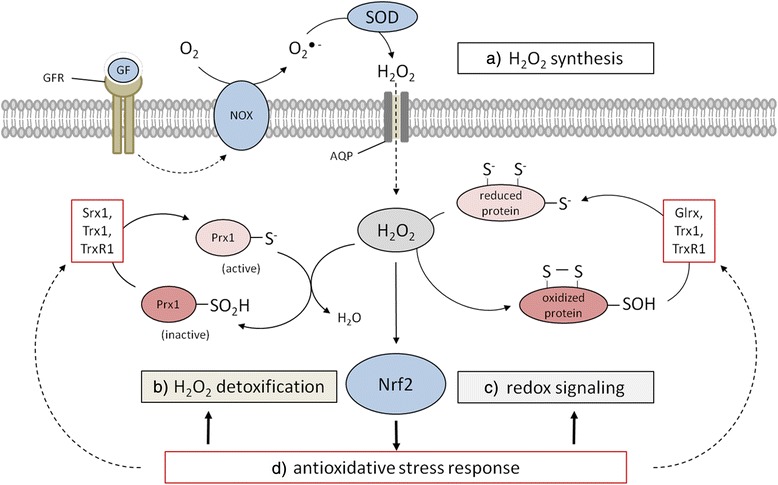
Interplay between physiological/pathophysiological H2O2 generation and the anti-oxidative response mechanism. a H2O2 is produced, e.g. in response to growth factors by the NOX/SOD system and enters cells through simple diffusion and facilitated diffusion through AQPs, respectively, leading to increased intracellular H2O2 levels. b Peroxiredoxins (Prx) act as highly active redox sensors and are part of one of the main H2O2 detoxifying systems. Hyperoxidation inactivates Prxs allowing c the oxidation of sensitive cysteine residues in cellular proteins including transcription factors. d The Nrf2 system is activated in response to increased H2O2 levels leading to the anti-oxidative response. AQP, aquaporin; GF, growth factor; GFR, growth factor receptor.
Transport and subcellular localization of hydrogen peroxide
In comparison to water, H2O2 possesses a reduced membrane permeability, which is influenced by the phosphorylation and glycosylation states of membrane proteins, the lipid composition (lipid rafts) and osmotic stretching of lipid bilayers [10–16]. Aquaporin (AQP) 8, but not the classical AQP1 facilitates the transport of H2O2 across membranes [17, 18]. Treatment of AQP3-overexpressing HeLa cells with H2O2 resulted in an enhanced phosphorylation of protein kinase B (AKT) [19], while overexpression of AQP8 increased intracellular H2O2 levels in leukemia cells in the presence of H2O2. Moreover, vascular endothelial growth factor (VEGF) signaling results in increased intracellular H2O2 levels, which can be reduced by silencing AQP8 [20]. Furthermore, silencing of AQP8 can inhibit the epidermal growth factor (EGF) mediated stimulation of tyrosine kinases. [21]. Thus, AQPs not only play important roles in the diffusion of H2O2 across membranes, but also on downstream signaling cascades. Furthermore, H2O2 detoxifying enzymes, such as glutathione peroxidases (GPxs), catalases and Prxs, can lead to rapidly decreasing intracellular H2O2 concentrations [9] thereby establishing the formation of H2O2 gradients resulting in selective and localized H2O2 signaling events. The inactivation of scavenger enzymes by H2O2 represents a mechanism that allows the selective enrichment (“flooding”) of a cellular area by H2O2 thereby promoting the H2O2-mediated oxidation of specific thiols within target proteins at this site [22, 23].
Features of H2O2 – second messenger like characteristics and principles of redox modifications
Since H2O2 is produced, enzymatically removed and exerts a low overall reactivity, but a relatively high selectivity towards certain proteins, in particular proteins containing thiol groups [24, 25], it is postulated to act as a second messenger. H2O2 mediates chemical modifications of specific cysteine residues, which are overrepresented in functionally relevant regions of some proteins [26]. Approximately 10 % of free cysteines are ionized at pH 7.4 due to their low pKa and thus are more susceptible to H2O2 than protonated cysteine thiol groups [27]. Although H2O2 detoxification enzymes, like GPxs, Prxs and catalase, are more abundantly expressed than proteins involved in the redox signaling [28], cysteine residues of the ubiquitously expressed Prxs are prone to be oxidized at even relatively low H2O2 levels [29–31]. In contrast, signaling molecules e.g. protein tyrosine phosphatases (PTPs) require extremely high concentrations of H2O2 to undergo oxidation [9]. Furthermore, if Prxs are inactivated by over-oxidation or phosphorylation this might lead to localized H2O2 accumulation thereby triggering redox signaling [22] (Fig. 1). The first step of oxidative thiol/thiolate modification by H2O2 is the formation of sulfonate or sulfenic acid (R-SOH), which might reacts with any thiol in the vicinity, e.g. glutathione (GSH) to form inter- and intramolecular disulfide bonds or protein-SSGs, respectively. In some cases, e.g. with electron-rich amino groups they also form sulfenylamides (Fig. 2). These oxidized forms can be easily reduced to thiolate by the Trx- and GSH-based anti-oxidative systems thereby ensuring the reversibility of redox modifications caused by H2O2. In the presence of excessive concentrations of H2O2 further oxidation of sulfenic acids might occur thereby resulting in the formation of sulfinic (−SO2H), sulfonic acids (−SO3H) or their respective anions. In general these sulfur (IV) and (VI) oxidative states are irreversible oxidation products and some of these species can be linked to H2O2-mediated toxicity (Fig. 2). However, if these oxidative modifications occur in members of the Prx family their reduction can be mediated by sulfiredoxins (Srx) [32], which might represent an adaptive process of eukaryotic cells to cope with increased H2O2 levels [22, 23].
Fig. 2.
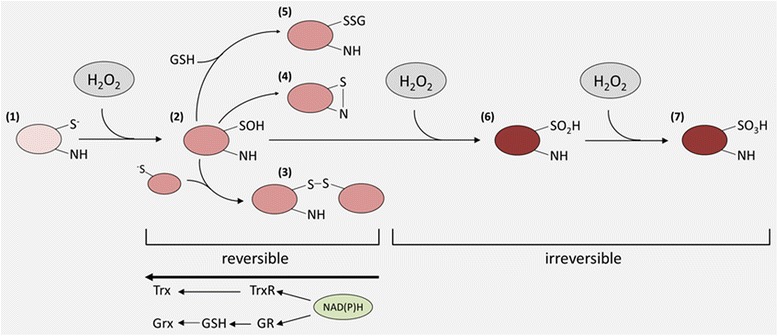
Redox modifications of reactive cysteine residues by H2O2. Redox-sensitive proteins contain cysteine residues, which are partially ionized under physiological pH. Oxidation of this thiolate anion (1) results in a sulfenic acid or rather its salt (2), which is relatively reactive and forms intra-/intermolecular disulfide bonds in the presence of thiolate. This sulfenylation can be intramolecular or intermolecular (3), the latter predominantly with GSH to form glutathionylated intermediates (5), or sulfenylamides with oxidizable amines (4) and glutathionylated intermediates (5), respectively. These redox modifications result in altered functions of their target proteins and can be reversed by the Trx- or GSH-based anti-oxidative systems. Under excessive H2O2 concentration the sulfonate or sulfonamide intermediates can be further irreversibly oxidized to sulfinic (6) and sulfonic acids (7) forming the respective anions under physiological pH thus also shifting the isoelectric points of affected proteins.
The anti-oxidative response – factors that maintain redox signaling
Whereas intracellular O2− concentrations are tightly controlled by the activity of SODs and thus kept at very low levels [33], the metabolite H2O2 is a rather stable ROS compound. Cells have developed distinct mechanisms to maintain the production and clearance of such reactive species in a homeostatic state in order to properly proliferate and to differentiate. Tumor cells are metabolically hyperactive resulting in the production of excessive ROS levels including H2O2. To cope with enhanced H2O2 concentrations and to protect cells from oxidative damage, anti-oxidative defense systems are upregulated, which results in a shift of the redox balance towards an upregulation of pro-survival signaling pathways as summarized in Table 1 for a set of Nrf2-regulated anti-oxidative proteins and their correlation to cancer.
Table 1.
Nrf2 targets and their correlation to cancer
| target | name | relation to cancer |
|---|---|---|
| Trx1 | thioredoxin 1 | proto-oncogene expressed in many cancers [224] |
| elevated serum levels in patients with ovarian cancer [225] | ||
| overexpression in HCC, CRC, liver metastasis [224] and | ||
| in breast cancer [226] | ||
| TrxR1 | thioredoxin reductase1 | overexpressed in many cancer cells and cell lines of distinct histology [227] |
| TXNIP | thioredoxin interacting protein | negatively interferes with bladder carcinogenesis [228] |
| tumor suppressor in thyroid cancer [229] | ||
| Prx1 | peroxiredoxin 1 | increased expression in pancreatic cancer [230], breast cancer [226, 231] and HCC [232] |
| promotion of lung cancer progression [233] | ||
| related to tumor angiogenesis in pancreatic cancer [230] | ||
| Prx6 | peroxiredoxin 6 | promotion of lung tumor growth [234, 235] repressed expression in papillary thyroid carcinomas [236] |
| Srx | sulfiredoxin | oncogenic role in skin tumorigenesis [237] |
| overexpression in squamous cell carcinoma [56] | ||
| promotion of lung cancer progression [69] |
CRC colorectal carcinoma, HCC hepatic cell carcinoma
Transcription factor Nrf2 as regulator of the anti-oxidative response
Nuclear factor-erythroid 2 p45-related factor 2 (Nrf2) is a transcription factor (TF) that plays a key role in controlling the response to oxidative stress by its regulation of anti-oxidative enzymes, phase II enzymes and enzymes of the glutathione biosynthesis. Under physiological conditions the constitutive abundance of active Nrf2 is relatively low due to its continuous proteasomal degradation, but can be modified at the post-translational level to ensure rapid and efficient adaption to metabolic alterations in particular to oxidative stress. The best characterized repressor of Nrf2 is the kelch-like ECH-associated protein (Keap1), which serves as a substrate adapter protein within the RBX1 E3 ubiquitin ligase complex (CRLKeap1) [34]. Keap1 contains multiple highly reactive cysteine residues, which can act as stress sensors, if modified by electrophiles or oxidants, e.g. from food [35]. This results in an altered conformation of Keap1 and an impaired binding capacity to Nrf2 thereby preventing Nrf2 from proteasomal degradation. Thus, Nrf2 accumulates in the nucleus leading to the induction of genes by binding to the anti-oxidant response element (ARE) in their promoter regions (Fig. 3). In addition Nrf2 undergoes post-translational modifications such as PKC-dependent phosphorylation on Ser-40, phosphorylation through the MAPK/ERK signaling pathways in response to endoplasmic reticulum/unfolded protein stress or by casein kinase and CBP/p300 also promoting its binding to such ARE sites. The activation of Nrf2 can be also mediated by additional signal transduction pathways, e.g. ERK, c-Jun amino-terminal kinase (JNK), AMP-activated protein kinase (AMPK) or PI3K/AKT promoting anti-oxidative effects, which mediate enhanced resistance to oxidative stress as well as to further oxidative insults [36–38]. Constitutive stabilization of Nrf2 is found in several human cancers [39–41] and is associated with increased cancer chemotherapy resistance, enhanced tumor progression [42, 43] and poor prognosis and/or survival for patients [40, 44, 45]. Mechanisms by which the Nrf2 signaling pathway is constitutively activated in several types of cancer include (i) somatic mutations of Keap1 disrupting the binding capacity to Nrf2, (ii) epigenetic silencing of Keap1 and (iii) transcriptional induction of Nrf2 by oncogenes such as K-ras, B-raf or c-myc [46] (Fig. 3). Furthermore, increased levels of ROS (H2O2) and increased Nrf2 activity in tumor cells, result in an enhanced anaerobic glycolysis and utilization of the pentose phosphate pathway activity to generate NAD(P)H equivalents necessary for the Trx- and GSH-based anti-oxidative systems [47]. Since NAD(P)H generating enzymes are Nrf2 targets, the energy metabolism is directly connected with the redox homeostasis (Fig. 4). This is confirmed by an increased metabolic oxidative stress and cytotoxicity in response to the inhibition of glycolysis and/or the pentose phosphate pathways in combination with an inhibition of the Trx metabolism [48]. In contrast, knock down of Nrf2 suppresses tumor growth, inhibits cell proliferation and promotes increased apoptosis [44, 49]. The fact, that several cancers exhibit induced Nrf2 levels associated with enhanced tumor progression and chemotherapy resistance, whereas the lack of Nrf2 has opposite effects, Nrf2 represents a promising target for cancer therapies.
Fig. 3.
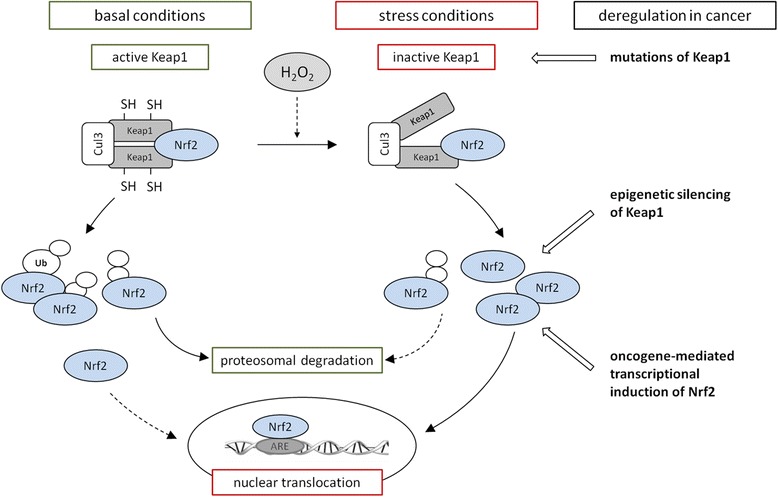
The Nrf2/Keap1 signaling pathway. Under basal conditions Nrf2 is bound by two molecules of Keap1, poly-ubiquitinylated by the Cul3 system and thereby marked for proteasomal degradation. Only a small portion of Nrf2 escapes from this degradation process and translocates to the nucleus to maintain the basal expression of anti-oxidant response genes. Under stress conditions like elevated levels of H2O2 Keap1 is modified at redox sensitive cysteine residues leading to an impaired conformation and inactivation of Keap1. Newly translated Nrf2 escapes ubiquitinylation, translocates to the nucleus and induces the anti-oxidative stress response. Mechanisms for the continuously accumulation of Nrf2 in the nucleus of several cancer cells can be triggered by (i) mutations of Keap1 associated with its inactivation, (ii) epigenetic silencing of Keap1 and (iii) mutations of oncogenes such as K-ras, B-raf and c-myc leading to the transcriptional induction of Nrf2.
Fig. 4.
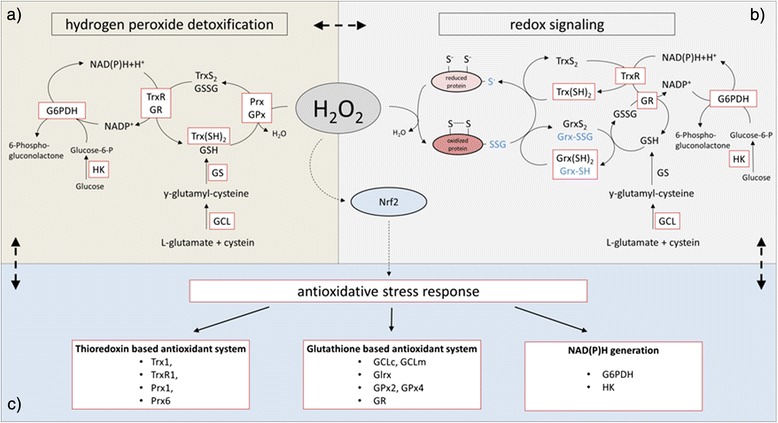
Maintenance of redox homeostasis by Nrf2. Nrf2 induces the expression of genes coding for enzymes involved in (a) hydrogen peroxide detoxification and (b) redox signaling. (c) High levels of H2O2 activate Nrf2 resulting in the induction of the anti-oxidative stress response. The red boxes symbolize Nrf2 inducible enzymes
Targets of Nrf2
Peroxiredoxins
Prxs represent members of the so called thiol-based anti-oxidant system [50] that act as redox switches to modulate homeostasis [51]. As important H2O2 scavenging enzymes Prxs are involved in the anti-oxidative response and in the regulation of redox-dependent signaling pathways by converting H2O2 into water [52, 53]. In mammals, the family of Prxs consists of 6 members located either in the cytosol (Prx1, Prx2, Prx4, Prx5, Prx6), mitochondria (Prx3, Prx5) or in other cellular compartments (Prx1, nucleus; Prx2, membrane; Prx4, Golgi apparatus, extracellular space, endoplasmic reticulum; Prx5, peroxisomes) [9, 54]. Prxs are upregulated under conditions of oxidative stress [55–57] and it could be shown that Prx1 and Prx6 are direct targets of Nrf2 [58, 59]. Prx1 – Prx5 are 2-Cys-Prx and utilize Trx as electron donor for their catalytic activity, while Prx6 is a 1-Cys-Prx and depends on GSH instead of Trx for its reduction [54, 60]. The hyper-oxidation of 2-Cys Prx, in particular of Prx1, adds further chaperone function to these Prxs, but depends on certain motif elements downstream of the peroxidatic cysteine residue (GGLG and YF motifs) [23, 61]. The chaperone function is based on the formation of stack like higher molecular weight complexes, thereby preventing the denaturation of proteins from external stresses like heat shock or oxidative stress. This multimeric complex can be subsequently dissolved into low molecular weight species by Srx [61]. Whereas in some species more distant cysteine residues might act as redox sensors, human Prxs are known to gain such a chaperone function only after the peroxidatic cysteine is hyper-oxidized [51]. At the transcriptional level Nrf2 and to some degree also focal adhesion kinase (FAK) have been demonstrated to activate the expression of Prxs [62, 63]. However, there is also evidence that modifications at the post-translational level have an impact on the function of Prxs. For example nitrosylation of the tyrosine residue within the YF motif of Prx2 plays a crucial role in the regulation of disulfide bond formation under oxidative stress conditions resulting in a more active and robust peroxidase [64]. In addition, its glutathionylation may affect its localization to the extracellular compartment, along with Trx, thereby inducing TNFα production leading to an oxidative stress-dependent inflammatory reaction [65]. For Prx3 the complex formation of FoxO3a with the peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC1 alpha) is enhanced by sirtuin-1 (SirT1), which is similar to the regulation of other anti-oxidant proteins [66]. The Prx4, which is mainly expressed in the endoplasmic reticulum compartment can be enhanced at the post-transcriptional level by calpain [67]. Due to its high susceptibility to undergo hyperoxidation even at low levels of oxidative stress its chaperone function is frequently involved in the oxidative folding of various ER resident proteins, likely in cooperation with protein disulfide isomerase (PDI) [68]. There is also evidence that Prx4 in addition to Srx plays a crucial role in enhancing RAS-RAF-MEK signaling to control cancer cell proliferation and metastasis formation [69].
Sulfiredoxins
Srxs reduce double oxidized catalytic cysteine (sulfinic acid) residues of 2-Cys-Prxs [70] thereby restoring their peroxidase function [32, 71]. Based on studies in yeast, the rate constant for the reduction of oxidized Prx by Trx (about 106 M−1 s−1) is much faster than the rate of reduction of hyperoxidized Prx by Srx [72, 73]. Thus, the reduction of hyperoxidized Prx by Srx might be considered as a rate limiting step. Moreover Srxs are involved in deglutathionylation processes [74] and can regulate the chaperone function of Prx1 by controlling its glutathionylation levels at position cysteine 83 [75]. In contrast to its anti-oxidant function, which is highly specific for Prxs, the deglutathionylation activity of Srx appears much less restricted [51]. The Srx promoter contains a sequence resembling the consensus sequence for ARE, which is important for its regulation [76]. In response to cigarette smoke and under hypoxic conditions, Srx expression is transcriptionally controlled in a Nrf2-dependent manner [77, 78]. By using overexpression and knock out model systems it has been demonstrated that upon treatment with the chemopreventive Nrf2 inducer 3H-1,2-dithiole-3-thione (D3T) the expression of Srx is upregulated and thus prevents the double oxidation of Prx in neurons [79]. Moreover, hyperoxia has been shown to induce the degradation of mitochondrial double oxidized Prx3 in Nrf2-deficient, but not in WT mice. Thus, in the absence of Srx hyperoxidized Prx becomes susceptible to proteolysis [78]. In addition, the disparate resistance of colon carcinoma cells to ROS has been linked to higher basal levels of Nrf2 and Srx as well as to their distinct cellular localizations [56, 80].
Thioredoxin / thioredoxin reductase / TXNIP system
Trxs are small ubiquitously expressed proteins maintaining the cellular environment in a reduced state [81]. Trxs are involved in the catalysis of redox-dependent reactions, display oxidoreductase activity, serve as electron donors for enzymes with biosynthetic properties [82] and are involved in the transcriptional control of diverse physiologic and pathophysiologic processes such as cell growth [83], proliferation [84], apoptosis [85] and inflammation [86]. Under physiological conditions Trx is fully reduced and interacts with pro-apoptotic proteins, such as apoptosis signaling kinase 1 (ASK1), the tumor suppressor phosphatase and tensin homologue deleted on chromosome 10 (PTEN), activator protein 1 (AP-1) and p53 [87–89]. In general reversible oxidized redox sensitive cysteine residues of proteins are largely dependent on Trx, which restores their reduced state. However, the enzymatic activity of Trxs relies on the activity of thioredoxin reductases (TrxR), which are selenoproteins and reduce oxidized Trxs under consumption of NAD(P)H [90–93]. Thus, at least baseline activities of TrxR are necessary for cell survival (Fig. 5). As a consequence TrxR might serve as a potential target for cancer treatment by its targeting with electrophilic compounds, which might interact with the redox-active moiety of TrxR [94]. In contrast, the thioredoxin interacting protein (TXNIP) inhibits Trx by binding to its catalytic site thereby competing with other proteins such as ASK1 resulting in an increased susceptibility to undergo apoptosis. In addition, low TXNIP expression correlates with an enhanced tumorigenicity [95] and increased metastasis formation [96].
Fig. 5.
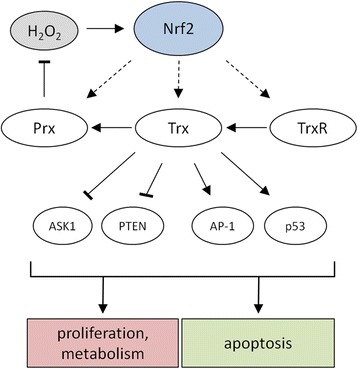
Trx-based upregulation of anti-oxidative systems by Nrf2. Oxidized Trxs are reduced by TrxRs and maintained in their active form. Reduced Trxs can reduce oxidized Prxs, which under physiological conditions detoxify H2O2. Reduced Trxs can interact with redox-sensitive proteins, such as ASK1, PTEN, AP-1 and p53 suggesting that different cellular processes such as proliferation, the cellular metabolism and apoptosis and might be regulated by Trxs.
Glutathione system
The glutathione (GSH) system is a major thiol-based defense system against oxidative and electrophilic stress in mammals and functions as co-substrate for the GPxs, which efficiently remove H2O2 thereby preventing oxidative insults and influencing together with glutaredoxin (Grx) the redox state of proteins via reversible S-glutathionylation [97]. Thus GSH plays an important role in redox-signaling and in the regulation of protein functions. In addition key enzymes of the GSH biosynthesis can be upregulated by Nrf2 [98].
The specific role of H2O2 in cancer
Whereas low ROS levels seem to be relevant for the maintenance of cellular homeostasis in normal cells, most cancer cells show metabolic alterations resulting in significantly higher ROS levels, which can trigger either pro- as well as anti-tumorigenic processes. The increased levels of ROS can promote pro-survival and pro-proliferative pathways as well as the metabolic adaption of tumor cells to the tumor environment [99]. The latter includes phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) resulting at least in part in an increased mitochondrial metabolism [100] along with the inhibition of the anti-oxidative response by phosphorylating members of the fork head box O transcription factor (FOXO) family [101, 102], of the mitogen-activated kinase (MAPK/ERK) as well as of the hypoxia-inducible factor (HIF) signaling cascades [103–105]. Moreover, several oncogenes linked to these pathways, such as RAS, MYC and AKT as well as mutations or loss of tumor suppressors like p53, are associated with increased ROS levels [106, 107]. Yet, increased spatially localized ROS levels can also promote cell toxicity thereby leading to the activation of cell cycle arrest or cell death-inducing pathways resulting in the inhibition of cancer progression [108, 109]. Thus cancer cells do not only have to cope with higher ROS levels [110, 111], but also have to maintain their redox balance, which is frequently accomplished by up-regulating anti-oxidants [112]. In addition, the master regulator of the cellular anti-oxidant response Nrf2 can be activated and stabilized by a number of oncogenes, for example PI3K, K-ras or MYC [47, 113], known to drive signaling cascades that mediate cancer cell proliferation and/or survival. Furthermore, primary tumor cells exert not only higher expression levels of ROS scavengers, including Prxs, SODs and GPxs, but also structural alterations of the Nrf2 inhibitor Keap1 suggesting that an imbalanced redox status promotes tumorigenicity [114–116] (Fig. 6). This is in accordance with an enhanced tumor progression rate in response to treatment with anti-oxidants [117] and an increased resistance to chemotherapeutic drugs via the activation of the Nrf2 [118]. In this context it is noteworthy that several hallmarks of cancer can be directly linked to an increased ROS production [119], such as sustained proliferative signaling [99], resistance to cell death [120], activation of invasion and metastasis [121] as well as induction of angiogenesis [122]. The role of H2O2 as a promoter of neoplastic transformation is supported by the modulation of the PI3K/AKT signaling pathway due to oxidation of the PTP1B [123, 124] and of PTEN [125] and supported by the inhibition of its induction in the presence of anti-oxidant scavengers [126]. Regarding the tumor cell survival next to the hyperactivation of the PI3K/AKT and K-ras signaling pathways the activation and stabilization of Nrf2 is important for the protection of cancer cells from oxidative stress.
Fig. 6.
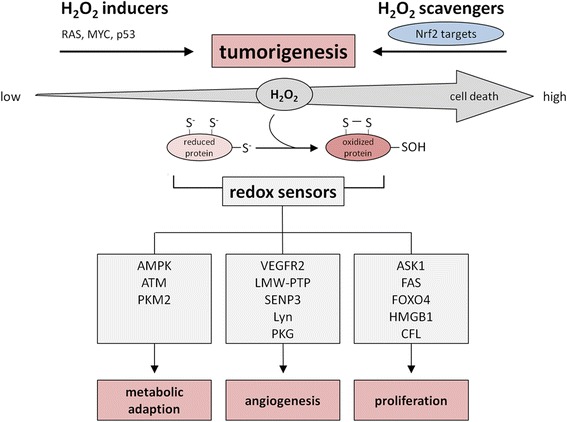
Components of anti-oxidative systems involved in tumor development. Cancer cells are characterized by high levels of ROS (H2O2). To prevent cell damage and cell death cancer cells induce the expression of anti-oxidative enzymes via the activation of the transcription factor Nrf2. Despite high H2O2 levels cancer cells maintain the capacity to promote cell survival, differentiation and proliferation by undergoing metabolic adaption processes thereby relying on the redox regulation of cancer-related redox sensors.
Correlation of redox-sensitive proteins with neoplastic transformation
H2O2-mediated signaling events have been reported to affect major features of the cancer cell behavior. Since H2O2 is involved in the regulation of apoptosis, cell cycle progression and proliferation, the energy metabolism and angiogenesis, specific redox-sensitive targets with redox-sensor functions are necessary (Fig. 6, Table 2).
Table 2.
Redox-sensitive proteins involved in the regulation of cell metabolism, angiogenesis and cell death
| protein | category | modificationa | cysteine residue | effect |
|---|---|---|---|---|
| anti-oxidative defense-related redox sensors | ||||
| Keap1 | others | S-S | C151 | inactivation [238] |
| 2-cys Prx | peroxidase | sulfonic acid | C51 | inactivation [239] |
| Trx | others | S-S | C32/C35 | inactivation [240] |
| metabolism-related redox sensors | ||||
| AMPK | kinase | G | C299, C304 | activation [241] |
| S-S | C130, C174 | inactivation [132] | ||
| ATM | kinase | S-S | C2291 | activation [138, 142] |
| PKM2 | kinase | C358 | inactivation [146] | |
| angiogenesis-related redox sensors | ||||
| VEGFR | receptor | S-S | C1206/C1199 | inactivation [147, 148] |
| LMW-PTP | phosphatase | S-S | C12/C17 | inactivation [242] |
| SENP3 | sumo | C243 and/or C274 | stabilization [158] | |
| protease | C243 | HIF-1 transactivation [160, 161] | ||
| C532 | inactivation, suppressed HIF-1 activity [160, 161] | |||
| Lyn | kinase | C466 | recruitment of leukocytes [168, 169] | |
| PKG | kinase | S-S | C42 | activation [172] |
| cell death-related redox sensors | ||||
| ASK1 | kinase | S-S | C250 | inactivation [178, 179] |
| Fas | receptor | G | C294 | aggregation, FasL binding [186] |
| FOXO4 | TF | S-S | C477 | inactivation [189] |
| HMGB1 | others | S-S | C23/C45 | induction of apoptosis [193] |
| CFL | others | S-S, G | C139/C147, C39/C80 | inactivation, inhibition of apoptosis [208–210] |
a C cysteine, S-S intra-/intermolecular disulfide, G glutathionylation
Redox control of the cellular energy metabolism with the relation to cellular growth
In comparison to non-malignant normal cells, cancer cells shift their metabolism to anaerobic glycolysis, which is driven by multiple oncogenic pathways. The PI3K-driven AKT activation leads to a direct regulation of glycolytic enzymes and activation of mTOR. This has an effect on (i) glycolytic enzymes by activation of HIF and/or (ii) induction of the glucose transporter GLUT1, enzymes of the glycolysis as well as the mitochondrial PDK, which inhibits the flux of pyruvate into the TCA [110] (Fig. 7). AMPK can act as an energy sensor protein kinase and opposes this effect by blocking the mTOR activity. Therefore AMPK regulates the energy metabolism by activating energy-producing pathways and inhibiting energy-consuming processes in response to low intracellular ATP levels thereby also linking cellular metabolism to growth control and cell polarity [127]. This was further confirmed by the AMPK inducer 5-aminoimidazole-4-carboxyamide ribonucleoside, which inhibits tumor growth in vitro and in vivo, suggesting that the AMPK signaling pathway might contribute to the suppression of (tumor) growth by acting as a metabolic checkpoint resulting in cell growth arrest in the G1/S phase in the presence of low intracellular ATP levels [128–130]. Thus, AMPK has a strong impact on the proliferation rate of both non-malignant as well as tumor cells [131].
Fig. 7.
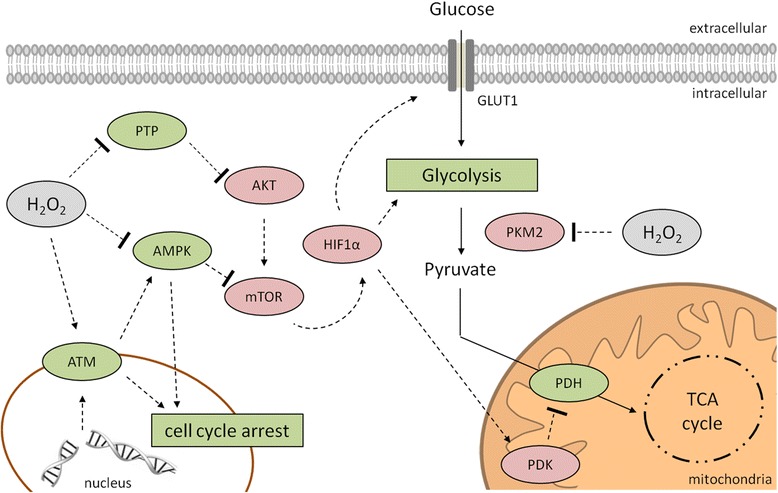
Redox control of the cellular energy metabolism. In cancer cells the shift of the metabolism into anaerobic glycolysis is mainly mediated by the PI3K/AKT pathway. AKT activates mTOR, which subsequently activates HIF1α resulting in an induction of GLUT1, enzymes of the glycolysis and the mitochondrial PDK, which inhibits the pyruvate flux into the TCA. The AMPK is able to block this mechanism by inhibition of mTOR to conserve energy. Cancer cells exhibit high ROS (H2O2) levels leading to an inhibition of the AMPK and of PTPs, which can inactivate AKT. Even through high H2O2 levels DSBs could occur leading to the activation of ATM accompanied with cell cycle arrest. The interaction of ATM and AMPK might enhance the DNA damage response. In addition H2O2 might inactivate the PKM2 leading to an altered flux of glucose in the pentose phosphate pathway for the generation of reductions equivalents to detoxify ROS. PDK, pyruvate dehydrogenase kinase; PKM2, pyruvate kinase M2; TCA, tricarboxylic acid.
In addition to oncogenic mutations and signaling pathways [128] the AMPK activity can be suppressed by oxidation of cysteine residues within the catalytic subunit alpha at positions 130 and 174 promoting its aggregation. In contrast, the reduction of these sites is required for the successful activation of the AMPK complex during energy starvation, which is mediated by Trx thereby providing evidence that oxidative stress and metabolism can be linked via AMPK [132]. Furthermore, AMPK can function as a sensor of genomic stress and interacts/enhances the DNA damage response by interaction with the serine/threonine protein kinase ATM [133] a redox sensor for the regulation of DNA repair processes. Under physiologic conditions ATM is recruited and activated by DNA double-strand breaks (DSBs) via the formation of MRE11-Rad50-Nibrin (MRN) DNA repair complexes. This results in the phosphorylation of various key proteins involved in DNA repair processes, such as p53, the serine/threonine-protein kinase Chk2 (CHK2) and the histone H2AX (H2AX) [134–137]. In the presence of H2O2 ATM forms a disulfide-cross-linked dimer resulting in its direct activation independent from the MRN complex formation thereby supporting its redox sensor function [138]. Furthermore, ATM is involved in the regulation of mitochondrial function and metabolic control by interaction with p53, AMPK, mTOR and HIF1α [139–141], which is independent of DSBs [142]. In addition, the redox status of tumors functions as a major determinant of the ATM-dependent molecular switch of resistance to apoptosis. At low ROS levels apoptosis was blocked, whereas increased cellular ROS levels restored ATM/JNK-mediated apoptotic signaling [143]. There is also evidence that pathological neoangiogenesis requires ATM-mediated oxidative defense, since agents promoting excessive ROS generation have beneficial effects in the treatment of neovascular diseases [144]. Not only AMPK, but also the pyruvate kinase isoform M2 (PKM2), known to be over-expressed in tumors [110], represents a switch between glycolysis and gluconeogenesis. Inhibition of PKM2 caused by oxidative modification of the cysteine residue at position 358 [145] contributes to maintain cellular anti-oxidant responses by diverting the glucose flux into the pentose phosphate pathway thereby generating sufficient reducing potential for the detoxification of ROS [146].
Redox control of cellular signaling processes in association with angiogenesis and cell death
ROS, which are generated in response to various stimuli including growth factors, have been shown to modulate cellular growth and angiogenesis. A major source for ROS are NOX enzymes that can be activated by various growth factors, e.g. vascular endothelial growth factor (VEGF) and angiopoietin-1, leading to the induction of genes involved in angiogenesis and thus represent therapeutic targets for the inhibition of tumor angiogenesis [122]. H2O2 derived from NOX activities can affect the vascular endothelial growth factor receptor (VEGFR) 2, which regulates angiogenesis, vascular development, vascular permeability and embryonic hematopoiesis, but also promotes cell proliferation, survival, migration, and differentiation of vascular endothelial cells. Despite VEGFR1 and VEGFR2 can bind VEGFA, VEGFR2 plays the major role in modulating these processes. Its activation depends not only on the autophosphorylation of defined tyrosine residues, but is also regulated by oxidative modifications [147, 148]. Increased cellular H2O2 levels promote the formation of an intracellular disulfide bond thereby blocking the receptor activity, whereas the presence of Prx2 effectively prevents this oxidative modification leaving the receptor responsive to VEGFA stimulation [147, 148]. Furthermore, extracellular H2O2 generated by extracellular SOD promotes VEGFR2 signaling via oxidative inactivation of protein tyrosine phosphatases (PTPs) in mice [149]. Moreover, the expression of TXNIP is required for the VEGF-mediated VEGFR2 activation and angiogenic response in vivo and in vitro by regulating VEGFR2 phosphorylation via S-glutathionylation of the low molecular weight protein tyrosine phosphatase (LMW-PTP) in endothelial cells [150]. In addition the interaction of TXNIP with the poly-ADP-ribose polymerase 1 (PARP1) is a relevant regulator for its translocalization and function leading to the activation of VEFGR2 signaling in human umbilical vein endothelial cells [151]. Furthermore, H2O2 was shown to induce the expression levels of the VEGFR2 ligand VEGF by inducing the transcription factors NFκB or AP-1 [152]. Under hypoxic conditions VEGF expression is upregulated by HIF1α which is over-expressed in many tumors and its activity levels influence angiogenesis as well as tumorigenesis [153]. Under normoxic conditions HIF1α is hydroxylated and subsequently ubiquitinated for proteasomal degradation, whereas under hypoxic conditions its hydroxylation is blocked leading to its accumulation, dimerization with its beta subunit and subsequent translocation into the nucleus, where it regulates the expression of genes linked to cellular transformation, cell proliferation and angiogenesis [154–156]. The transcriptional activity of HIF1α depends on the translocation of sentrin/SUMO-specific protease 3 (SENP3) from the nucleoli to the nucleoplasm [157]. ROS seem to be involved in limiting its proteasomal degradation. The complex formation with either the heat shock protein 90 (Hsp90) or the co-chaperone/ubiquitin ligase carboxyl terminus of Hsc70-interacting protein (CHIP) leads to the stabilization or degradation of SENP3. Under mild oxidative stress the oxidation of thiol residues favors the recruitment of Hsp90 thereby protecting SENP3 from binding to CHIP, which results in its ubiquitination and subsequent elimination via proteasomal degradation. Thus, the redox status of SENP3 is a decisive factor for its stabilization or degradation [158] and can regulate the expression of the EMT-inducing transcription factor fork head box C2 (FOXC2) that is de-SUMOylated and thereby activated in response to increased ROS levels. As a result the expression of the mesenchymal marker protein N-cadherin is induced [159]. In HeLa cells ROS levels are involved in the activation of HIF1α by modifying cysteine residues at positions 243 and 532 of SENP3 thereby controlling the interaction of SENP3 with p300, the co-activator of HIF1α. This is accompanied by SUMOylation of p300 resulting in the transcriptional silencing of HIF1α. The shift of HIF1α transactivation by ROS depends on the biphasic redox sensing of SENP3. Whereas low ROS levels lead to SENP3 accumulation and therefore enhanced HIF1α transcriptional activity, high concentrations of ROS inactivated SENP3 resulting in the suppression of HIF1α transcriptional activity. Thus SENP3 is an example for a redox sensitive protein with cysteine residues that can sense different ROS levels [160, 161]. VEGF can also promote endothelial permeability through the activation of the Src family non-receptor tyrosine kinases (SFKs) [162]. Lyn, a member of the SFK family, has been shown to be amplified and upregulated in tumor cells, which is associated with resistance to chemotherapy [163] and plays an important role in the regulation of both innate and adaptive anti-tumoral immune responses. Since NOX-expressing tumors are able to efficiently produce H2O2, the tumor stroma can mimic features of ‘unhealed’ wounds [164]. Using distinct model systems, extracellular H2O2 levels have been linked to the recruitment of leukocytes, such as neutrophils, representing the first line of innate immune responses [165–167]. In addition, Lyn serves as a redox sensor for neutrophils monitoring the redox state of wounds. The oxidation-specific modification site was defined as cysteine residue 466, which directly triggered the wound response and calcium signaling [168, 169]. In response to treatment with chromium (V) complexes the formation of ROS and activation of Lyn were found in lymphocytes leading to the activation of caspase-3 and subsequently to the induction of apoptosis [170]. Another kinase with redox-sensor function and involvement in angiogenesis is the cGMP-dependent protein kinase (PKG). PKG represents a member of a serine/threonine-specific protein kinase family that acts as a key mediator of the nitric oxide (NO)/cGMP signaling pathway. GMP binding has been shown to activate PKG resulting in the phosphorylation of serine and threonine residues on many cellular proteins [171] involved in modulating cellular calcium. Besides this activation mechanism it is also known that PKG can be activated under oxidative stress independent of the respective cGMP or NO levels [172]. PKG controls the regulation of platelet activation and adhesion, smooth muscle contraction, cardiac function, gene expression and the feedback of the NO-signaling pathway amongst others. While the expression of PKG in metastatic colon carcinoma blocks tumor angiogenesis by down-regulating the expression level of beta-catenin [173], PKG signaling can also mediate cytoprotective and anti-apoptotic function in various tissues including non-small-cell lung carcinoma. Thus, PKG inhibitors might be of therapeutic relevance and have been suggest for treatment in combination with cisplatin chemotherapy of solid tumors [174]. PKG-inhibitors limit the migration and invasion capacity of colorectal carcinoma cells [175]. Moreover, pro-apoptotic effects of PKG signaling have been reported for various colon carcinoma as well as breast cancer cell lines, which is in line with the hypothesis that the loss of PKG expression in colon carcinoma cell lines may contribute their resistance to undergo anoikis [176, 177].
Redox control of cellular signaling processes in association with apoptosis
By acting as a mitogen-activated protein (MAP) kinase kinase kinase (MAPKKK) ASK1 can activate two distinct sets of MAPKK. Whereas the tumor necrosis factor alpha (TNF-α)-mediated activation of MKK4 (SEK1) via its downstream target JNK leads to the induction of apoptotic cell death, the activation of MKK6 activates p38 subgroups of MAPK, which phosphorylate a wide range of potential targets in response to inflammatory cytokines and cellular stress. A key role in the ASK1-mediated induction of apoptosis via MKK is its dimer formation, known to be induced by exposure to H2O2, but blocked by Trx supporting its role as a redox sensor. Moreover, the interaction of ASK1 and Trx is based on the formation of a disulfide bond at the N-terminal domain of ASK1 leading to its ubiquitination and subsequent proteasomal degradation. However, high levels of H2O2 caused a loss of the protective function of Trx due to the formation of an intramolecular disulfide bond resulting in its release from ASK1, which is accompanied by its activation [178, 179]. Furthermore, the selective inhibition of TrxR by the drug MC3 or by electrophilic pollutants leads to the induction of apoptosis via the Trx-ASK1-p39 signal cascade by blocking the interaction of Trx with ASK1 [180, 181]. In addition, redox alterations induced by selective inhibition of the glucose metabolism leading to massive oxidative stress might serve as a molecular switch that activates the ASK1-JNK/p38 MAPK signaling pathways accompanied by promotion of the radiosensitization of malignant cells [182]. Similar effects have been reported in response to treatment with iron chelators, which also resulted in reduced ASK1-Trx complex formation [183]. The genetic inhibition of ASK1 resulted not only in the inhibition of JNK activation, but also in decreased expression of Fas ligand (FasL) and subsequent apoptosis, whereas the inhibition of p38 did not alter the FasL expression [184]. The activation of Fas upon ligand engagement leads to the formation of a death-inducing signaling complex accompanied by caspase 8-mediated apoptosis [185]. The Fas/FasL interaction results in the S-glutathionylation of Fas at cysteine residue 294 [186], which not only increases the binding to its ligand, but also its aggregation and recruitment into lipid rafts. This oxidative modification can be linked to the activity of Grx1 [187], since the depletion of Grx1 results in an increased S-glutathionylation rate along with the induction of apoptosis, while Grx1 overexpression causes opposite effects. The level of oxidative stress mediated by exogenous sources or endogenously generated upon receptor stimulation regulates the sensitivity to Fas-mediated apoptosis [188]. Additionally FOXO4, a TF involved in the regulation of the insulin signaling pathway, can be activated by oxidative stress due to the formation of an intermolecular disulfide bond between cysteine residue 477 and histone acetyltransferase p300 resulting in the formation of a covalently linked heterodimer. The redox modification of FOXO4 is essential for its subsequent CREB-binding protein (CBP)-mediated acetylation [189]. However, the activity of the heterodimeric complex is regulated by the Trx system, which has a strong impact on the turnover of this interaction by reducing the cysteine-dependent heterodimer of FOXO4 and p300 thereby providing evidence that Trx might be a key regulator of ROS-dependent FOXO4 signaling [189]. In addition, the efficient nuclear translocation and subsequent activation of FOXO4 in response to ROS depends on disulfide formation with the nuclear import receptor transportin-1 (TNPO1), whereas its insulin signaling-dependent nuclear shuttling is not dependent on TNPO1 [190]. Although high-mobility group box 1 protein (HMGB1) might act as a redox-sensitive switch between autophagy and apoptosis. HMGB1 is a DNA-binding protein that associates with chromatin, but can also bind single stranded DNA linking the assembly of transcriptional active protein complexes on specific targets. Its reduced form interacts with the receptor for advanced glycation end products (RAGE) thereby inducing beclin1-dependent autophagy [191]. In the presence of higher ROS levels HMGB1 can undergo oxidative modification leading to the formation of a disulfide bond between cysteine residues 23 and 45 [192], which induces apoptosis via the intrinsic pathway [193]. When released in its partially oxidized status, HMGB1 functions as a pro-inflammatory cytokine [194], whereas in its fully oxidized form (sulfonylated) all biologic activities are lost. Furthermore, HMGB1 can be released from both activated and dying cells thereby acting as a damage-associated molecular pattern molecule [195]. However, its biochemical and immunological properties depend on both its cellular localization as well as its release mechanism [196]. Due to different intracellular and extracellular functions HMGB1 is a central mediator in inflammation and immunity, but its activity depends on the state of its redox-sensitive cysteine residues at positions 23, 45 and 106 ranging from DNA binding, to induction of chemotaxis and transcription of chemokines [197, 198] suggesting its classification as an “alarmin” for sepsis and cancer [199]. Different diseases, such as cancer, are often accompanied by T cell hyporesponsiveness, which is mediated by ROS. The release of H2O2 produced by tumor-infiltrating macrophages leads to the suppression of potentially tumor reactive T cells [200]. Cofilin (CFL), a member of the actin-depolymerizing factor protein family, binds to F-actin and plays an important role in the regulation of the actin cytoskeleton dynamics as well as in the mitochondrial apoptosis. Its translocation from the cytoplasm into the mitochondria leads to cytochrome c release and activation of caspase signaling, thus representing an early step in the induction of apoptosis [201, 202]. Since CFL is also associated with invasion and metastatic capacity of tumors [203–206], it is a key therapeutic target for tumors [207]. CFL might function as a redox sensor [208] and its dephosphorylation-dependent glutathionylation [209, 210] not only leads to a loss of its actin binding affinity, but also blocks its translocation to the mitochondria thereby preventing apoptosis induction. The oxidation-mediated inactivation of CFL can also provoke T cell hyporesponsiveness or the necrotic-like programmed cell death, which modulates the T cell activation processes including the duration of the effectors phase [211]. In contrast, knockdown of CFL could protect T cells from fatal effects of long-term oxidative stress [212] suggesting that oxidation and mitochondrial localization of CFL represents a check point for necrotic-like cell death. Therefore the oxidation of CFL might provide a molecular explanation for the T cell hyporesponsiveness found in diseases such as cancer under oxidative stress conditions [200].
Components of redox regulating processes as therapeutic targets
Tumor cells take the advantage of upregulating anti-oxidant systems to protect themselves against ROS-induced cell damage. The upregulation of anti-oxidant molecules is often associated with an increased cell proliferation, survival and chemotherapy resistance. Therefore components of the anti-oxidant processes including the Trx system represent potential therapeutic targets for the treatment of cancer patients to trigger ROS mediated cell death (Table 3). This is in line with the reduced tumor cell proliferation, induced apoptosis and increased sensitivity of tumor cells to anti-cancer therapy in the presence of Trx and TrxR1 inhibitors [213, 214]. Since a cross-talk between different anti-oxidant molecules has been shown, a combinatorial targeting of these molecules is essential for complete inhibition of the anti-oxidant defense system. Indeed inhibition of TrxR in combination with the disruption of the GSH biosynthesis, caused a selective cell death of human head, neck, and lung cancer cells by inducing oxidative stress [215, 216]. In addition to targeting the Trx system other molecules, e.g. the inhibition of the PTEN axis [217] or the protein deglycase DJ-1 (DJ-1) known to exhibit anti-oxidative and cyto-protective functions in other diseases [218–220] might have therapeutic potential, since DJ-1 is often upregulated in cancer cells and involved in the regulation of various redox stress responsive signaling pathways (PI3K/AKT/PKB; Trx1/ASK1) [217, 221]. Thus, a combined targeting of Trx and DJ-1 result in a complete loss of the anti-oxidant defense system [217].
Table 3.
Current therapeutic strategies for the treatment of cancer patients to trigger ROS-mediated cell death
| substance | target/mechanism | treatment of |
|---|---|---|
| Auranofin | TrxR inhibitor | leukemia, solid cancer, melanoma [213] |
| + BSO | inhibition of GSH biosynthesis | head and neck cancer cells, malignant B cells [215, 216] |
| + Selenocystin | lung cancer [214] | |
| MJ25 | TrxR inhibitor | melanoma [213] |
| Imexon | prooxidant, cysteine depletion | B cell non-Hodgkin lymphoma [243, 244] |
| Calmangafodipin | SOD mimetica | metastatic colorectal cancer [245] |
| Molexafin gadolinum | TrxR inhibitor, superoxide formation | glioblastoma multiform [246] |
| Arsenic trioxide | inhibitor of mitochondrial chain, GPx and TrxR | melanoma [247] |
| + GSH depletion |
However, the interaction between different anti-oxidant molecules in distinct tumor models requests further analysis to increase the insights of the underlying molecular mechanisms of these interactions and the identification of additional molecular targets for cancer therapy. In addition, a better understanding of the role of the intracellular redox state balance and the redox-regulated signaling cascades might enhance the therapeutic options for the treatment of various human cancer types.
Conclusions
Many cancer cells are characterized by an increased intrinsic formation of ROS as a result of their malignant transformation process. Yet, they have to adapt to this challenge in order to maintain the capacity for tumor progression. ROS, in particular H2O2, play an important role in facilitating both cell proliferation and cell survival of tumor cells by triggering the redox signaling cascades. New therapeutic approaches are currently developed that aim towards altering the tumor cell redox state, including (i) the selective inhibition of cellular ROS sources [222, 223], e.g. NOX, (ii) the hyperactivation of anti-oxidant enzymes to lower intracellular ROS levels and (iii) the modulation of the anti-oxidant response system towards increasing ROS levels thereby further promoting the induction of apoptosis. So far, the underlying molecular mechanisms of the interactions between different redox signaling compounds and the tumor progression processes are not fully understood. In addition, there is still a need to define additional redox sensors. Therefore, further research is required to gain additional insights into these signaling networks and sensors, which then might lead to the identification and subsequent design of novel targeted therapies for the treatment of cancer patients.
Acknowledgements
This work was funded/supported by an interdisciplinary DFG grant (grant numbers: MU3275/3-1, WE1467/13-1 and LI1527/3-1).
Abbreviations
- AMPK
AMP-activated protein kinase
- AKT
Protein kinase B
- AQP
Aquaporin
- ARE
Anti-oxidant response element
- ASK1
Apoptosis signal-regulating kinase 1
- ATM
Ataxia telangiectasia mutated
- CFL
Cofilin
- EMT
Epithelial-mesenchymal transition
- ERK
Extracellular signal regulated kinase
- Fas
Tumor necrosis factor receptor superfamily member 6
- GPx
Glutathione peroxidase
- Grx
Glutaredoxin
- GSH
Glutathione
- GST
Glutathione S transferase
- H2O2
Hydrogen peroxide
- HIF
Hypoxia inducible factor
- HMGB1
High-mobility group box 1 protein
- JNK
c-Jun amino-terminal kinase
- Keap1
Kelch-like ECH-associated protein 1
- MAPK
Mitogen-activated protein kinase
- mTOR
Mammalian target of rapamycin
- NO
Nitric oxide
- NOX
NAD(P)H oxidase
- NFκB
Nuclear factor-kappaB
- Nrf2
Nuclear factor-erythroid 2 p45-related factor 2
- O2−
Superoxide anion
- PI3K
Phosphatidylinositol 3-kinase
- PKG
cGMP-dependent protein kinase
- PKM2
Pyruvate kinase M2
- Prx
Peroxiredoxin
- PTEN
Phosphatase and tensin homologue deleted on chromosome 10
- PTP
Protein tyrosine phosphatase
- ROS
Reactive oxygen species
- SENP3
Sentrin/SUMO-specific protease 3
- SOD
Superoxide dismutase
- Srx
Sulfiredoxin
- TF
Transcription factor
- TNF-α
Tumor necrosis factor alpha
- Trx
Thioredoxin
- TrxR
Thioredoxin reductase
- TXNIP
Thioredoxin interacting protein
- VEGF
Vascular endothelial growth factor
- VEGFR2
Vascular endothelial growth factor receptor 2
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CL conceptualized the review and prepared the figures; CL, JR, RL and LAW co-wrote the manuscript; BS was involved in the design of the review and in preparing the manuscript; all authors have read and approved the final manuscript.
References
- 1.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–62. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411–21. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 3.Glasauer A, Chandel NS. Targeting antioxidants for cancer therapy. Biochem Pharmacol. 2014;92:90–101. doi: 10.1016/j.bcp.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 4.Gañán-Gómez I, Wei Y, Yang H, Boyano-Adánez MC, García-Manero G. Oncogenic functions of the transcription factor Nrf2. Free Radic Biol Med. 2013;65:750–64. doi: 10.1016/j.freeradbiomed.2013.06.041. [DOI] [PubMed] [Google Scholar]
- 5.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–9. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 6.Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466–72. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–73. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 8.Fisher AB. Redox signaling across cell membranes. Antioxid Redox Signal. 2009;11:1349–56. doi: 10.1089/ars.2008.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–61. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Mizoguchi H, Hara S. Effect of fatty acid saturation in membrane lipid bilayers on simple diffusion in the presence of ethanol at high concentrations. J Ferment Bioengeneering. 1996;81:406–11. doi: 10.1016/0922-338X(96)85141-5. [DOI] [Google Scholar]
- 11.Branco MR, Marinho HS, Cyrne L, Antunes F. Decrease of H2O2 plasma membrane permeability during adaptation to H2O2 in Saccharomyces cerevisiae. J Biol Chem. 2004;279:6501–6. doi: 10.1074/jbc.M311818200. [DOI] [PubMed] [Google Scholar]
- 12.Mathai JC, Sitaramam V. Stretch sensitivity of transmembrane mobility of hydrogen peroxide through voids in the bilayer. Role of cardiolipin J Biol Chem. 1994;269:17784–93. [PubMed] [Google Scholar]
- 13.Folmer V, Pedroso N, Matias AC, Lopes SCDN, Antunes F, Cyrne L, et al. H2O2 induces rapid biophysical and permeability changes in the plasma membrane of Saccharomyces cerevisiae. Biochim Biophys Acta. 1778;2008:1141–7. doi: 10.1016/j.bbamem.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 14.Cordeiro RM. Reactive oxygen species at phospholipid bilayers: distribution, mobility and permeation. Biochim Biophys Acta. 2014;1838(1 Pt B):438–44. doi: 10.1016/j.bbamem.2013.09.016. [DOI] [PubMed] [Google Scholar]
- 15.Seaver LC, Imlay JA. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J Bacteriol. 2001;183:7182–9. doi: 10.1128/JB.183.24.7182-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sousa-Lopes A, Antunes F, Cyrne L, Marinho HS. Decreased cellular permeability to H2O2 protects Saccharomyces cerevisiae cells in stationary phase against oxidative stress. FEBS Lett. 2004;578:152–6. doi: 10.1016/j.febslet.2004.10.090. [DOI] [PubMed] [Google Scholar]
- 17.Bienert GP, Møller ALB, Kristiansen KA, Schulz A, Møller IM, Schjoerring JK, et al. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007;282:1183–92. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 18.Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochim Biophys Acta. 1758;2006:994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 19.Miller EW, Dickinson BC, Chang CJ. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc Natl Acad Sci U S A. 2010;107:15681–6. doi: 10.1073/pnas.1005776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vieceli Dalla Sega F, Zambonin L, Fiorentini D, Rizzo B, Caliceti C, Landi L, et al. Specific aquaporins facilitate Nox-produced hydrogen peroxide transport through plasma membrane in leukaemia cells. Biochim Biophys Acta. 2014;1843:806–14. doi: 10.1016/j.bbamcr.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 21.Bertolotti M, Bestetti S, García-Manteiga JM, Medraño-Fernandez I, Dal Mas A, Malosio ML, et al. Tyrosine kinase signal modulation: a matter of H2O2 membrane permeability? Antioxid Redox Signal. 2013;19:1447–51. doi: 10.1089/ars.2013.5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woo HA, Yim SH, Shin DH, Kang D, Yu DY, Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell. 2010;140:517–28. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 23.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–3. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 24.Mahadev K, Wu X, Zilbering A, Zhu L, Lawrence JT, Goldstein BJ. Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3 T3-L1 adipocytes. J Biol Chem. 2001;276:48662–9. [DOI] [PubMed]
- 25.Suzuki Y, Ono Y, Hirabayashi Y. Rapid and specific reactive oxygen species generation via NADPH oxidase activation during Fas-mediated apoptosis. FEBS Lett. 1998;425:209–12. doi: 10.1016/S0014-5793(98)00228-2. [DOI] [PubMed] [Google Scholar]
- 26.Marino SM, Gladyshev VN, Marino SM, Gladyshev VN. Cysteine function governs its conservation and degeneration and restricts its utilization on protein surface. J Mol Biol. 2010;404:902–16. doi: 10.1016/j.jmb.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrer-Sueta G, Manta B, Botti H, Radi R, Trujillo M, Denicola A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem Res Toxicol. 2011;24:434–50. doi: 10.1021/tx100413v. [DOI] [PubMed] [Google Scholar]
- 28.Marinho HS, Real C, Cyrne L, Soares H, Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014;2:535–62. doi: 10.1016/j.redox.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox AG, Hampton MB. Bcl-2 over-expression promotes genomic instability by inhibiting apoptosis of cells exposed to hydrogen peroxide. Carcinogenesis. 2007;28:2166–71. doi: 10.1093/carcin/bgm093. [DOI] [PubMed] [Google Scholar]
- 30.Low FM, Hampton MB, Peskin AV, Winterbourn CC. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood. 2007;109:2611–7. doi: 10.1182/blood-2006-09-048728. [DOI] [PubMed] [Google Scholar]
- 31.Zeida A, Reyes AM, Lebrero MCG, Radi R, Trujillo M, Estrin DA. The extraordinary catalytic ability of peroxiredoxins: a combined experimental and QM/MM study on the fast thiol oxidation step. Chem Commun (Camb) 2014;50:10070–3. doi: 10.1039/C4CC02899F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woo HA, Jeong W, Chang TS, Park KJ, Park SJ, Yang JS, et al. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J Biol Chem. 2005;280:3125–8. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- 33.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–55. [PubMed] [Google Scholar]
- 34.Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD, Lau A, et al. Dual roles of Nrf2 in cancer. Pharmacol Res. 2008;58:262–70. [DOI] [PMC free article] [PubMed]
- 35.Haack M, Löwinger M, Lippmann D, Kipp A, Pagnotta E, Iori R, et al. Breakdown products of neoglucobrassicin inhibit activation of Nrf2 target genes mediated by myrosinase-derived glucoraphanin hydrolysis products. Biol Chem. 2010;391:1281–93. doi: 10.1515/bc.2010.134. [DOI] [PubMed] [Google Scholar]
- 36.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–22. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 37.Itoh K, Mimura J, Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal. 2010;13:1665–78. doi: 10.1089/ars.2010.3222. [DOI] [PubMed] [Google Scholar]
- 38.Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31:90–9. doi: 10.1093/carcin/bgp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. 2010;16:3743–53. doi: 10.1158/1078-0432.CCR-09-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3 doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135:1358–68. doi: 10.1053/j.gastro.2008.06.082. [DOI] [PubMed] [Google Scholar]
- 42.Li K, Zhong C, Wang B, He J, Bi J. Nrf2 expression participates in growth and differentiation of endometrial carcinoma cells in vitro and in vivo. J Mol Histol. 2014;45:161–7. doi: 10.1007/s10735-013-9538-z. [DOI] [PubMed] [Google Scholar]
- 43.Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M, et al. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther. 2010;9:336–46. doi: 10.1158/1535-7163.MCT-09-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ji XJ, Chen SH, Zhu L, Pan H, Zhou Y, Li W, et al. Knockdown of NF-E2-related factor 2 inhibits the proliferation and growth of U251MG human glioma cells in a mouse xenograft model. Oncol Rep. 2013;30:157–64. doi: 10.3892/or.2013.2476. [DOI] [PubMed] [Google Scholar]
- 45.Soini Y, Eskelinen M, Juvonen P, Kärjä V, Haapasaari KM, Saarela A, et al. Nuclear Nrf2 expression is related to a poor survival in pancreatic adenocarcinoma. Pathol Res Pract. 2014;210:35–9. doi: 10.1016/j.prp.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 46.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–9. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 48.Li L, Fath MA, Scarbrough PM, Watson WH, Spitz DR. Combined inhibition of glycolysis, the pentose cycle, and thioredoxin metabolism selectively increases cytotoxicity and oxidative stress in human breast and prostate cancer. Redox Biol. 2014;4C:127–35. doi: 10.1016/j.redox.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ji X, Wang H, Zhu J, Zhu L, Pan H, Li W, et al. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Int J Cancer. 2014;135:574–84. doi: 10.1002/ijc.28699. [DOI] [PubMed] [Google Scholar]
- 50.Groitl B, Jakob U. Thiol-based redox switches. Biochim Biophys Acta. 1844;2014:1335–43. doi: 10.1016/j.bbapap.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mishra M, Jiang H, Wu L, Chawsheen HA, Wei Q. The sulfiredoxin-peroxiredoxin (Srx-Prx) axis in cell signal transduction and cancer development. Cancer Lett. 2015;366:150–9. doi: 10.1016/j.canlet.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flohé L, Harris JR. Introduction. History of the peroxiredoxins and topical perspectives. Subcell Biochem. 2007;44:1–25. doi: 10.1007/978-1-4020-6051-9_1. [DOI] [PubMed] [Google Scholar]
- 53.Trujillo M, Ferrer-Sueta G, Thomson L, Flohé L, Radi R. Kinetics of peroxiredoxins and their role in the decomposition of peroxynitrite. Subcell Biochem. 2007;44:83–113. doi: 10.1007/978-1-4020-6051-9_5. [DOI] [PubMed] [Google Scholar]
- 54.Wood ZA, Schröder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/S0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 55.Yazheng L, Kitts DD. Activation of antioxidant response element (ARE)-dependent genes by roasted coffee extracts. Food Funct. 2012;3:950–4. doi: 10.1039/c2fo30021d. [DOI] [PubMed] [Google Scholar]
- 56.Kim YS, Lee HL, Lee KB, Park JH, Chung WY, Lee KS, et al. Nuclear factor E2-related factor 2 dependent overexpression of sulfiredoxin and peroxiredoxin III in human lung cancer. Korean J Intern Med. 2011;26:304–13. doi: 10.3904/kjim.2011.26.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miyamoto N, Izumi H, Miyamoto R, Kondo H, Tawara A, Sasaguri Y, et al. Quercetin induces the expression of peroxiredoxins 3 and 5 via the Nrf2/NRF1 transcription pathway. Invest Ophthalmol Vis Sci. 2011;52:1055–63. [DOI] [PubMed]
- 58.Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, et al. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40:7416–29. doi: 10.1093/nar/gks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chowdhury I, Mo Y, Gao L, Kazi A, Fisher AB, Feinstein SI. Oxidant stress stimulates expression of the human peroxiredoxin 6 gene by a transcriptional mechanism involving an antioxidant response element. Free Radic Biol Med. 2009;46:146–53. doi: 10.1016/j.freeradbiomed.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2007;292:H1227–36. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 61.Jang HH, Lee KO, Chi YH, Jung BG, Park SK, Park JH, et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–35. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 62.Kim YJ, Ahn JY, Liang P, Ip C, Zhang Y, Park YM. Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: implication to tumor biology. Cancer Res. 2007;67:546–54. doi: 10.1158/0008-5472.CAN-06-2401. [DOI] [PubMed] [Google Scholar]
- 63.McKean DM, Sisbarro L, Ilic D, Kaplan-Alburquerque N, Nemenoff R, Weiser-Evans M, et al. FAK induces expression of Prx1 to promote tenascin-C-dependent fibroblast migration. J Cell Biol. 2003;161:393–402. doi: 10.1083/jcb.jcb.200302126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Randall LM, Manta B, Hugo M, Gil M, Batthyàny C, Trujillo M, et al. Nitration transforms a sensitive peroxiredoxin 2 into a more active and robust peroxidase. J Biol Chem. 2014;289:15536–43. doi: 10.1074/jbc.M113.539213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salzano S, Checconi P, Hanschmann EM, Lillig CH, Bowler LD, Chan P, et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc Natl Acad Sci U S A. 2014;111:12157–62. doi: 10.1073/pnas.1401712111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Olmos Y, Sánchez-Gómez FJ, Wild B, García-Quintans N, Cabezudo S, Lamas S, et al. SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1α complex. Antioxid Redox Signal. 2013;19:1507–21. doi: 10.1089/ars.2012.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roumes H, Pires-Alves A, Gonthier-Maurin L, Dargelos E, Cottin P. Investigation of peroxiredoxin IV as a calpain-regulated pathway in cancer. Anticancer Res. 2010;30:5085–9. [PubMed] [Google Scholar]
- 68.Zhu L, Yang K, Wang X, Wang X, Wang C. A novel reaction of peroxiredoxin 4 towards substrates in oxidative protein folding. PLoS One. 2014;9 doi: 10.1371/journal.pone.0105529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wei Q, Jiang H, Xiao Z, Baker A, Young MR, Veenstra TD, et al. Sulfiredoxin-Peroxiredoxin IV axis promotes human lung cancer progression through modulation of specific phosphokinase signaling. Proc Natl Acad Sci U S A. 2011;108:7004–9. doi: 10.1073/pnas.1013012108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–4. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 71.Woo HA, Kang SW, Kim HK, Yang KS, Chae HZ, Rhee SG. Reversible oxidation of the active site cysteine of peroxiredoxins to cysteine sulfinic acid. Immunoblot detection with antibodies specific for the hyperoxidized cysteine-containing sequence. J Biol Chem. 2003;278:47361–4. doi: 10.1074/jbc.C300428200. [DOI] [PubMed] [Google Scholar]
- 72.Roussel X, Béchade G, Kriznik A, Van Dorsselaer A, Sanglier-Cianferani S, Branlant G, et al. Evidence for the formation of a covalent thiosulfinate intermediate with peroxiredoxin in the catalytic mechanism of sulfiredoxin. J Biol Chem. 2008;283:22371–82. doi: 10.1074/jbc.M800493200. [DOI] [PubMed] [Google Scholar]
- 73.Tairum CA, de Oliveira MA, Horta BB, Zara FJ, Netto LES. Disulfide biochemistry in 2-cys peroxiredoxin: requirement of Glu50 and Arg146 for the reduction of yeast Tsa1 by thioredoxin. J Mol Biol. 2012;424:28–41. doi: 10.1016/j.jmb.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 74.Findlay VJ, Townsend DM, Morris TE, Fraser JP, He L, Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006;66:6800–6. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hall A, Nelson K, Poole LB, Karplus PA. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid Redox Signal. 2011;15:795–815. doi: 10.1089/ars.2010.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jeong W, Bae SH, Toledano MB, Rhee SG. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic Biol Med. 2012;53:447–56. doi: 10.1016/j.freeradbiomed.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 77.Singh A, Ling G, Suhasini AN, Zhang P, Yamamoto M, Navas-Acien A, et al. Nrf2-dependent sulfiredoxin-1 expression protects against cigarette smoke-induced oxidative stress in lungs. Free Radic Biol Med. 2009;46:376–86. doi: 10.1016/j.freeradbiomed.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bae SH, Woo HA, Sung SH, Lee HE, Lee SK, Kil IS, et al. Induction of sulfiredoxin via an Nrf2-dependent pathway and hyperoxidation of peroxiredoxin III in the lungs of mice exposed to hyperoxia. Antioxid Redox Signal. 2009;11:937–48. doi: 10.1089/ars.2008.2325. [DOI] [PubMed] [Google Scholar]
- 79.Soriano FX, Léveillé F, Papadia S, Higgins LG, Varley J, Baxter P, et al. Induction of sulfiredoxin expression and reduction of peroxiredoxin hyperoxidation by the neuroprotective Nrf2 activator 3H-1,2-dithiole-3-thione. J Neurochem. 2008;107:533–43. doi: 10.1111/j.1471-4159.2008.05648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ishaq M, Evans MDM, Ostrikov KK. Atmospheric pressure gas plasma-induced colorectal cancer cell death is mediated by Nox2-ASK1 apoptosis pathways and oxidative stress is mitigated by Srx-Nrf2 anti-oxidant system. Biochim Biophys Acta. 1843;2014:2827–37. doi: 10.1016/j.bbamcr.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 81.Mahmood DFD, Abderrazak A, El Hadri K, Simmet T, Rouis M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid Redox Signal. 2013;19:1266–303. doi: 10.1089/ars.2012.4757. [DOI] [PubMed] [Google Scholar]
- 82.Holmgren A. Thioredoxin and glutaredoxin systems. J Biol Chem. 1989;264:13963–6. [PubMed] [Google Scholar]
- 83.Yoshioka J, Schreiter ER, Lee RT. Role of thioredoxin in cell growth through interactions with signaling molecules. Antioxid Redox Signal. 2006;8:2143–51. doi: 10.1089/ars.2006.8.2143. [DOI] [PubMed] [Google Scholar]
- 84.Gallegos A, Gasdaska JR, Taylor CW, Paine-Murrieta GD, Goodman D, Gasdaska PY, et al. Transfection with human thioredoxin increases cell proliferation and a dominant-negative mutant thioredoxin reverses the transformed phenotype of human breast cancer cells. Cancer Res. 1996;56:5765–70. [PubMed] [Google Scholar]
- 85.Lu J, Holmgren A. Thioredoxin system in cell death progression. Antioxid Redox Signal. 2012;17:1738–47. doi: 10.1089/ars.2012.4650. [DOI] [PubMed] [Google Scholar]
- 86.Yoshihara E, Masaki S, Matsuo Y, Chen Z, Tian H, Yodoi J. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front Immunol. 2014;4:514. doi: 10.3389/fimmu.2013.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Meuillet EJ, Mahadevan D, Berggren M, Coon A, Powis G. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN’s lipid phosphatase activity and membrane binding: a mechanism for the functional loss of PTEN's tumor suppressor activity. Arch Biochem Biophys. 2004;429:123–33. doi: 10.1016/j.abb.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 88.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Das KC, Muniyappa H. c-Jun-NH2 terminal kinase (JNK)-mediates AP-1 activation by thioredoxin: phosphorylation of cJun, JunB, and Fra-1. Mol Cell Biochem. 2010;337:53–63. doi: 10.1007/s11010-009-0285-0. [DOI] [PubMed] [Google Scholar]
- 90.Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, et al. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. 1999;274:21645–50. doi: 10.1074/jbc.274.31.21645. [DOI] [PubMed] [Google Scholar]
- 91.Junn E, Han SH, Im JY, Yang Y, Cho EW, Um HD, et al. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J Immunol. 2000;164:6287–95. doi: 10.4049/jimmunol.164.12.6287. [DOI] [PubMed] [Google Scholar]
- 92.Brandt W, Wessjohann LA. The functional role of selenocysteine (Sec) in the catalysis mechanism of large thioredoxin reductases: proposition of a swapping catalytic triad including a Sec-His-Glu state. Chembiochem. 2005;6:386–94. doi: 10.1002/cbic.200400276. [DOI] [PubMed] [Google Scholar]
- 93.Gromer S, Wessjohann LA, Eubel J, Brandt W. Mutational studies confirm the catalytic triad in the human selenoenzyme thioredoxin reductase predicted by molecular modeling. Chembiochem. 2006;7:1649–52. doi: 10.1002/cbic.200600080. [DOI] [PubMed] [Google Scholar]
- 94.Powis G, Kirkpatrick DL. Thioredoxin signaling as a target for cancer therapy. Curr Opin Pharmacol. 2007;7:392–7. doi: 10.1016/j.coph.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 95.Masutani H, Yoshihara E, Masaki S, Chen Z, Yodoi J. Thioredoxin binding protein (TBP)-2/Txnip and α-arrestin proteins in cancer and diabetes mellitus. J Clin Biochem Nutr. 2012;50:23–34. doi: 10.3164/jcbn.11-36SR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Masaki S, Masutani H, Yoshihara E, Yodoi J. Deficiency of thioredoxin binding protein-2 (TBP-2) enhances TGF-β signaling and promotes epithelial to mesenchymal transition. PLoS One. 2012;7 doi: 10.1371/journal.pone.0039900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA, Shelton MD. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signal. 2008;10:1941–88. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lu SC. Glutathione synthesis. Biochim Biophys Acta. 1830;2013:3143–53. doi: 10.1016/j.bbagen.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Weinberg F, Chandel NS. Reactive oxygen species-dependent signaling regulates cancer. Cell Mol Life Sci. 2009;66:3663–73. doi: 10.1007/s00018-009-0099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nemoto S, Takeda K, Yu ZX, Ferrans VJ, Finkel T. Role for mitochondrial oxidants as regulators of cellular metabolism. Mol Cell Biol. 2000;20:7311–8. doi: 10.1128/MCB.20.19.7311-7318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hedrick SM. The cunning little vixen: Foxo and the cycle of life and death. Nat Immunol. 2009;10:1057–63. doi: 10.1038/ni.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kops GJPL, Dansen TB, Polderman PE, Saarloos I, Wirtz KWA, Coffer PJ, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–21. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 103.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 104.Bell EL, Emerling BM, Chandel NS. Mitochondrial regulation of oxygen sensing. Mitochondrion. 2005;5:322–32. doi: 10.1016/j.mito.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 105.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158–67. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Behrend L, Henderson G, Zwacka R. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans. 2003;31(Pt 6):1141–4. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- 107.Attardi LD, Donehower LA. Probing p53 biological functions through the use of genetically engineered mouse models. Mutat Res. 2005;576:4–21. doi: 10.1016/j.mrfmmm.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 108.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–4. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 109.Kamata H, Honda SI, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–61. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 110.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 111.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–8. [PubMed] [Google Scholar]
- 112.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–47. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 113.Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013;73:4158–68. doi: 10.1158/0008-5472.CAN-12-4499. [DOI] [PubMed] [Google Scholar]
- 114.Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105:13568–73. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hu Y, Rosen DG, Zhou Y, Feng L, Yang G, Liu J, et al. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J Biol Chem. 2005;280:39485–92. doi: 10.1074/jbc.M503296200. [DOI] [PubMed] [Google Scholar]
- 116.Saydam N, Kirb A, Demir O, Hazan E, Oto O, Saydam O, et al. Determination of glutathione, glutathione reductase, glutathione peroxidase and glutathione S-transferase levels in human lung cancer tissues. Cancer Lett. 1997;119:13–9. doi: 10.1016/S0304-3835(97)00245-0. [DOI] [PubMed] [Google Scholar]
- 117.Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6:221ra15. doi: 10.1126/scitranslmed.3007653. [DOI] [PubMed] [Google Scholar]
- 118.Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–43. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 120.Clerkin JS, Naughton R, Quiney C, Cotter TG. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett. 2008;266:30–6. doi: 10.1016/j.canlet.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 121.Nishikawa M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008;266:53–9. doi: 10.1016/j.canlet.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 122.Ushio-Fukai M, Nakamura Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008;266:37–52. doi: 10.1016/j.canlet.2008.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, et al. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–73. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 124.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–72. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 125.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. 2004;101:16419–24. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Policastro L, Molinari B, Larcher F, Blanco P, Podhajcer OL, Costa CS, et al. Imbalance of antioxidant enzymes in tumor cells and inhibition of proliferation and malignant features by scavenging hydrogen peroxide. Mol Carcinog. 2004;39:103–13. doi: 10.1002/mc.20001. [DOI] [PubMed] [Google Scholar]
- 127.Shirwany NA, Zou MH. AMPK a cellular metabolic and redox sensor. A minireview. Front Biosci. 2014;19:447–74. doi: 10.2741/4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–78. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 130.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–93. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 131.Motoshima H, Goldstein BJ, Igata M, Araki E. AMPK and cell proliferation-AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol. 2006;574(Pt 1):63–71. doi: 10.1113/jphysiol.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shao D, Oka SI, Liu T, Zhai P, Ago T, Sciarretta S, et al. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014;19:232–45. doi: 10.1016/j.cmet.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sanli T, Steinberg GR, Singh G, Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol Ther. 2014;15:156–69. doi: 10.4161/cbt.26726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–9. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 135.Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–7. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 136.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 137.Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A. 2000;97:10389–94. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–21. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 139.Ditch S, Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci. 2012;37:15–22. doi: 10.1016/j.tibs.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Krüger A, Ralser M. ATM is a redox sensor linking genome stability and carbon metabolism. Sci Signal. 2011;4:pe17. doi: 10.1126/scisignal.2001959. [DOI] [PubMed] [Google Scholar]
- 141.Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107:4153–8. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tang S, Yang L, Tang X, Liu M. The role of oxidized ATM in the regulation of oxidative stress-induced energy metabolism reprogramming of CAFs. Cancer Lett. 2014;353:133–44. doi: 10.1016/j.canlet.2014.07.028. [DOI] [PubMed] [Google Scholar]
- 143.Mohanty S, Saha S, Md S, Hossain D, Adhikary A, Mukherjee S, et al. ROS-PIASγ cross talk channelizes ATM signaling from resistance to apoptosis during chemosensitization of resistant tumors. Cell Death Dis. 2014;5, e1021. [DOI] [PMC free article] [PubMed]
- 144.Okuno Y, Nakamura-Ishizu A, Otsu K, Suda T, Kubota Y. Pathological neoangiogenesis depends on oxidative stress regulation by ATM. Nat Med. 2012;18:1208–16. [DOI] [PubMed]
- 145.Wang J, Jin L, Li X, Deng H, Chen Y, Lian Q, et al. Gossypol induces apoptosis in ovarian cancer cells through oxidative stress. Mol Biosyst. 2013;9:1489–97. doi: 10.1039/c3mb25461e. [DOI] [PubMed] [Google Scholar]
- 146.Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–83. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kang DH, Lee DJ, Lee KW, Park YS, Lee JY, Lee SH, et al. Peroxiredoxin II is an essential antioxidant enzyme that prevents the oxidative inactivation of VEGF receptor-2 in vascular endothelial cells. Mol Cell. 2011;44:545–58. doi: 10.1016/j.molcel.2011.08.040. [DOI] [PubMed] [Google Scholar]
- 148.Kaplan N, Urao N, Furuta E, Kim SJ, Razvi M, Nakamura Y, et al. Localized cysteine sulfenic acid formation by vascular endothelial growth factor: role in endothelial cell migration and angiogenesis. Free Radic Res. 2011;45:1124–35. doi: 10.3109/10715762.2011.602073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Oshikawa J, Urao N, Kim HW, Kaplan N, Razvi M, McKinney R, et al. Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PLoS One. 2010;5 doi: 10.1371/journal.pone.0010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Abdelsaid MA, Matragoon S, El-Remessy AB. Thioredoxin-interacting protein expression is required for VEGF-mediated angiogenic signal in endothelial cells. Antioxid Redox Signal. 2013;19:2199–212. doi: 10.1089/ars.2012.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Spindel ON, Yan C, Berk BC. Thioredoxin-interacting protein mediates nuclear-to-plasma membrane communication: role in vascular endothelial growth factor 2 signaling. Arterioscler Thromb Vasc Biol. 2012;32:1264–70. doi: 10.1161/ATVBAHA.111.244681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chua CC, Hamdy RC, Chua BH. Upregulation of vascular endothelial growth factor by H2O2 in rat heart endothelial cells. Free Radic Biol Med. 1998;25:891–7. doi: 10.1016/S0891-5849(98)00115-4. [DOI] [PubMed] [Google Scholar]
- 153.Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425–37. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230–8. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007;67:10823–30. doi: 10.1158/0008-5472.CAN-07-0783. [DOI] [PubMed] [Google Scholar]
- 156.Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–31. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Huang C, Han Y, Wang Y, Sun X, Yan S, Yeh ETH, et al. SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. EMBO J. 2009;28:2748–62. doi: 10.1038/emboj.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yan S, Sun X, Xiang B, Cang H, Kang X, Chen Y, et al. Redox regulation of the stability of the SUMO protease SENP3 via interactions with CHIP and Hsp90. EMBO J. 2010;29:3773–86. doi: 10.1038/emboj.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ren Y, Liu K, Wang M, Yu Y, Yang K, Chen Q, et al. De-SUMOylation of FOXC2 by SENP3 promotes the epithelial-mesenchymal transition in gastric cancer cells. Oncotarget. 2014;5:7093–104. doi: 10.18632/oncotarget.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang Y, Yang J, Yang K, Cang H, Huang X, Li H, et al. The biphasic redox sensing of SENP3 accounts for the HIF-1 transcriptional activity shift by oxidative stress. Acta Pharmacol Sin. 2012;33:953–63. [DOI] [PMC free article] [PubMed]
- 161.Wang Y, Yang J, Yi J. Redox sensing by proteins: oxidative modifications on cysteines and the consequent events. Antioxid Redox Signal. 2012;16:649–57. doi: 10.1089/ars.2011.4313. [DOI] [PubMed] [Google Scholar]
- 162.Han J, Zhang G, Welch EJ, Liang Y, Fu J, Vogel SM, et al. A critical role for Lyn kinase in strengthening endothelial integrity and barrier function. Blood. 2013;122:4140–9. doi: 10.1182/blood-2013-03-491423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F, et al. Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proc Natl Acad Sci U S A. 2009;106:7131–6. doi: 10.1073/pnas.0902232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 165.Klyubin IV, Kirpichnikova KM, Gamaley IA. Hydrogen peroxide-induced chemotaxis of mouse peritoneal neutrophils. Eur J Cell Biol. 1996;70:347–51. [PubMed] [Google Scholar]
- 166.Feng Y, Santoriello C, Mione M, Hurlstone A, Martin P. Live imaging of innate immune cell sensing of transformed cells in zebrafish larvae: parallels between tumor initiation and wound inflammation. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Moreira S, Stramer B, Evans I, Wood W, Martin P. Prioritization of competing damage and developmental signals by migrating macrophages in the Drosophila embryo. Curr Biol. 2010;20:464–70. doi: 10.1016/j.cub.2010.01.047. [DOI] [PubMed] [Google Scholar]
- 168.Yoo SK, Starnes TW, Deng Q, Huttenlocher A. Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature. 2011;480:109–12. [DOI] [PMC free article] [PubMed]
- 169.Yoo SK, Freisinger CM, LeBert DC, Huttenlocher A. Early redox, Src family kinase, and calcium signaling integrate wound responses and tissue regeneration in zebrafish. J Cell Biol. 2012;199:225–34. doi: 10.1083/jcb.201203154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vasant C, Rajaram R, Ramasami T. Apoptosis of lymphocytes induced by chromium(VI/V) is through ROS-mediated activation of Src-family kinases and caspase-3. Free Radic Biol Med. 2003;35:1082–100. doi: 10.1016/S0891-5849(03)00471-4. [DOI] [PubMed] [Google Scholar]
- 171.Münzel T, Feil R, Mülsch A, Lohmann SM, Hofmann F, Walter U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3′,5’-cyclic monophosphate-dependent protein kinase [corrected] Circulation. 2003;108:2172–83. doi: 10.1161/01.CIR.0000094403.78467.C3. [DOI] [PubMed] [Google Scholar]
- 172.Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schröder E, et al. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317:1393–7. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- 173.Kwon IK, Schoenlein PV, Delk J, Liu K, Thangaraju M, Dulin NO, et al. Expression of cyclic guanosine monophosphate-dependent protein kinase in metastatic colon carcinoma cells blocks tumor angiogenesis. Cancer. 2008;112:1462–70. doi: 10.1002/cncr.23334. [DOI] [PubMed] [Google Scholar]
- 174.Wong JC, Bathina M, Fiscus RR. Cyclic GMP/protein kinase G type-Iα (PKG-Iα) signaling pathway promotes CREB phosphorylation and maintains higher c-IAP1, livin, survivin, and Mcl-1 expression and the inhibition of PKG-Iα kinase activity synergizes with cisplatin in non-small cell lung cancer cells. J Cell Biochem. 2012;113:3587–98. doi: 10.1002/jcb.24237. [DOI] [PubMed] [Google Scholar]
- 175.Babykutty S, Suboj P, Srinivas P, Nair AS, Chandramohan K, Gopala S. Insidious role of nitric oxide in migration/invasion of colon cancer cells by upregulating MMP-2/9 via activation of cGMP-PKG-ERK signaling pathways. Clin Exp Metastasis. 2012;29:471–92. doi: 10.1007/s10585-012-9464-6. [DOI] [PubMed] [Google Scholar]
- 176.Hou Y, Wong E, Martin J, Schoenlein PV, Dostmann WR, Browning DD. A role for cyclic-GMP dependent protein kinase in anoikis. Cell Signal. 2006;18:882–8. doi: 10.1016/j.cellsig.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 177.Fallahian F, Karami-Tehrani F, Salami S, Aghaei M. Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor-positive and -negative breast cancer cell lines. FEBS J. 2011;278:3360–9. doi: 10.1111/j.1742-4658.2011.08260.x. [DOI] [PubMed] [Google Scholar]
- 178.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–90. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Katagiri K, Matsuzawa A, Ichijo H. Regulation of apoptosis signal-regulating kinase 1 in redox signaling. Methods Enzymol. 2010;474:277–88. doi: 10.1016/S0076-6879(10)74016-7. [DOI] [PubMed] [Google Scholar]
- 180.Cheng X, Holenya P, Can S, Alborzinia H, Rubbiani R, Ott I, et al. A TrxR inhibiting gold(I) NHC complex induces apoptosis through ASK1-p38-MAPK signaling in pancreatic cancer cells. Mol Cancer. 2014;13:221. doi: 10.1186/1476-4598-13-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Myers CR, Myers JM, Kufahl TD, Forbes R, Szadkowski A. The effects of acrolein on the thioredoxin system: implications for redox-sensitive signaling. Mol Nutr Food Res. 2011;55:1361–74. doi: 10.1002/mnfr.201100224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sharma PK, Dwarakanath BS, Varshney R. Radiosensitization by 2-deoxy-D-glucose and 6-aminonicotinamide involves activation of redox sensitive ASK1-JNK/p38MAPK signaling in head and neck cancer cells. Free Radic Biol Med. 2012;53:1500–13. doi: 10.1016/j.freeradbiomed.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 183.Yu Y, Richardson DR. Cellular iron depletion stimulates the JNK and p38 MAPK signaling transduction pathways, dissociation of ASK1-thioredoxin, and activation of ASK1. J Biol Chem. 2011;286:15413–27. doi: 10.1074/jbc.M111.225946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Su JL, Lin MT, Hong CC, Chang CC, Shiah SG, Wu CW, et al. Resveratrol induces FasL-related apoptosis through Cdc42 activation of ASK1/JNK-dependent signaling pathway in human leukemia HL-60 cells. Carcinogenesis. 2005;26:1–10. doi: 10.1093/carcin/bgh220. [DOI] [PubMed] [Google Scholar]
- 185.Wajant H. The Fas signaling pathway: more than a paradigm. Science. 2002;296:1635–6. doi: 10.1126/science.1071553. [DOI] [PubMed] [Google Scholar]
- 186.Anathy V, Aesif SW, Guala AS, Havermans M, Reynaert NL, Ho YS, et al. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J Cell Biol. 2009;184:241–52. doi: 10.1083/jcb.200807019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Allen EMG, Mieyal JJ. Protein-thiol oxidation and cell death: regulatory role of glutaredoxins. Antioxid Redox Signal. 2012;17:1748–63. doi: 10.1089/ars.2012.4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Devadas S, Hinshaw JA, Zaritskaya L, Williams MS. Fas-stimulated generation of reactive oxygen species or exogenous oxidative stress sensitize cells to Fas-mediated apoptosis. Free Radic Biol Med. 2003;35:648–61. doi: 10.1016/S0891-5849(03)00391-5. [DOI] [PubMed] [Google Scholar]
- 189.Dansen TB, Smits LMM, van Triest MH, de Keizer PLJ, van Leenen D, Koerkamp MG, et al. Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat Chem Biol. 2009;5:664–72. doi: 10.1038/nchembio.194. [DOI] [PubMed] [Google Scholar]
- 190.Putker M, Madl T, Vos HR, de Ruiter H, Visscher M, van den Berg MCW, et al. Redox-dependent control of FOXO/DAF-16 by transportin-1. Mol Cell. 2013;49:730–42. doi: 10.1016/j.molcel.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 191.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 192.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209:1519–28. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29:5299–310. doi: 10.1038/onc.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yang H, Lundbäck P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1) Mol Med. 2012;18:250–9. doi: 10.2119/molmed.2011.00389. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 195.Castellani P, Balza E, Rubartelli A. Inflammation, DAMPs, tumor development, and progression: a vicious circle orchestrated by redox signaling. Antioxid Redox Signal. 2014;20:1086–97. doi: 10.1089/ars.2012.5164. [DOI] [PubMed] [Google Scholar]
- 196.Li G, Tang D, Lotze MT. Ménage à Trois in stress: DAMPs, redox and autophagy. Semin Cancer Biol. 2013;23:380–90. doi: 10.1016/j.semcancer.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Antoine DJ, Harris HE, Andersson U, Tracey KJ, Bianchi ME. A systematic nomenclature for the redox states of high mobility group box (HMGB) proteins. Mol Med. 2014;20:135–7. doi: 10.2119/molmed.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Magna M, Pisetsky DS. The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol Med. 2014;20:138–46. doi: 10.2119/molmed.2013.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Diener KR, Al-Dasooqi N, Lousberg EL, Hayball JD. The multifunctional alarmin HMGB1 with roles in the pathophysiology of sepsis and cancer. Immunol Cell Biol. 2013;91:443–50. doi: 10.1038/icb.2013.25. [DOI] [PubMed] [Google Scholar]
- 200.Klemke M, Wabnitz GH, Funke F, Funk B, Kirchgessner H, Samstag Y. Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity. 2008;29:404–13. doi: 10.1016/j.immuni.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 201.Zhu B, Fukada K, Zhu H, Kyprianou N. Prohibitin and cofilin are intracellular effectors of transforming growth factor beta signaling in human prostate cancer cells. Cancer Res. 2006;66:8640–7. doi: 10.1158/0008-5472.CAN-06-1443. [DOI] [PubMed] [Google Scholar]
- 202.Chua BT, Volbracht C, Tan KO, Li R, Yu VC, Li P. Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat Cell Biol. 2003;5:1083–9. doi: 10.1038/ncb1070. [DOI] [PubMed] [Google Scholar]
- 203.Lu LI, Fu NI, Luo XU, Li X-Y, Li X-P. Overexpression of cofilin 1 in prostate cancer and the corresponding clinical implications. Oncol Lett. 2015;9:2757–61. doi: 10.3892/ol.2015.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Collazo J, Zhu B, Larkin S, Martin SK, Pu H, Horbinski C, et al. Cofilin drives cell-invasive and metastatic responses to TGF-β in prostate cancer. Cancer Res. 2014;74:2362–73. doi: 10.1158/0008-5472.CAN-13-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Madak-Erdogan Z, Ventrella R, Petry L, Katzenellenbogen BS. Novel roles for ERK5 and cofilin as critical mediators linking ERα-driven transcription, actin reorganization, and invasiveness in breast cancer. Mol Cancer Res. 2014;12:714–27. doi: 10.1158/1541-7786.MCR-13-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Chung H, Kim B, Jung SH, Won KJ, Jiang X, Lee CK, et al. Does phosphorylation of cofilin affect the progression of human bladder cancer? BMC Cancer. 2013;13:45. doi: 10.1186/1471-2407-13-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tang Q, Ji Q, Tang Y, Chen T, Pan G, Hu S, et al. Mitochondrial translocation of cofilin-1 promotes apoptosis of gastric cancer BGC-823 cells induced by ursolic acid. Tumour Biol. 2014;35:2451–9. doi: 10.1007/s13277-013-1325-7. [DOI] [PubMed] [Google Scholar]
- 208.Klamt F, Zdanov S, Levine RL, Pariser A, Zhang Y, Zhang B, et al. Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat Cell Biol. 2009;11:1241–6. doi: 10.1038/ncb1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci U S A. 2002;99:3505–10. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kim JS, Huang TY, Bokoch GM. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol Biol Cell. 2009;20:2650–60. doi: 10.1091/mbc.E09-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Samstag Y, John I, Wabnitz GH. Cofilin: a redox sensitive mediator of actin dynamics during T-cell activation and migration. Immunol Rev. 2013;256:30–47. doi: 10.1111/imr.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wabnitz GH, Goursot C, Jahraus B, Kirchgessner H, Hellwig A, Klemke M, et al. Mitochondrial translocation of oxidized cofilin induces caspase-independent necrotic-like programmed cell death of T cells. Cell Death Dis. 2010;1 doi: 10.1038/cddis.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sachweh MCC, Stafford WC, Drummond CJ, McCarthy AR, Higgins M, Campbell J, et al. Redox effects and cytotoxic profiles of MJ25 and auranofin towards malignant melanoma cells. Oncotarget. 2015;6:16488–506. doi: 10.18632/oncotarget.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Fan C, Zheng W, Fu X, Li X, Wong YS, Chen T. Enhancement of auranofin-induced lung cancer cell apoptosis by selenocystine, a natural inhibitor of TrxR1 in vitro and in vivo. Cell Death Dis. 2014;5, e1191. [DOI] [PMC free article] [PubMed]
- 215.Sobhakumari A, Love-Homan L, Fletcher EVM, Martin SM, Parsons AD, Spitz DR, et al. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS One. 2012;7 doi: 10.1371/journal.pone.0048175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kiebala M, Skalska J, Casulo C, Brookes PS, Peterson DR, Hilchey SP, et al. Dual targeting of the thioredoxin and glutathione antioxidant systems in malignant B cells: a novel synergistic therapeutic approach. Exp Hematol. 2015;43:89–99. doi: 10.1016/j.exphem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Raninga PV, Di Trapani G, Tonissen KF. Cross Talk between Two Antioxidant Systems, Thioredoxin and DJ-1: Consequences for Cancer. Oncoscience. 2014;1:95–110. doi: 10.18632/oncoscience.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–9. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 219.Aleyasin H, Rousseaux MWC, Phillips M, Kim RH, Bland RJ, Callaghan S, et al. The Parkinson’s disease gene DJ-1 is also a key regulator of stroke-induced damage. Proc Natl Acad Sci. 2007;104:18748–53. doi: 10.1073/pnas.0709379104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Jain D, Jain R, Eberhard D, Eglinger J, Bugliani M, Piemonti L, et al. Age- and diet-dependent requirement of DJ-1 for glucose homeostasis in mice with implications for human type 2 diabetes. J Mol Cell Biol. 2012;4:221–30. doi: 10.1093/jmcb/mjs025. [DOI] [PubMed] [Google Scholar]
- 221.Im JY, Lee KW, Woo JM, Junn E, Mouradian MM. DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Hum Mol Genet. 2012;21:3013–24. doi: 10.1093/hmg/dds131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Doering M, Ba LA, Lilienthal N, Nicco C, Scherer C, Abbas M, et al. Synthesis and selective anticancer activity of organochalcogen based redox catalysts. J Med Chem. 2010;53:6954–63. doi: 10.1021/jm100576z. [DOI] [PubMed] [Google Scholar]
- 223.Busch C, Jacob C, Anwar A, Burkholz T, Aicha Ba L, Cerella C, et al. Diallylpolysulfides induce growth arrest and apoptosis. Int J Oncol. 2010;36:743–9. doi: 10.3892/ijo_00000550. [DOI] [PubMed] [Google Scholar]
- 224.An N, Kang Y. Thioredoxin and hematologic malignancies. Adv Cancer Res. 2014;122:245–79. doi: 10.1016/B978-0-12-420117-0.00007-4. [DOI] [PubMed] [Google Scholar]
- 225.Park BJ, Cha MK, Kim IH. Thioredoxin 1 as a serum marker for ovarian cancer and its use in combination with CA125 for improving the sensitivity of ovarian cancer diagnoses. Biomarkers. 2014;19:604–10. doi: 10.3109/1354750X.2014.956793. [DOI] [PubMed] [Google Scholar]
- 226.Park BJ, Cha MK, Kim IH. Thioredoxin 1 as a serum marker for breast cancer and its use in combination with CEA or CA15-3 for improving the sensitivity of breast cancer diagnoses. BMC Res Notes. 2014;7:7. doi: 10.1186/1756-0500-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Hatfield DL, Yoo MH, Carlson BA, Gladyshev VN. Selenoproteins that function in cancer prevention and promotion. Biochim Biophys Acta. 1790;2009:1541–5. doi: 10.1016/j.bbagen.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Nishizawa K, Nishiyama H, Matsui Y, Kobayashi T, Saito R, Kotani H, et al. Thioredoxin-interacting protein suppresses bladder carcinogenesis. Carcinogenesis. 2011;32:1459–66. doi: 10.1093/carcin/bgr137. [DOI] [PubMed] [Google Scholar]
- 229.Morrison JA, Pike LA, Sams SB, Sharma V, Zhou Q, Severson JJ, et al. Thioredoxin interacting protein (TXNIP) is a novel tumor suppressor in thyroid cancer. Mol Cancer. 2014;13:62. doi: 10.1186/1476-4598-13-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Cai CY, Zhai LL, Wu Y, Tang ZG. Expression and clinical value of peroxiredoxin-1 in patients with pancreatic cancer. Eur J Surg Oncol. 2015;41:228–35. doi: 10.1016/j.ejso.2014.11.037. [DOI] [PubMed] [Google Scholar]
- 231.Noh DY, Ahn SJ, Lee RA, Kim SW, Park IA, Chae HZ. Overexpression of peroxiredoxin in human breast cancer. Anticancer Res. 2001;21:2085–90. [PubMed] [Google Scholar]
- 232.Sun QK, Zhu JY, Wang W, Lv Y, Zhou HC, Yu JH, et al. Diagnostic and prognostic significance of peroxiredoxin 1 expression in human hepatocellular carcinoma. Med Oncol. 2014;31:786. doi: 10.1007/s12032-013-0786-2. [DOI] [PubMed] [Google Scholar]
- 233.Jiang H, Wu L, Mishra M, Chawsheen HA, Wei Q. Expression of peroxiredoxin 1 and 4 promotes human lung cancer malignancy. Am J Cancer Res. 2014;4:445–60. [PMC free article] [PubMed] [Google Scholar]
- 234.Yun HM, Park KR, Lee HP, Lee DH, Jo M, Shin DH, et al. PRDX6 promotes lung tumor progression via its GPx and iPLA2 activities. Free Radic Biol Med. 2014;69:367–76. doi: 10.1016/j.freeradbiomed.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 235.Jo M, Yun HM, Park KR, Hee Park M, Myoung Kim T, Ho Pak J, et al. Lung tumor growth-promoting function of peroxiredoxin 6. Free Radic Biol Med. 2013;61:453–63. doi: 10.1016/j.freeradbiomed.2013.04.032. [DOI] [PubMed] [Google Scholar]
- 236.Nicolussi A, D’Inzeo S, Mincione G, Buffone A, Di Marcantonio MC, Cotellese R, et al. PRDX1 and PRDX6 are repressed in papillary thyroid carcinomas via BRAF V600E-dependent and -independent mechanisms. Int J Oncol. 2014;44:548–56. doi: 10.3892/ijo.2013.2208. [DOI] [PubMed] [Google Scholar]
- 237.Wu L, Jiang H, Chawsheen HA, Mishra M, Young MR, Gerard M, et al. Tumor promoter-induced sulfiredoxin is required for mouse skin tumorigenesis. Carcinogenesis. 2014;35:1177–84. doi: 10.1093/carcin/bgu035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J Biol Chem. 2010;285:8463–71. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Chae HZ, Chung SJ, Rhee SG. Thioredoxin-dependent peroxide reductase from yeast. J Biol Chem. 1994;269:27670–8. [PubMed] [Google Scholar]
- 240.Lee S, Kim SM, Lee RT. Thioredoxin and thioredoxin target proteins: from molecular mechanisms to functional significance. Antioxid Redox Signal. 2013;18:1165–207. doi: 10.1089/ars.2011.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem. 2010;285:33154–64. doi: 10.1074/jbc.M110.143685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Chiarugi P, Fiaschi T, Taddei ML, Talini D, Giannoni E, Raugei G, et al. Two vicinal cysteines confer a peculiar redox regulation to low molecular weight protein tyrosine phosphatase in response to platelet-derived growth factor receptor stimulation. J Biol Chem. 2001;276:33478–87. doi: 10.1074/jbc.M102302200. [DOI] [PubMed] [Google Scholar]
- 243.Barr PM, Miller TP, Friedberg JW, Peterson DR, Baran AM, Herr M, et al. Phase 2 study of imexon, a prooxidant molecule, in relapsed and refractory B-cell non-Hodgkin lymphoma. Blood. 2014;124:1259–65. doi: 10.1182/blood-2014-04-570044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Remers WA, Dorr RT. Chemistry and pharmacology of imexon and related cyanoaziridines. Curr Med Chem. 2012;19:5745–53. doi: 10.2174/092986712803988802. [DOI] [PubMed] [Google Scholar]
- 245.Karlsson JOG, Ignarro LJ, Lundström I, Jynge P, Almén T. Calmangafodipir [Ca4Mn(DPDP)5], mangafodipir (MnDPDP) and MnPLED with special reference to their SOD mimetic and therapeutic properties. Drug Discov Today. 2015;20:411–21. doi: 10.1016/j.drudis.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 246.Brachman DG, Pugh SL, Ashby LS, Thomas TA, Dunbar EM, Narayan S, et al. Phase 1/2 trials of Temozolomide, Motexafin Gadolinium, and 60-Gy fractionated radiation for newly diagnosed supratentorial glioblastoma multiforme: final results of RTOG 0513. Int J Radiat Oncol Biol Phys. 2015;91:961–7. doi: 10.1016/j.ijrobp.2014.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Venè R, Castellani P, Delfino L, Lucibello M, Ciriolo MR, Rubartelli A. The cystine/cysteine cycle and GSH are independent and crucial antioxidant systems in malignant melanoma cells and represent druggable targets. Antioxid Redox Signal. 2011;15:2439–53. doi: 10.1089/ars.2010.3830. [DOI] [PubMed] [Google Scholar]