

Abstract
IMPORTANCE
Individuals in the presymptomatic stage of Alzheimer disease (AD) are increasingly being targeted for AD secondary prevention trials. How early during the normal life span underlying AD pathologies begin to develop, their patterns of change over time, and their relationship with future cognitive decline remain to be determined.
OBJECTIVE
To characterize the within-person trajectories of cerebrospinal fluid (CSF) biomarkers of AD over time and their association with changes in brain amyloid deposition and cognitive decline in cognitively normal middle-aged individuals.
DESIGN, SETTING, AND PARTICIPANTS
As part of a cohort study, cognitively normal (Clinical Dementia Rating [CDR] of 0) middle-aged research volunteers (n = 169) enrolled in the Adult Children Study at Washington University, St Louis, Missouri, had undergone serial CSF collection and longitudinal clinical assessment (mean, 6 years; range, 0.91–11.3 years) at 3-year intervals at the time of analysis, between January 2003 and November 2013. A subset (n = 74) had also undergone longitudinal amyloid positron emission tomographic imaging with Pittsburgh compound B (PiB) in the same period. Serial CSF samples were analyzed for β-amyloid 40 (Aβ40), Aβ42, total tau, tau phosphorylated at threonine 181 (P-tau181), visinin-like protein 1 (VILIP-1), and chitinase-3-like protein 1 (YKL-40). Within-person measures were plotted according to age and AD risk defined by APOE genotype (ε4 carriers vs noncarriers). Linear mixed models were used to compare estimated biomarker slopes among middle-age bins at baseline (early, 45–54 years; mid, 55–64 years; late, 65–74 years) and between risk groups. Within-person changes in CSF biomarkers were also compared with changes in cortical PiB binding and progression to a CDR higher than 0 at follow-up.
MAIN OUTCOMES AND MEASURES
Changes in Aβ40, Aβ42, total tau, P-tau181, VILIP-1, and YKL-40 and, in a subset of participants, changes in cortical PiB binding.
RESULTS
While there were no consistent longitudinal patterns in Aβ40 (P = .001–.97), longitudinal reductions in Aβ42 were observed in some individuals as early as early middle age (P ≤ .05) and low Aβ42 levels were associated with the development of cortical PiB-positive amyloid plaques (area under receiver operating characteristic curve = 0.9352; 95% CI, 0.8895–0.9808), especially in mid middle age (P < .001). Markers of neuronal injury (total tau, P-tau181, and VILIP-1) dramatically increased in some individuals in mid and late middle age (P ≤ .02), whereas the neuroinflammation marker YKL-40 increased consistently throughout middle age (P ≤ .003). These patterns were more apparent in at-risk ε4 carriers (Aβ42 in an allele dose-dependent manner) and appeared to be associated with future cognitive deficits as determined by CDR.
CONCLUSIONS AND RELEVANCE
Longitudinal CSF biomarker patterns consistent with AD are first detectable during early middle age and are associated with later amyloid positivity and cognitive decline. Such measures may be useful for targeting middle-aged, asymptomatic individuals for therapeutic trials designed to prevent cognitive decline.
Alzheimer disease (AD) is the most common cause of dementia in elderly individuals, accounting for up to 70% of all dementia cases, and is now estimated to be the third-leading cause of death after heart disease and cancer.1 To date, clinical trials of potential disease-modifying therapies for AD have met with little success in halting or slowing cognitive decline in patients who already have cognitive symptoms or dementia.2 However, clinicopathologic and more recent biomarker data suggest that AD pathology begins to accrue approximately 10 to 20 years before any cognitive signs or symptoms (termed asymptomatic or pre clinical AD),3–11 thus providing a window of opportunity for the initiation of secondary prevention trials that aim to prevent the development of symptoms in individuals while they are still cognitively normal.12 How early during the normal life span such pathologies begin to develop, their patterns of change over time, and their relationship with future cognitive decline remain to be determined.
Because, by definition, preclinical AD eludes detection by current clinical measures, disease-specific biomarkers are necessary to identify individuals in this asymptomatic stage. To this end, the Adult Children Study (ACS) of the Knight Alzheimer’s Disease Research Centerat Washington University, St Louis, Missouri, was initiated. The ACS is a longitudinal clinical and biomarker research study of cognitively normal, middle-aged adults exhibiting different AD risk profiles in cluding age, family history of AD, and APOE genotype (APOE ε4 carriers vs noncarriers).13 Participants undergo comprehensive, longitudinal clinical and psychometric assessments and evaluation of biomarkers in cerebrospinal fluid (CSF) and plasma, along with several imaging modalities. We hypothesized that biomarker patterns indicative of underlying AD pathology would be evident in a subset of cognitively normal individuals during middle age, at a greater frequency in those at higher risk for AD (ie, older and/or carrying the ε4 allele of APOE), and would increase in severity over time, ultimately culminating in cognitive decline.
The 3 CSF biomarker analytes that reflect the core neuropathologies in AD, β-amyloid 42 (Aβ42; the primary constituent of amyloid plaques), total tau (a marker of neuronal injury and/or death), and hyperphosphorylated tau (P-tau; forms intraneuronal neurofibrillary tangles), demonstrate excellent diagnostic and prognostic utility in research cohorts.10,14,15 Other recently identified biomarkers, including visinin-like protein 1 (VILIP-1) and chitinase-3-like protein 1 (YKL-40) (markers of neuronal death and gliosis/neuroinflammation, respectively) have also demonstrated clinical utility in AD, especially when combined in an algorithm with CSF Aβ42.16–20 This first report of longitudinal biomarker changes in the ACS cohort describes the within-person trajectories of these CSF biomarkers over time and their association with longitudinal changes on in vivo amyloid imaging and future cognitive decline as a function of risk conferred by APOE genotype.
Methods
Participants
Participants were cognitively normal, community-dwelling research volunteers enrolled in the ACS at the Knight Alzheimer’s Disease Research Center at Washington University. Inclusion criteria include the following: (1) positive family history (≥1 biological parent with age at AD dementia onset <80 years) or negative family history (both biological parents living to age ≥70 years in the absence of AD dementia); (2) aged 45 to 74 years at study entry (1 enrollee was aged 43 years, 3 were aged 75 years, 3 were aged 76 years, and 1 was aged 81 years); (3) availability of an informant who knows the participant well; (4) normal cognition at study entry (defined as having a Clinical Dementia Rating [CDR]21 of 0); and (5) willingness in principle to complete all study procedures at baseline and longitudinally. Exclusion criteria include the following: (1) presence of a neurological, psychiatric, or systemic illness that might affect cognition or interfere with longitudinal follow-up; (2) a known deterministic mutation for AD; and (3) medical contraindication to lumbar puncture for CSF collection or imaging.
Specific inclusion criteria for the present analyses included the availability of data from at least 2 serial clinical assessments and CSF collection procedures (mean [SD] interval between clinical assessment and CSF collection, 3.3 [3.8] years) as of September 2013; thus, this cohort represents a subset (n = 169) of ACS participants to date. All procedures were approved by the Human Research Protection Office at Washing-ton University, and written informed consent was obtained from all participants and their informants.
Clinical and Cognitive Assessments
The presence or absence of dementia (and, when present, its severity) was operationalized with the CDR in accordance with standard protocols and criteria.22 A CDR of 0 indicates cognitive normality, whereas CDRs of 0.5, 1, 2, and 3 are indicative of very mild, mild, moderate, and severe dementia, respectively.21
Genotyping
Using standard procedures, DNA was extracted from peripheral blood samples. Genotyping of APOE was performed by the Knight Alzheimer’s Disease Research Center Genetics Core as previously described.23
CSF Collection and Processing
A sample of CSF (20–30 mL) was collected by routine lumbar puncture at 8 AM after overnight fasting as described.24 Samples were processed into 500–μL aliquots and immediately frozen at −80°C.
CSF Biomarker Analyses
The eTable in the Supplement shows the details of the kit specifications and general assay performance. The CSF samples were analyzed for Aβ and tau proteins using single-analyte enzyme-linked immunosorbent assays (ELISAs; research use only) from 2 different vendors. Samples were analyzed for Aβ1–40 (Aβ40), Aβ1–42 (Aβ42), total tau, and tau phosphorylated at threonine 181 (P-tau181) using the Improved INNOTEST ELISA (Fujirebio Europe), a modified version of the assay most widely used in the field. In parallel, Aβ40, Aβ42, and total tau were measured at the same time (from the same sample aliquot) using a set of second-generation (precision-based and accuracy-based) EUROIMMUN ELISAs (EUROIMMUN). The Aβ42 to Aβ40 ratio was calculated to normalize the Aβ42 production concentrations to the total amount of Aβ (Aβ40 is the most abundant Aβ species in CSF).25–27 The ratio of total tau (or P-tau181) to Aβ42 was also evaluated because it has been shown to be a predictor of future cognitive decline in elderly cohorts.17,28–30 It must be stated at the outset that the focus of this study is on the clinical utility of the biomarker and that conclusions drawn from one assay can be confirmed or qualified with data derived from another immunoassay. The well-studied INNOTEST ELISA was considered a priori to be the reference assay; therefore, INNOTEST data are shown.
The VILIP-1 concentration was measured using a 2-site immunoassay (Singulex).17 The YKL-40 concentration was measured with the MicroVue ELISA (Quidel).16
Longitudinal CSF samples from a given individual were run on the same assay plate (and same lot number) to minimize potential interplate and interlot methodological variability. Samples underwent a single freeze-thaw cycle prior to assay, were thawed on wet ice (approximately 3 hours) prior to analysis, and were all run in duplicate. Values had to pass quality control criteria, including coefficients of variation of 25% or lower, kit controls within the expected range as defined by the manufacturer (where applicable), and measurement consistency of 2 common pooled CSF samples that were included on each plate.
In Vivo Amyloid Imaging
A subset (n = 74) of the 169 participants with longitudinal CSF analysis had also undergone longitudinal in vivo amyloid imaging via positron emission tomography (PET) with Pittsburgh compound B (PiB)31–33 within approximately 12 months of CSF collection (mean [SD], 84.3 [92] days). The PiB PET imaging was conducted with a Siemens 962 HR+ Emission Computer-Aided Tomograph PET or Biograph 40 scanner (Siemens/CTI). Magnetic resonance imaging using magnetization-prepared rapid-acquisition gradient-echo T1-weighted images (1 × 1 × 1.25 mm) was obtained for anatomical reference.
Deposition of PiB in brain regions of interest was determined using FreeSurfer version 5.1 software (Martinos Center for Biomedical Imaging),32,34,35 and a standardized uptake value ratio (SUVR) corrected for partial volume effects36 was calculated for each region of interest. The mean cortical SUVR was calculated from FreeSurfer regions within the prefrontal cortex, precuneus, and temporal cortex. Cerebellar cortex served as the reference region. Based on a study of 77 symptomatic and asymptomatic Knight Alzheimer’s Disease Research Center participants,32 PiB positivity was defined as an SUVR of 1.42, commensurate with a mean cortical binding potential of 0.18 defined previously for PiB positivity.31
Statistical Analysis
Baseline demographic characteristics were summarized as mean (standard deviation) for continuous variables or number (percentage) for categorical variables. Demographic variables were compared across 3 age bins within the 2 APOE ε4 groups and between the ε4 carriers and noncarriers within each age bin using post hoc t tests within analysis of variance for continuous variables or logistic regression for dichotomous variables. To quantify the within-person annual rate of change in CSF biomarkers, general linear mixed models with random intercepts and random time slopes at the participant level were used to regress the concentrations on time from study entry (baseline). These models incorporated baseline age category, APOE category, and time from study entry as fixed effects as well as all possible higher-order interactions among these factors. This facilitated the estimation of average baseline CSF biomarker concentrations as well as their change over time separately in each of the 6 participant groups (cross-classification of 3 baseline age categories by 2 APOE categories). The resulting estimated average within-person annual rates of change in CSF biomarkers were compared among the 6 groups with model-derived approximate t tests with the approximate denominator df based on the Satterthwaite approximation.37 Baseline comparisons between CSF biomarkers among the groups in Table 1 were also carried out within these general linear mixed models by testing the estimated average concentrations when time from study entry was equal to 0. These CSF biomarker comparisons, at baseline and on the longitudinal rate of change, were also reexamined after adjusting for family history, sex, and education by including fixed effects for these factors and their interactions with time from study entry. The general linear mixed model assumptions were evaluated via analyses of residuals. Owing to the preliminary nature of hypotheses examined in this cohort, no adjustment was made for multiplicity. For exploratory purposes, an optimal CSF Aβ42 cutoff was determined using the Youden Index after receiver operating characteristic analysis for discriminating between PiB-positive and PiB-negative individuals at baseline. For each biomarker, baseline and longitudinal comparisons between PiB-positive (PiB SUVR ≥ 1.42) and PiB-negative individuals were performed using general linear mixed models with fixed effects included for PiB category, time from study entry, and their interaction. We used SAS version 9.3 statistical software (SAS Institute, Inc) for all statistical analyses, with statistical significance defined as P < .05.
Table 1.
Demographic Characteristics and Baseline Cerebrospinal Fluid Biomarkersa
|
APOE ε4 Noncarriers (n = 108)
|
APOE ε4 Carriers (n = 61)
|
|||||
|---|---|---|---|---|---|---|
| Variable | Early (n = 26) | Mid (n = 44) | Late (n = 38) | Early (n = 19) | Mid (n = 17) | Late (n = 25) |
| Baseline age, mean (SD), y | 50.1 (3.0) | 59.4 (2.9)b | 69.9 (3.5)b,c | 49.6 (2.9) | 59.3 (3.1)b | 69.4 (3.6)b,c |
|
| ||||||
| Female, No. (%) | 17 (65) | 32 (73) | 22 (58) | 14 (74) | 11 (65) | 16 (64) |
|
| ||||||
| Positive family history, No. (%) | 12 (46) | 22 (50) | 15 (39) | 15 (79)d | 13 (76) | 17 (68)d |
|
| ||||||
| APOE genotype, No. | ||||||
|
| ||||||
| ε2/ε2 | 0 | 1 | 1 | 0 | 0 | 0 |
|
| ||||||
| ε2/ε3 | 3 | 8 | 6 | 0 | 0 | 0 |
|
| ||||||
| ε3/ε3 | 23 | 35 | 31 | 0 | 0 | 0 |
|
| ||||||
| ε2/ε4 | 0 | 0 | 0 | 2 | 2 | 2 |
|
| ||||||
| ε3/ε4 | 0 | 0 | 0 | 14 | 12 | 20 |
|
| ||||||
| ε4/ε4 | 0 | 0 | 0 | 3 | 3 | 3 |
|
| ||||||
| Education, mean (SD), y | 16.1 (2.10) | 16.9 (2.27) | 15.6 (2.64)c | 15.8 (1.95) | 15.4 (3.45)d | 16.3 (2.23) |
|
| ||||||
| Baseline MMSE score, mean (SD)e | 29.5 (0.65) | 29.3 (1.10) | 28.8 (1.22)b | 29.8 (0.38) | 28.9 (1.52)b | 28.9 (1.39)b |
|
| ||||||
| Received ≥1 CDR >0 at follow-up, No.f | 0 | 1 | 4 | 1 | 3 | 5 |
|
| ||||||
| Participants with 2/3/4 serial LPs, No. | 12/13/1 | 21/19/4 | 25/13/0 | 11/8/0 | 12/5/0 | 18/7/0 |
|
| ||||||
| LP interval, mean (SD), mo | 3.3 (0.76) | 3.3 (0.91) | 3.1 (0.77) | 3.3 (0.73) | 3.6 (1.4) | 3.2 (0.77) |
|
| ||||||
| Baseline biomarkers, mean (IQR) | ||||||
|
| ||||||
| Improved INNOTEST ELISA | ||||||
|
| ||||||
| Aβ40, pg/mL | 12 657 (10 461–14 480) | 14 319 (12 185–16 371) | 15 382 (12 417–17 906)b | 14 555 (12 984–16 638) | 13 103 (10 629–15 838) | 14 343 (12 199–16 748) |
|
| ||||||
| Aβ42, pg/mL | 1293 (1046–1525) | 1340 (1132–1544) | 1270 (1021–1608) | 1306 (1193–1498) | 937 (671–1116)b,d | 970 (733–1225)b,d |
|
| ||||||
| Aβ42 to Aβ40 ratio | 0.1052 (0.0900–0.1225) | 0.0972 (0.0800–0.1100) | 0.0871 (0.0700–0.1000)b,c | 0.0924 (0.0800–0.1000) | 0.0719 (0.0600–0.0850)b,d | 0.0709 (0.0550–0.0900)b,d |
|
| ||||||
| Total tau, pg/mL | 202.3 (146.0–243.2) | 259.0 (182.6–278.7) | 324.3 (205.2–389.3)b,c | 257.7 (194.4–314.6) | 298.0 (210.2–391.6) | 321.4 (198.6–413.2) |
|
| ||||||
| P-tau181, pg/mL | 39.8 (27.7–50.3) | 51.2 (37.2–55.4) | 58.8 (41.7–68.5)b | 47.7 (38.5–55.4) | 54.4 (37.9–67.8) | 55.4 (38.2–69.8) |
|
| ||||||
| Total tau to Aβ42 ratio | 0.1541 (0.1200–0.1725) | 0.1908 (0.1400–0.2200) | 0.3054 (0.1500–0.3100)b,c | 0.1986 (0.1600–0.2300) | 0.4207 (0.1900–0.4550)b,d | 0.3816 (0.2150–0.5200)b |
|
| ||||||
| EUROIMMUN ELISA | ||||||
|
| ||||||
| Aβ40, pg/mL | 4857 (3525–6101) | 5408 (4305–6220) | 5569 (4347–6224) | 5535 (4816–6433) | 5266 (3966–7043) | 5257 (4119–5942) |
|
| ||||||
| Aβ42, pg/mL | 616.1 (438.1–683.1) | 616.1 (495.9–741.8) | 590.1 (459.4–701.1) | 676.0 (462.3–797.6) | 449.5 (349.9–564.3)b,d | 487.5 (365.3–601.8)b,d |
|
| ||||||
| Total tau, pg/mL | 254.7 (194.4–304.8) | 310.3 (230.3–344.6) | 362.5 (255.6–430.3)b,c | 299.1 (234.1–351.6) | 380.5 (313.6–469.6)d | 395.8 (274.1–487.4)b |
|
| ||||||
| Total tau to Aβ42 ratio | 0.4050 (0.3341–0.4851) | 0.5022 (0.3900–0.5192) | 0.7023 (0.4065–0.6874)b,c | 0.4563 (0.3675–0.5390) | 1.073 (0.4680–1.1100)b,d | 0.9342 (0.5383–1.2250)b,d |
|
| ||||||
| VILIP-1, pg/mL | 140.8 (102.3–169.8) | 154.4 (116.7–166.4) | 179.8 (133.6–218.9)b,c | 155.6 (128.8–175.4) | 153.2 (105.4–193.8) | 154.7 (117.0–180.2) |
|
| ||||||
| YKL-40, ng/mL | 180.3 (124.2–220.3) | 231.3 (192.3–259.7)b | 301.1 (221.7–368.2)b,c | 188.4 (135.3–238.7) | 240.6 (165.5–297.9)b | 281.5 (201.8–353.8)b |
Abbreviations: Aβ, β-amyloid; CDR, Clinical Dementia Rating; ELISA, enzyme-linked immunosorbent assay; IQR, interquartile range; LP, lumbar puncture; MMSE,Mini-Mental State Examination; P-tau181, tau phosphorylated at threonine 181; VILIP-1; visinin-like protein 1; YKL-40, chitinase-3-like protein 1.
Age groups indicate the ages within middle age: early, ages 45 to 54 years; mid, ages 55 to 64 years; and late, ages 65 to 74 years.
Significantly different from early within the same APOE ε4 group (P < .05).
Significantly different from mid within the same APOE ε4 group (P < .05).
Significantly different from the same age group of the other APOE ε4 group (P < .05).
The MMSE scores can range from 0 to 30, with 30 as a perfect score.
A CDR of 0 indicates cognitively normal; a CDR higher than 0, cognitively abnormal.
Results
Baseline data are presented in Table 1 and grouped into 6 bins: the absence (n = 108) or presence (n = 61) of at least 1 APOE ε4 allele (as an indicator of neutral and high AD risk, respectively) and middle-age bin at baseline (early [45–54 years], mid [55–64 years], or late [65–74 years]). Ninety-nine participants underwent 2 serial CSF collections, 65 underwent 3 serial CSF collections, and 5 underwent 4 serial CSF collections, at intervals of approximately 3 years. Forty-five of the 61 ε4 carriers (74%) and 49 of the 108 ε4 noncarriers (45%) reported a positive family history.
Comparison of the CSF Aβ40, Aβ42, and Total Tau Assays
Concentrations of Aβ40, Aβ42, and total tau obtained with the 2 assays were positively correlated (Aβ40, n = 412, Pearson r = 0.772 [95% CI, 0.730–0.808], P < .001; Aβ42, n = 394, Pearson r = 0.879 [95% CI, 0.855–0.900], P < .001; total tau, n = 410, Pearson r = 0.958 [95% CI, 0.949–0.965], P < .001). Although the absolute values for Aβ40 and Aβ42 differed between the assays (roughly 2- to 3-fold higher with EUROIMMUN compared with INNOTEST), absolute values for total tau were similar. Patterns of within-person biomarker changes over time were virtually identical between the 2 kits for Aβ42, total tau, and the total tau to Aβ42 ratio. However, baseline comparisons and longitudinal patterns for Aβ40 were slightly different between the kits and thus are difficult to interpret. Data for Aβ40, Aβ42, and total tau are presented for the reference assay, INNOTEST, whereas EUROIMMUN data are presented as supplementary data. The clinical observations were confirmed in both immunoassays for Aβ42 and total tau.
Baseline and Slope Analyses: CSF Biomarker Changes Occur in Middle Age
Baseline biomarker levels (Table 1) and slopes of change within individuals (Table 2) were evaluated in the 6 bins defined earlier. Slopes were calculated as the representative mean of all annual individual slopes per age bin (extrapolated to 9 years for illustrative purposes) and superimposed on the spaghetti plots of the associated individual trajectories (Figure 1 shows the INNOTEST assay data for Aβ40, Aβ42, Aβ42 to Aβ40 ratio, total tau, P-tau181, and total tau to Aβ42 ratio; Figure 2 shows the data for VILIP-1 and YKL-40; eFigure 1 in the Supplement shows the EUROIMMUN assay data). Controlling for family history, sex, and education did not substantially influence the comparisons between age and ε4 categories.
Table 2.
Mean Annual Slopes of Within-Individual Longitudinal Change in Cerebrospinal Fluid Biomarkers During Middle Agea
| APOE ε4 Noncarriers (n = 108) | APOE ε4 Carriers (n = 61) | |||||
|---|---|---|---|---|---|---|
| Variable | Early (n = 26) | Mid (n = 44) | Late (n = 38) | Early (n = 19) | Mid (n = 17) | Late (n = 25) |
| Aβ40 | ||||||
|
| ||||||
| Estimated annual slope, mean (SE), pg/mL | −163.59 (80.50) | −153.82 (61.81) | −130.39 (77.20) | 30.63 (102.86) | 3.31 (107.75) | 110.77 (97.06) |
|
| ||||||
| Different from 0, P value | .04b | .01b | .09 | .77 | .98 | .26 |
|
| ||||||
| APOE ε4carriers vs noncarriers, P value | … | … | … | .14 | .21 | .05 |
|
| ||||||
| Aβ42 | ||||||
|
| ||||||
| Estimated annual slope, mean (SE), pg/mL | −14.81 (5.83) | −19.34 (4.48) | −22.80 (5.56) | −14.99 (7.42) | −29.22 (7.79) | −26.76 (6.99) |
|
| ||||||
| Different from 0, P value | .01b | <.001b | <.001b | .045b | <.001b | <.001b |
|
| ||||||
| APOE ε4 carriers vs noncarriers, P value | … | … | … | .98 | .27 | .66 |
|
| ||||||
| Aβ42 to Aβ40 ratio | ||||||
|
| ||||||
| Estimated annual slope, mean (SE) | 0.00027 (0.00042) | −0.00023 (0.00032) | −0.00068 (0.00042) | −0.00090 (0.00055) | −0.00202 (0.00057) | −0.00220 (0.00052) |
|
| ||||||
| Different from 0, P value | .52 | .47 | .10 | .10 | <.001b | <.001b |
|
| ||||||
| APOE ε4 carriers vs noncarriers, P value | … | … | … | .09 | .007b | .02b |
|
| ||||||
| Total tau | ||||||
|
| ||||||
| Estimated annual slope, mean (SE), pg/mL | 0.96 (3.44) | 2.84 (2.68) | 14.58 (3.08)c,d | 5.40 (4.20) | 22.28 (4.45)c | 18.45 (3.85)c |
|
| ||||||
| Different from 0, P value | .78 | .29 | <.001b | .20 | <.001b | <.001b |
|
| ||||||
| APOE ε4 carriers vs noncarriers, P value | … | … | … | .42 | <.001b | .43 |
|
| ||||||
| P-tau181 | ||||||
|
| ||||||
| Estimated annual slope, mean (SE), pg/mL | 0.23 (0.51) | 0.32 (0.40) | 1.84 (0.47)c,d | 1.08 (0.63) | 3.41 (0.67)c | 1.92 (0.58) |
|
| ||||||
| Different from 0, P value | .66 | .43 | <.001b | .09 | <.001b | .001b |
|
| ||||||
| APOE ε4 carriers vs noncarriers, P value | … | … | … | .30 | <.001b | .91 |
|
| ||||||
| Total tau to Aβ42 ratio | ||||||
|
| ||||||
| Estimated annual slope, mean (SE) | 0.0026 (0.0084) | 0.0081 (0.0066) | 0.0268 (0.0071)c | 0.0076 (0.0100) | 0.0538 (0.0106)c | 0.0478 (0.0088)c |
|
| ||||||
| Different from 0, P value | .76 | .22 | <.001b | .45 | <.001b | <.001b |
|
| ||||||
| APOE ε4 carriers vs noncarriers, P value | … | … | … | .70 | <.001b | .07 |
|
| ||||||
| VILIP-1 | ||||||
|
| ||||||
| Estimated annual slope, mean (SE), pg/mL | −0.18 (1.03) | −0.48 (0.80) | 2.39 (1.01)d | 0.79 (1.34) | 5.17 (1.39)c | 1.42 (1.27)d |
|
| ||||||
| Different from 0, P value | .86 | .55 | .02b | .55 | <.001b | .26 |
|
| ||||||
| APOE ε4 carriers vs noncarriers, P value | … | … | … | .56 | <.001b | .55 |
|
| ||||||
| YKL-40 | ||||||
|
| ||||||
| Estimated annual slope, mean (SE), ng/mL | 4.80 (1.29) | 4.26 (0.99) | 6.91 (1.27) | 6.25 (1.68) | 10.83 (1.75) | 4.90 (1.60)d |
|
| ||||||
| Different from 0, P value | <.001b | <.001b | <.001b | <.001b | <.001b | .002b |
|
| ||||||
| APOE ε4 carriers vs noncarriers, P value | … | … | … | .50 | .001b | .32 |
Abbreviations: Aβ, β-amyloid; P-tau181, tau phosphorylated at threonine 181; VILIP-1; visinin-like protein 1; YKL-40, chitinase-3-like protein 1; ellipses, not applicable.
Age groups indicate the ages within middle age: early, ages 45 to 54 years; mid, ages 55 to 64 years; and late, ages 65 to 74 years. Results for Aβ40,
Aβ42, Aβ42 to Aβ40 ratio, total tau, P-tau181, and total tau to Aβ42 ratio are from the improved INNOTEST enzyme-linked immunosorbent assay.
Statistically significant at P < .05.
Significantly different from early within the same APOE ε4 group (P < .05).
Significantly different frommid within the same APOE ε4 group (P < .05).
Figure 1. Longitudinal Change in Cerebrospinal Fluid Biomarkers β-Amyloid 40 (Aβ40), Aβ42, Aβ42 to Aβ40 Ratio, Total Tau, Tau Phosphorylated at Threonine 181 (P-tau181), and Total Tau to Aβ42 Ratio During Middle Age.

Estimated group slopes and within-person changes for Aβ40 (A), Aβ42 (B), Aβ42 to Aβ40 ratio (C), total tau (D), tau phosphorylated at threonine 181 (P-tau181) (E), and total tau to Aβ42 ratio (F) are shown in the 3 age bins for APOE ε4 noncarriers (top graph of each panel; n = 108 participants) and ε4 carriers (bottom graph of each panel; n = 61 participants). Annual slopes have been extrapolated to 9 years, and each slope begins at the mean baseline biomarker value from individuals in each age bin. Group baseline values and slopes represent the estimates reported in Table 1 and Table 2, respectively, for the different cohorts defined by baseline age in which biomarker concentrations were regressed on time from study entry. Data are from the INNOTEST enzyme-linked immunosorbent assay (Fujirebio Europe).
aSlope significantly different from 0 (P < .05).
bSlope significantly different between APOE ε4 groups within a given age group (P < .05).
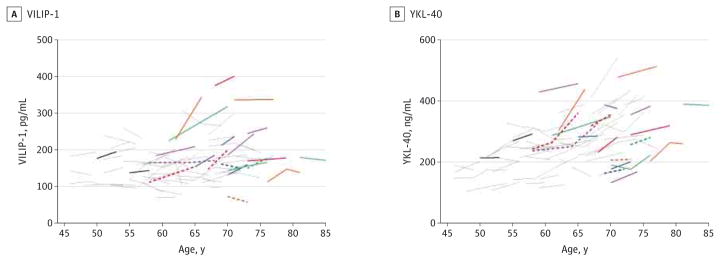
Figure 2. Longitudinal Change in Cerebrospinal Fluid Biomarkers Visinin-Like Protein 1 (VILIP-1) and Chitinase-3-Like Protein 1 (YKL-40) During Middle Age.

Estimated group slopes and within-person changes for VILIP-1 (A) and YKL-40 (B) are shown in the 3 age bins for APOE ε4 noncarriers (top graph of each panel; n = 108 participants) and ε4 carriers (bottom graph of each panel; n = 61 participants). Annual slopes have been extrapolated to 9 years, and each slope begins at the mean baseline biomarker value from individuals in each age bin. Group baseline values and slopes represent the estimates reported in Table 1 and Table 2, respectively, for the different cohorts defined by baseline age in which biomarker concentrations were regressed on time from study entry.
aSlope significantly different from 0 (P < .05).
bSlope significantly different between APOE ε4 groups within a given age group (P < .05).
Aβ40, Aβ42, and Aβ42 to Aβ40 Ratio
Baseline levels of CSF Aβ40 (INNOTEST) were significantly higher in the late middle-aged group compared with the early middle-aged group in ε4 noncarriers (P = .004) (Table 1) but decreased significantly within individuals in the early (P = .04) and mid (P = .01) middle-aged groups over time (Table 2 and Figure 1A). In contrast, no significant differences were observed in ε4 carriers at baseline or longitudinally (Table 1 and Table 2).
In contrast to Aβ40, robust decreases within individuals in all age groups were observed for Aβ42 in both risk groups (Figure 1B and Table 2), and this pattern was detectable in many participants as early as 45 to 54 years of age. While baseline concentrations did not differ among the age groups in the ε4 noncarriers, levels in ε4 carriers were significantly lower in the mid (P < .001) and late (P < .001) middle-aged groups compared with the early middle-aged group and also significantly lower than the levels in the mid (P < .001) and late (P < .001) middle-aged ε4 noncarriers (Table 1).
Similar to the patterns observed for Aβ42 alone, the ratios of Aβ42 to Aβ40 were significantly lower in the mid (P = .02) and late (P = .005) middle-aged groups compared with the early middle-aged group in ε4 carriers (Table 1), and the within-person values significantly decreased over time in the 2 older age groups (both P < .001) (Figure 1C and Table 2). Although baseline ratios in the ε4 noncarriers were significantly lower in the late middle-aged group compared with the mid (P = .05) and early (P = .004) middle-aged groups (Table 1), they did not change significantly within these low-risk individuals at any age (Figure 1C and Table 2).
Total Tau and P-tau181
Baseline total tau was higher in late middle-aged participants compared with early middle-aged participants in both risk groups, with intermediate levels in the mid middle-aged participants, although differences were statistically significant only in the ε4 noncarriers (P < .001 and P = .02, respectively) (Table 1). Within ε4 noncarriers, total tau increased significantly over time during late middle age (P < .001), while increases were observed earlier (mid and late middle age) in the higher-risk ε4 carriers (both P < .001) (Figure 1D and Table 2). Interestingly, the annual mean (SE) increase in total tau in mid middle age was significantly higher in ε4 carriers (22.28 [4.45] pg/mL) compared with ε4 noncarriers (2.84 [2.68] pg/mL) (P < .001) (Table 2). Results for P-tau181 were virtually identical to those for total tau, including more robust elevations in the ε4 carriers during mid middle age (Figure 1E and Table 2).
Ratios of Total Tau and P-tau181 to Aβ42
In ε4 noncarriers, the baseline total tau to Aβ42 ratio was significantly higher in late middle age compared with both early (P = .005) and mid (P = .01) middle age (Table 1). In at-risk ε4 carriers, significantly higher ratios were observed even earlier (mid [P = .002] and late [P = .004] middle age) compared with early middle age (Table 1). Longitudinal patterns for the total tau to Aβ42 ratio were virtually identical to those of total tau, with significant within-person increases in the late middle-aged group in ε4 noncarriers (P < .001) and even earlier (mid and late middle age) in the ε4 carriers (both P < .001) (Figure 1F and Table 2). Patterns for the P-tau181 to Aβ42 ratio were virtually identical to those of the total tau to Aβ42 ratio (data not shown).
Other Biomarkers of Neuronal Injury and Gliosis/ Neuroinflammation
VILIP-1 Concentration |
The concentration of VILIP-1 was positively correlated with total tau during middle age (INNOTEST total tau: n = 401, Pearson r = 0.763 [95% CI, 0.719–0.801], P < .001; EUROIMMUN total tau: n = 403, Pearson r = 0.743 [95% CI, 0.696–0.784], P < .001), consistent with earlier reports in elderly cohorts. Similar to total tau, mean baseline VILIP-1 concentration increased with age, with significantly higher levels in late middle age compared with early (P = .008) and mid (P = .03) middle age in the ε4 noncarriers (Table 1) and within-person increases over time in late middle age (P = .02) (Figure 2A and Table 2). While baseline levels of VILIP-1 in the at-risk ε4 carriers at baseline were not significantly different among the age groups (Table 1), they significantly increased longitudinally within individuals at an earlier age (mid middle age [P < .001]) compared with the ε4 noncarriers (late middle age [P = .02]) (Figure 2A and Table 2). Also similar to total tau, the annual mean increase in VILIP-1 concentration in mid middle age was greater in ε4 carriers compared with ε4 non-carriers (P < .001).
YKL-40 Concentration
Baseline CSF YKL-40 concentration was significantly higher in mid and late middle age compared with early middle age in both ε4 groups (all P ≤ .04) as well as in late middle age compared with mid middle age in the ε4 noncarriers (P < .001) (Table 1). In both groups, YKL-40 concentration significantly increased within individuals over time in all age bins (P = .002 in late middle age among ε4 noncarriers; all others, P < .001) (Figure 2B and Table 2). In mid middle age, YKL-40 concentration increased at a significantly higher rate in the ε4 carriers compared with ε4 noncarriers (P = .001) (Table 2), similar to what was observed for the injury markers.
APOE ε4 Gene Dose Influences CSF Biomarker Patterns Consistent With the Presence of Preclinical AD During Middle Age
Given the known APOE ε4 gene dosage effects on the risk of AD and age at dementia onset, we evaluated biomarker trajectories as a function of ε4 allele number. The majority (82%) of ε4 noncarriers had the ε3/ε3 genotype, whereas the majority (75%) of ε4 carriers had the ε3/ε4 genotype (Table 1). Nine participants were ε4 homozygotes (ε4/ε4 genotype). Trajectory patterns for Aβ40 did not differ as a function of ε4 allele dose (eFigure 2A in the Supplement). In contrast, patterns differed dramatically for Aβ42 (eFigure 2B in the Supplement) and the Aβ42 to Aβ40 ratio (data not shown) across the entire age range, with ε4 homozygotes falling among the lowest values, ε4 noncarriers typically falling among the highest, and heterozygotes falling in the middle range (although overlapping with many of the ε4 noncarriers). The longitudinal patterns for total tau, total tau to Aβ42 ratio, VILIP-1, and YKL-40 in ε4 carriers appeared to overlap to a greater extent with those for ε4 noncarriers (eFigure 2C–F in the Supplement). However, the number of ε4 homozygotes is too small to perform rigorous statistical analyses in the current cohort.
Association of CSF Aβ42 and In Vivo Amyloid Imaging During Middle Age
Because studies to date evaluating the concordance of CSF Aβ42 concentrations with in vivo amyloid load have focused on elderly cohorts, it was of interest to characterize this association in middle age, a time during which a subset of individuals are expected to be in the very earliest stages of preclinical AD. This analysis used data from a subset of 74 participants (n = 50 ε4 noncarriers; n = 24 ε4 carriers) within the longitudinal CSF cohort who had also undergone longitudinal in vivo PiB PET imaging within 376 days (mean [SD], 84.3 [92] days) of CSF collection. Twenty of these individuals were considered PiB positive (mean cortical SUVR ≥ 1.42) at baseline, follow-up, or both (Figure 3A). Of these 20 individuals, 10 (50%) were ε4 noncarriers and 10 (50%) were ε4 carriers. Although there was no significant association between the cross-sectional patterns (P = .12) or longitudinal trajectories (P = .65) of Aβ40 and cortical PiB binding (Figure 3B), PiB positivity was associated with low baseline levels of CSF Aβ42 (P < .001) but not longitudinal change (P = .37) (Figure 3C). However, 15 PiB-negative individuals (20%) had concentrations of Aβ42 that were as low as those who were PiB positive. Because low Aβ42 values could conceivably reflect low production of all Aβ species rather than an amyloidosis-specific decrease in Aβ42, we also evaluated the relationship between PiB and the Aβ42 to Aβ40 ratio (Figure 3D). Twelve of the PiB-negative participants (16%) had Aβ42 to Aβ40 ratios at some point that were as low as those who were PiB positive. Notably, all 4 ε4 homozygotes in this subcohort had a low Aβ42 concentration and a low Aβ42 to Aβ40 ratio at both baseline and follow-up (Figure 3C and D), including the 2 young participants (aged <55 years at baseline) who were PiB negative (Figure 3C and D, solid black lines). The PiB-positive individuals typically had higher baseline (P < .001) and longitudinally increasing (P < .001) levels of total tau (and P-tau181 [scatterplots not shown]) compared with those who were PiB negative (Figure 3E). The PiB associations with baseline (P = .04) and longitudinal (P = .004) VILIP-1 concentrations were similar to total tau but less concordant (Figure 4A). Being PiB positive was not significantly associated with YKL-40 levels at baseline (P = .08) but was associated with greater longitudinal increases (P = .04) (Figure 4B). Overall, Aβ42, Aβ42 to Aβ40 ratio, total tau, and P-tau181 appeared to be more strongly associated with PiB positivity than were Aβ40, VILIP-1, and YKL-40.
Figure 3. Association Between Longitudinal Patterns of Cerebrospinal Fluid Biomarkers Cortical Pittsburgh Compound B (PiB) Standardized Uptake Value Ratio (SUVR), β-Amyloid 40 (Aβ40), Aβ42, Aβ42 to Aβ40 Ratio, and Total Tau, Cortical Amyloid, and Age.

A subset (n = 74) of Adult Children Study participants had undergone longitudinal amyloid imaging via PiB positron emission tomographic imaging within 376 days (mean [SD], 84.3 [92] days) of cerebrospinal fluid collection. Biomarker measures include cortical PiB SUVR (A), Aβ40 (B), Aβ42 (C), Aβ42 to Aβ40 ratio (D), and total tau (E). The Aβ40, Aβ42, and total tau were analyzed by INNOTEST enzyme-linked immunosorbent assay (Fujirebio Europe). Being PiB positive was defined as having a mean cortical PiB SUVR higher than 1.42 and is represented by the dashed horizontal line in panel A. Gray lines indicate PiB negative at baseline and follow-up (n = 52); solid colored lines, PiB positive at both baseline and follow-up (n = 14); dashed colored lines, PiB negative at baseline but positive at follow-up (n = 6); and solid black lines, PiB negative with discordant (low) cerebrospinal fluid Aβ measures at baseline and follow-up (n = 2). Colored solid and dashed lines are each differently colored only to facilitate visual comparisons across all analytes for each PiB-positive individual.
Figure 4. Association Between Longitudinal Patterns of Cerebrospinal Fluid Biomarkers Visinin-Like Protein 1 (VILIP-1) and Chitinase-3-Like Protein 1 (YKL-40), Cortical Amyloid, and Age.
A subset (n = 74) of Adult Children Study participants had undergone longitudinal amyloid imaging via Pittsburgh compound B (PiB) positron emission tomographic imaging within 376 days (mean [SD], 84.3 [92] days) of cerebrospinal fluid collection. Biomarker measures include VILIP-1 (A) and YKL-40 (B). Being PiB positive was defined as having a mean cortical PiB standardized uptake value ratio higher than 1.42 (see dashed horizontal line in Figure 3A). Gray lines indicate PiB negative at baseline and follow-up (n = 52); solid colored lines, PiB positive at both baseline and follow-up (n = 14); dashed colored lines, PiB negative at baseline but positive at follow-up (n = 6); and solid black lines, PiB negative with discordant (low) cerebrospinal fluid β-amyloid measures at baseline and follow-up (n = 2). Colored solid and dashed lines are each differently colored only to facilitate visual comparisons across all analytes for each PiB-positive individual.
Aβ42 Cutoff as Estimated Using PiB at Baseline
Using only baseline CSF and PiB obtained with in 376 days(mean [SD],89.9[95]days),a slightly larger subcohort of 105 participants was used to calculate a cutoff for CSF Aβ42 (INNOTEST) based on PiB positivity. The optimal cutoff in this cohort is 1041 pg/mL (sensitivity = 1; specificity = 0.82),with an area under the receiver operating characteristic curve of 0.9352(95% CI, 0.8895–0.9808).
Case Study of Participants Who Received a CDR Higher Than 0 at Clinical Follow-up
Biomarker studies in cognitively normal elderly cohorts have demonstrated prognostic utility of baseline CSF measures for predicting future cognitive decline. To assess whether this relationship exists even earlier in the preclinical stages (during middle age), as a preliminary analysis we compared the biomarker trajectories in participants who received a CDR higher than 0 at some point during clinical follow-up with those who retained a CDR of 0. Of the 169 participants evaluated, all of whom were cognitively normal (CDR of 0) at the time of baseline CSF collection, 14 received a CDR of 0.5 at some point during follow-up (mean [SD], 6.55 [1.94] years; median, 6.15 years; range, 4.21–10.28 years), and 3 of these progressed further to a CDR of 1. The remaining 155 participants had a CDR of 0 at all follow-up (mean [SD], 6.01 [1.94] years; median, 6.21 years; range, 0.98–11.32 years). The duration of follow-up did not differ significantly between the groups (P > .05). All individuals who progressed to a CDR higher than 0 were older than 61 years at baseline. There was no apparent relationship between baseline or longitudinal trajectories of Aβ40 and cognitive status (Figure 5A). In contrast, the majority of progressors exhibited low Aβ42 (Figure 5B) and Aβ42 to Aβ40 ratio (data not shown) at baseline and follow-up and high total tau and total tau to Aβ42 ratio (Figure 5C and D). Patterns of VILIP-1 and YKL-40 did not appear to differ between the clinical groups (Figure 5E and F). However, the number of clinical progressors is too small to perform rigorous statistical analyses in the current cohort.
Figure 5. Cerebrospinal Fluid Biomarker Trajectories in Participants Receiving a Clinical Dementia Rating Higher Than 0 at Some Point During Clinical Follow-up.

Within-person trajectories of cerebrospinal fluid β-amyloid 40 (Aβ40) (A), Aβ42 (B), total tau (C), total tau to Aβ42 ratio (D), visinin-like protein 1 (VILIP-1) (E), and chitinase-3-like protein 1 (YKL-40) (F) are plotted as a function of age. The Aβ40, Aβ42, and total tau were analyzed by INNOTEST enzyme-linked immunosorbent assay (Fujirebio Europe). Fourteen individuals received a Clinical Dementia Rating of 0.5 or 1 at some point during follow-up (mean [SD], 6.55 [1.94] years; range, 4.21–10.28 years). Orange lines indicate individuals who received a Clinical Dementia Rating higher than 0 at available follow-up visits; gray lines, individuals who did not receive a Clinical Dementia Rating higher than 0.
Discussion
Our results demonstrate the following: (1) levels of CSF Aβ42 in some cognitively normal individuals decrease over time, starting as young as early middle age (45–54 years); (2) in mid middle age (55–64 years), reductions in Aβ42 are associated with the development of PiB-positive amyloid plaques; (3) elevations in neuronal injury markers total tau, P-tau181, and (to a lesser extent) VILIP-1 increase dramatically in some individuals in mid and late (65–74 years) middle age; (4) the gliosis/neuroinflammation marker YKL-40 increases throughout middle age; (5) these biomarker changes are observed in both risk groups defined by APOE genotype but are more evident in ε4 carriers and (for amyloid-related measures) in an allele dose-dependent manner; and (6) these AD-consistent trajectories are not clinically benign but instead are associated with future cognitive decline. These observations were confirmed in both evaluated immunoassays for Aβ42 and total tau.
Reductions in CSF Aβ42 concentration within certain individuals throughout middle age suggest an ongoing pathological process that for some people starts quite early (ages 45–54 years). Levels may begin to decrease even earlier, but additional investigation in younger cohorts is needed to test this hypothesis. During middle age, the timing of this decrease is influenced by ε4 allele dosage, consistent with studies demonstrating a major influence of APOE genotype on Aβ aggregation and clearance.38,39 Baseline and follow-up Aβ42 levels are among the lowest in ε4 homozygotes compared with heterozygotes and ε4 noncarriers, with reductions evident at earlier ages. Such effects are consistent with the ε4 dosage effects on age at dementia onset.40
Regardless of when Aβ42 levels begin to decrease during the preclinical period, these decreases did not coincide with the presence of amyloid detectable by PiB PET until mid middle age. The Aβ42 level was stably low or beginning to decline in some individuals while cortical PiB binding was still below the threshold of positivity, and PiB binding did not begin to increase until the CSF Aβ42 level was already relatively low. Thus, it seems likely that Aβ42 aggregation can be detected earlier with CSF analysis than with cortical PiB PET imaging, consistent with recent studies in autosomal dominant AD.9,41 This is highlighted by 2 high-risk early middle-aged ε4 homozygotes who had stable, low Aβ42 levels (and Aβ42 to Aβ40 ratios) in longitudinal samples but were PiB negative. This observation may reflect sequestration of Aβ42 into oligomeric forms undetectable with the current assays or its deposition in nonfibrillar (PiB-negative) diffuse plaques. In support of the latter, low CSF Aβ42 concentration in the absence of PiB positivity has been reported in a case in which numerous diffuse plaques, but few neuritic plaques, were observed at autopsy.42 However, the early middle-age bin of the longitudinal PiB subcohort is quite small; subregional PiB analyses and evaluation of future longitudinal PiB scans in ACS participants are necessary to rigorously evaluate PiB changes in early middle age.
The calculated CSF Aβ42 cutoff in this cohort is quite high at 1041 pg/mL, higher than previously reported using the INNOTEST kit (typically 450–650 pg/mL).24,43,44 This apparent discrepancy may reflect the younger age of the ACS cohort. Most likely it reflects the fact that we used a newer modified, improved INNOTEST assay. This cutoff is not suggested for clinical use but was instead provided to evaluate amyloid positivity using CSF measures—similar to protocols being considered for enrollment in AD prevention trials. Using this cutoff, 51 of the 169 participants (30%) would be considered amyloid positive and eligible for clinical trial enrollment based on baseline CSF Aβ42 concentration alone. Further longitudinal follow-up is needed to determine what percentage of these individuals will present with cognitive decline, which will in turn enable analysis of the efficacy of CSF Aβ42 concentration at baseline for determination of pre-clinical AD.
In contrast to the early changes in Aβ42, increases in total tau, P-tau181, and VILIP-1 are typically not apparent until later (ages ≥ 55 years). Notably, the rate of increase was significantly greater in the ε4-carrying at-risk group during mid middle age, coincident with continuing, robust decreases in Aβ42 level. It was in this age range that many participants with the AD biomarker pattern began to exhibit cognitive decline. Interestingly, the absolute slopes (ie, rates of increase) of these neuronal injury markers in the ε4 carriers actually decreased from mid to late middle age. This pattern is consistent with a potential slowing of an earlier robust phase of neuronal injury or perhaps reflects neuronal dysfunction that adversely affects the normal cellular secretion or release of these proteins. It will be interesting to determine whether this pattern is also observed in those at lower risk (ε4 noncarriers), albeit at older ages, how it compares with proposed early markers of synaptic function currently in development, and whether this proposed slowing continues into the symptomatic phase as has been reported in individuals with autosomal dominant AD10 and late-onset AD dementia.45 The rate accelerations in these markers at mid middle age observed here in the at-risk group are consistent with the concept of an age-related transition between stage 1 (amyloid alone) and stage 2 (amyloid plus neuronal injury) of preclinical AD proposed by the National Institute on Aging–Alzheimer’s Association Pre-clinical AD Working Group.46 Although these proposed stages are currently defined by biomarker measures obtained at a single point in time, it is possible that a longitudinal biomarker metric may have more utility. This hypothesis awaits further investigation.
The consistent pattern of increases in YKL-40 level in all age bins suggests that neuroinflammation/gliosis (the hypothesized cause of the increase in YKL-40 level) is a process that occurs normally with aging. However, the particularly robust increases observed in at-risk ε4 carriers during mid middle age suggest that this age-related process may be further exacerbated in the presence of insults including amyloid deposition and neuronal injury. Whether this neuroinflammatory process contributes to the concomitant increase in neuronal injury or is a result of such injury remains to be determined.
This study is not without limitations. As by design the ACS cohort enrolls participants with and without family history of AD for longitudinal imaging and CSF biomarker studies, participants may not be representative of the general population. Despite the large number of participants in this unique cohort, there are fewer in the ε4-carrying group, and most participants at the time of analysis had only 2 longitudinal samples available. While some individuals had 10 years of clinical follow-up, others had only 4. Although the results provide support for a scenario in which changes in amyloid-related processes precede those of tau or other neurodegeneration-related processes, additional analyses during a longer period are required to determine the precise sequence of biomarker changes within a given individual. Furthermore, as expected in such a young, asymptomatic cohort, relatively few participants in this initial report had received a CDR greater than 0 during follow-up. Continued evaluation of longer clinical follow-up will provide an opportunity to better elucidate the biomarker patterns in middle age that predict future cognitive decline.
Conclusions
The present group wide analyses are supportive of a preclinical period of AD in which biomarker patterns consistent with underlying disease pathology are first detectable during middle age, the timing of which is influenced by APOE genotype, with amyloid changes occurring prior to neuronal injury. However, proposals to use biomarkers in clinical settings require demonstration of their utility on a patient-by-patient basis. Importantly, our preliminary findings of an association between CSF biomarker positivity in specific individuals who go on to develop cognitive deficits within a few years provide support for such potential use.
Supplementary Material
Acknowledgments
Funding/Support: This work was supported by grants P01AG026276 (Dr Morris), 5P30 NS048056 (Dr Benzinger), and 5P30 NS048056 (Dr Benzinger) from the National Institutes of Health, the Barnes-Jewish Hospital Foundation (Dr Morris), the Fred Simmons and Olga Mohan fund (Dr Morris), and a grant from Eli Lilly and Co (Dr Holtzman).
Role of the Funder/Sponsor: The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Supplemental content at jamaneurology.com
Author Contributions: Ms Sutphen and Mr Jasielec had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Sutphen, Holtzman, Fagan.
Acquisition, analysis, or interpretation of data Sutphen, Jasielec, Shah, Macy, Xiong, Vlassenko, Benzinger, Stoops, Vanderstichele, Brix, Darby, Vandijck, Ladenson, Morris, Fagan.
Drafting of the manuscript: Sutphen, Fagan.
Critical revision of the manuscript for important intellectual content: All authors.
Statistical analysis: Jasielec, Xiong.
Obtained funding: Benzinger, Morris, Holtzman.
Administrative, technical, or material support: Shah, Stoops, Vanderstichele, Brix, Darby, Vandijck, Ladenson.
Study supervision: Xiong, Fagan.
Conflict of Interest Disclosures: Dr Benzinger reported being a member of the advisory board for Eli Lilly and Co (2011); receiving research funding from Avid Radiopharmaceuticals; providing expert testimony and receiving compensation from Kujawaski and Associates (2011); and participating in clinical trials sponsored by Eli Lilly and Co, Avid Radiopharmaceuticals, and Roche. Mr Stoops reported being a shareholder of ADx NeuroSciences and BioMARIC NV. Dr Vanderstichele reported being a cofounder and shareholder of ADx NeuroSciences and founder of Biomarkable bvba. Dr Ladenson reported being named on patents related to use of VILIP-1; these are being managed by Washington University in accordance with university policy. Dr Morris reported having participated in or currently participating in clinical trials of antidementia drugs sponsored by Janssen Immunotherapy, Pfizer, Eli Lilly and Co/Avid Radiopharmaceuticals, SNIFF (Study of Nasal Insulin to Fight Forgetfulness), and A4 Study (Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease) and serving as a consultant for Lilly USA, ISIS Pharmaceuticals, and the Charles Dana Foundation. Dr Holtzman reported being a cofounder of C2N Diagnostics LLC; serving on the scientific advisory boards of AstraZeneca, Genentech, Neurophage, and C2N Diagnostics; and serving as a consultant for Eli Lilly and Co. Washington University receives grants to the laboratory of Dr Holtzman from the Tau Consortium, Cure Alzheimer’s Fund, the JPB Foundation, Eli Lilly and Co, Janssen, and C2N Diagnostics. Dr Fagan reported serving on the scientific advisory boards of IBL International and Roche and serving as a consultant for AbbVie and Novartis. No other disclosures were reported.
Additional Contributions: Genotyping of APOE was performed under the direction of Alison Goate, DPhil, Genetics Core, Knight Alzheimer’s Disease Research Center, Washington University, St Louis, Missouri; she received no compensation. Matthew Amos, Elizabeth Grant, PhD, and Sushila Sathyan, MA, Knight Alzheimer’s Disease Research Center, Washington University, provided technical support; they received no compensation. We acknowledge the contributions of the ACS Administration, Clinical, Biomarker, Imaging, and Biostatistics Cores at the Knight Alzheimer’s Disease Research Center. We gratefully acknowledge the altruism of the ACS participants and their families.
References
- 1.James BD, Leurgans SE, Hebert LE, Scherr PA, Yaffe K, Bennett DA. Contribution of Alzheimer disease to mortality in the United States. Neurology. 2014;82(12):1045–1050. doi: 10.1212/WNL.0000000000000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schneider LS, Mangialasche F, Andreasen N, et al. Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J Intern Med. 2014;275(3):251–283. doi: 10.1111/joim.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gómez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996;16(14):4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braak H, Braak E. Diagnostic criteria for neuropathologic assessment of Alzheimer’s disease. Neurobiol Aging. 1997;18(4 suppl):S85–S88. doi: 10.1016/s0197-4580(97)00062-6. [DOI] [PubMed] [Google Scholar]
- 5.Hulette CM, Welsh-Bohmer KA, Murray MG, Saunders AM, Mash DC, McIntyre LM. Neuropathological and neuropsychological changes in “normal” aging: evidence for preclinical Alzheimer disease in cognitively normal individuals. J Neuropathol Exp Neurol. 1998;57(12):1168–1174. doi: 10.1097/00005072-199812000-00009. [DOI] [PubMed] [Google Scholar]
- 6.Morris JC, Price JL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci. 2001;17(2):101–118. doi: 10.1385/jmn:17:2:101. [DOI] [PubMed] [Google Scholar]
- 7.Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol. 2001;58(9):1395–1402. doi: 10.1001/archneur.58.9.1395. [DOI] [PubMed] [Google Scholar]
- 8.Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63(1):38–46. doi: 10.1001/archneur.63.1.38. [DOI] [PubMed] [Google Scholar]
- 9.Bateman RJ, Xiong C, Benzinger TL, et al. Dominantly Inherited Alzheimer Network. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367(9):795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fagan AM, Xiong C, Jasielec MS, et al. Dominantly Inherited Alzheimer Network. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer’s disease. Sci Transl Med. 2014;6(226):226ra30. doi: 10.1126/scitranslmed.3007901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villemagne VL, Burnham S, Bourgeat P, et al. Australian Imaging Biomarkers and Lifestyle (AIBL) Research Group. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 12.Rafii MS. Preclinical Alzheimer’s disease therapeutics. J Alzheimers Dis. 2014;42(suppl 4):S545–S549. doi: 10.3233/JAD-141482. [DOI] [PubMed] [Google Scholar]
- 13.Xiong C, Roe CM, Buckles V, et al. Role of family history for Alzheimer biomarker abnormalities in the adult children study. Arch Neurol. 2011;68(10):1313–1319. doi: 10.1001/archneurol.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosén C, Hansson O, Blennow K, Zetterberg H. Fluid biomarkers in Alzheimer’s disease: current concepts. Mol Neurodegener. 2013;8:20. doi: 10.1186/1750-1326-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bjerke M, Portelius E, Minthon L, et al. Confounding factors influencing amyloid beta concentration in cerebrospinal fluid. Int J Alzheimers Dis. doi: 10.4061/2010/986310. published online July 15, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Craig-Schapiro R, Perrin RJ, Roe CM, et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol Psychiatry. 2010;68(10):903–912. doi: 10.1016/j.biopsych.2010.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tarawneh R, D’Angelo G, Macy E, et al. Visinin-like protein-1: diagnostic and prognostic biomarker in Alzheimer disease. Ann Neurol. 2011;70(2):274–285. doi: 10.1002/ana.22448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tarawneh R, Lee JM, Ladenson JH, Morris JC, Holtzman DM. CSF VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology. 2012;78(10):709–719. doi: 10.1212/WNL.0b013e318248e568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo X, Hou L, Shi H, et al. CSF levels of the neuronal injury biomarker visinin-like protein-1 in Alzheimer’s disease and dementia with Lewy bodies. J Neurochem. 2013;127(5):681–690. doi: 10.1111/jnc.12331. [DOI] [PubMed] [Google Scholar]
- 20.Rosén C, Andersson CH, Andreasson U, et al. Increased levels of chitotriosidase and YKL-40 in cerebrospinal fluid from patients with Alzheimer’s disease. Dement Geriatr Cogn Dis Extra. 2014;4(2):297–304. doi: 10.1159/000362164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 22.Berg L, McKeel DW, Jr, Miller JP, et al. Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol. 1998;55 (3):326–335. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- 23.Pastor P, Roe CM, Villegas A, et al. Apolipoprotein Eε4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. Ann Neurol. 2003;54(2):163–169. doi: 10.1002/ana.10636. [DOI] [PubMed] [Google Scholar]
- 24.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59(3):512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 25.Wiltfang J, Esselmann H, Bibl M, et al. Amyloid beta peptide ratio 42/40 but not A beta 42 correlates with phosphotau in patients with low-and high-CSF A beta 40 load. J Neurochem. 2007;101(4):1053–1059. doi: 10.1111/j.1471-4159.2006.04404.x. [DOI] [PubMed] [Google Scholar]
- 26.Hansson O, Zetterberg H, Buchhave P, et al. Prediction of Alzheimer’s disease using the CSF Abeta42/Abeta40 ratio in patients with mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;23(5):316–320. doi: 10.1159/000100926. [DOI] [PubMed] [Google Scholar]
- 27.Spies PE, Verbeek MM, van Groen T, Claassen JA. Reviewing reasons for the decreased CSF Abeta42 concentration in Alzheimer disease. Front Biosci (Landmark Ed) 2012;17:2024–2034. doi: 10.2741/4035. [DOI] [PubMed] [Google Scholar]
- 28.Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5(3):228–234. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- 29.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64(3):343–349. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 30.Li G, Sokal I, Quinn JF, et al. CSF tau/Abeta42 ratio for increased risk of mild cognitive impairment: a follow-up study. Neurology. 2007;69 (7):631–639. doi: 10.1212/01.wnl.0000267428.62582.aa. [DOI] [PubMed] [Google Scholar]
- 31.Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67(3):446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 32.Su Y, D’Angelo GM, Vlassenko AG, et al. Quantitative analysis of PiB-PET with FreeSurfer ROIs. PLoS One. 2013;8(11):e73377. doi: 10.1371/journal.pone.0073377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vlassenko AG, Mintun MA, Xiong C, et al. Amyloid-beta plaque growth in cognitively normal adults: longitudinal [11C]Pittsburgh compound B data. Ann Neurol. 2011;70(5):857–861. doi: 10.1002/ana.22608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33(3):341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 35.Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31(3):968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 36.Su Y, Blazey TM, Snyder AZ, et al. Dominantly Inherited Alzheimer Network. Partial volume correction in quantitative amyloid imaging. Neuroimage. 2015;107:55–64. doi: 10.1016/j.neuroimage.2014.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Satterthwaite FE. Synthesis of variance. Psychometrika. 1941;16(5):309–316. doi: 10.1007/BF02288586. [DOI] [Google Scholar]
- 38.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63(3):287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castellano JM, Kim J, Stewart FR, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3 (89):89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 41.Fleisher AS, Chen K, Quiroz YT, et al. Associations between biomarkers and age in the presenilin 1 E280A autosomal dominant Alzheimer disease kindred: a cross-sectional study. JAMA Neurol. 2015;72(3):316–324. doi: 10.1001/jamaneurol.2014.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cairns NJ, Ikonomovic MD, Benzinger T, et al. Absence of Pittsburgh compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: a case report. Arch Neurol. 2009;66(12):1557–1562. doi: 10.1001/archneurol.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fagan AM, Mintun MA, Shah AR, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO Mol Med. 2009;1(8–9):371–380. doi: 10.1002/emmm.200900048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tapiola T, Alafuzoff I, Herukka SK, et al. Cerebrospinal fluid β-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66(3):382–389. doi: 10.1001/archneurol.2008.596. [DOI] [PubMed] [Google Scholar]
- 45.Toledo JB, Xie SX, Trojanowski JQ, Shaw LM. Longitudinal change in CSF tau and Aβ biomarkers for up to 48 months in ADNI. Acta Neuropathol. 2013;126(5):659–670. doi: 10.1007/s00401-013-1151-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.