

Abstract
As part of an effort to identify substrate analogs suitable for helping to resolve structural features important for terpene synthases, the inhibition of 5-epi-aristolochene biosynthesis from farnesyl diphosphate (FPP) by the tobacco 5-epi-aristolochene synthase incubated with anilinogeranyl diphosphate (AGPP) was examined. The apparent noncompetitive nature of the inhibition supported further assessment of how AGPP might be bound to crystallographic forms of the enzyme. Surprisingly, the bound form of the inhibitor appeared to have undergone a cyclization event consistent with the native mechanism associated with FPP catalysis. Biocatalytic formation of a novel 13-membered macrocyclic paracyclophane alkaloid was confirmed by high-resolution GC-MS and NMR analysis. This work provides insights into new biosynthetic means for generating novel, functionally diversified, medium-sized terpene alkaloids.
Introduction
Nicotiana tabacum (tobacco) 5-epi-aristolochene synthase (EAS) is a member of a superfamily of enzymes referred to as terpene cyclases or synthases.1,2 These enzymes catalyze complex cyclizations of linear allylic substrates of 10 (geranyl diphosphate, GPP), 15 (farnesyl diphosphate, FPP), or 20 (geranylgeranyl diphosphate, GGPP) carbons in precise regio- and stereochemical ways and are capable of generating hundreds of different carbon skeletons. In plants, the cyclized terpene hydrocarbon skeletons can undergo a number of additional modifications including rearrangements, hydroxylations, oxidations, methylations, acetylations, arylations, and halogenations generating an even more diverse array of compounds. To date, more than 100 000 different cyclic terpene compounds have been isolated from plant sources.3 Why plants require such a diverse array of compounds is not completely understood, but some of these compounds serve primary metabolic needs (i.e., growth regulators) while others mediate plant-environment interactions. Solanaceous plants, for instance, synthesize sesquiterpenes possessing antimicrobial activities when challenged by microbial pests. Capsidiol (3) is one such example compound synthesized by the cyclization of farnesyl diphosphate (FPP) (1) to 5-epi-aristolochene (2) by 5-epi-aristolochene synthase (EAS),4 followed by epi-aristolochene hydroxylase (EAH), a cytochrome P450, catalyzing the successive hydroxylation of 5-epi-aristolochene (Scheme 1).5
Scheme 1. A Proposed Catalytic Mechanism for the Cyclization of Farnesyl Diphosphate (1) to 5-epi-Aristolochene (2) by TEAS, a Tobacco Sesquiterpene Synthase, Adapted with permission from ref 18, copyright American Chemical Society 2000a.

aIonization of the diphosphate moiety from FPP is followed by attack of the C10–C11 double bond on the newly created carbocation at C1 and subsequent proton abstraction at the cis-methyl function to yield germacrene A. Protonation of C6 of the germacrene A intermediate followed by attack of the C2–C3 double bond results in the formation of an internal bond between C7 and C2, and generation of a new carbocation centered at C3. A hydride shift from C2 to this carbocation, followed by a methyl migration from C7 and final proton abstraction at C8 generates 5-epi-aristolochene (2). In planta, 5-epi-aristolochene formation is followed by its successive hydroxylation catalyzed by 5-epi-aristolochene hydroxylase, a cytochrome P450 enzyme,5 to the final end-product capsidiol (3).
While an enormous variety of complex natural products are formed by plants, the generation of additional chemical complexity is desirable for the discovery of new drugs and chemicals for other agricultural and industrial uses. Many of the modern medicines in use today, for instance, are inspired by or derived from plant natural products.6,7 Natural products, however, can often exhibit significant short-comings as drugs. They can variously be insoluble, chemically unstable, available in only vanishingly small amounts, components of complex mixtures, or show unacceptable toxicity and/or side effects. Because of these difficulties, drug discovery efforts in the recent past focused on the preparation and screening of large synthetic libraries of compounds. It is now recognized that these synthetic libraries only sparsely cover the available structural and conformational space, and as a result, a much smaller number of drug leads have arisen than anticipated.8,9 Drug discovery efforts are now being re-expanded to include the preparation and screening of novel scaffolds as potential lead compounds. Natural products are very useful in this regard, because of their intrinsic chemical and structural complexity. Medium sized rings are of great interest as potential leads, in part because of their unusual conformations as well as their ability to display functional groups in spatial orientations not found in the relatively easy to synthesize five- and six-membered rings.10–13 However, medium sized rings are more difficult to obtain synthetically. In particular, efficient cyclization of acyclic precursors into seven- to 15-membered ring systems is challenging using standard synthetic organic methods.14–17
In the recent past, the range of natural product complexity has been extended by utilizing enzymatic conversions of unnatural substrates to provide novel, difficult to obtain, complex organic structures.19 We report here the efficient formation of a 13-membered macrocycle alkaloid 3,7-dimethyl-trans,trans-3,7-aza[9]paracyclophane-diene “geraniline” via a carbon–carbon bond forming cyclization of AGPP catalyzed by the tobacco 5-epi-aristolochene synthase. This report highlights the utility of enzymatic cyclization approaches for the generation of medium sized ring systems under mild conditions of aqueous incubations at RT.
Results and Discussion
AGPP is a Noncompetitive Inhibitor of TEAS
AGPP (4) is a structural analogue of FPP (1) where the terminal isoprene unit is replaced by an aniline group. AGPP has previously been shown to be an efficient substrate for the mammalian protein farnesyl transferase (FTase),20–22 an inhibitor of breast cancer cell invasion,23 but it is neither a substrate nor an inhibitor of mammalian squalene synthase, a key branch point enzyme for sterol biosynthesis.20 We first sought to determine if AGPP might bind to and inhibit the catalytic activity of TEAS, representative of yet another class of terpene biosynthetic enzymes. If so, then AGPP could serve as a potential substrate analog to further refine structural features of the TEAS enzyme in crystallographic studies.24,25 The ability of AGPP to inhibit TEAS was characterized by measuring the conversion rate of [1-3H]-FPP to [1-3H]-5-epi-aristolochene in the presence of varying concentrations of AGPP. The radiolabeled assay was previously qualified by comparison of reaction product analysis by GC-MS.26,27 Expression of the experimental data in Michaelis–Menten (Figure 1) or Line-weaver–Burk (Supporting Information Figure S1) plots with nonlinear regression line fitting were best for AGPP acting as a noncompetitive inhibitor of the TEAS-catalyzed cyclization of FPP, although these determinations were complicated by an apparent inhibition of the TEAS activity by high concentrations of the native substrate, FPP. The calculated Ki for the inhibition of TEAS activity by AGPP was 0.38 μM (range of 0.025–1.24 μM, Supporting Information Table S2), which is significantly lower than the Km of 3–7 μM for FPP as measured by others4,18,26 and 4.47 μM calculated from the data shown in Figure 1 (range of 4.3–9.8 μM, Supporting Information Table S2). The turnover rate (kcat) of FPP by the TEAS enzyme was 0.032 s−1, also comparable to that of 0.048 s−1 reported previously by Rising et al.4,18,26 Thus, AGPP appeared to be an effective FPP analog and inhibitor of the TEAS enzymatic activity.
Figure 1.
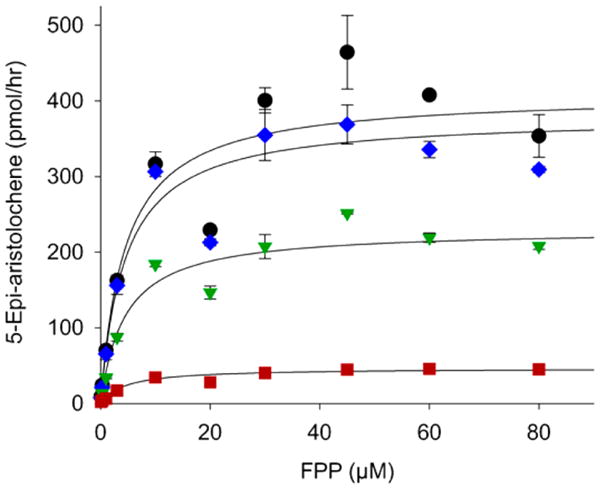
Catalytic conversion of FPP by 5-epi-aristolochene synthase to sesquiterpene hydrocarbons inhibited by AGPP. 5-Epi-aristolochene synthase (72 nM) was incubated at the indicated concentrations of 3H-FPP in the absence (●) or presence of 0.03 (blue ◆), 0.3 (green ▼), 3.0 (red ■) μM AGPP at RT for 10 min before determining the incorporation of radioactivity into hexane extractable products.
The Crystal Structure of TEAS with AGPP Reveals an Unexpected Cyclic Product
Noncompetitive inhibition often involves binding of the inhibitor to an allosteric binding site on the protein that reduces the rate of substrate turnover, but a noncompetitive inhibitor may also bind in the active site of the enzyme. An allosteric binding site was previously identified for the E. coli FPPS,28 a prenyl transferase that shares structural homology with TEAS, suggesting that an allosteric site could be involved in the present case. To determine which of these two possible noncompetitive inhibition mechanisms might be occurring, we soaked AGPP into crystals of WT TEAS and collected X-ray diffraction data.
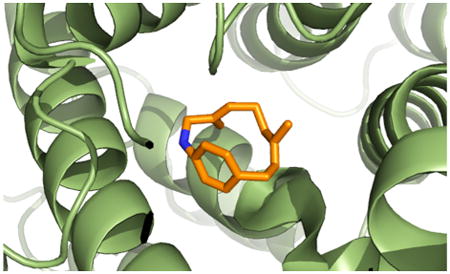
The overall structure of the TEAS–AGPP complex is similar to the apo-form and ligand-bound TEAS structures reported previously (Figure 2a).24,25 The enzyme has two structural domains, both of which consist entirely of helices and connecting loops. The single active site is a deep, hydrophobic, aromatic-rich pocket in the C-terminal domain, with bound magnesium ions and positively charged residues at its entrance. Several loops that surround the active site are disordered, resulting in an open, solvent-exposed pocket. In structures of TEAS soaked with AGPP, as in the previously examined TEAS–farnesyl hydroxyphosphonate complex (PDB code 5EAT), the active site is in a closed conformation, as evidenced by the strong electron density apparent for residues in the J/K loop (Figure 2a). This closed conformation is thought to sequester the hydrophobic active site from solvent and is likely adopted by the enzyme during catalysis. Beyond this region of the structure, the presence of AGPP in the soaking solution does not induce any large conformational changes in the protein structure. In addition, the only ligand-derived electron density visible in the solved crystal structure was found in the active site, providing another indication that the AGPP inhibition mechanism may not involve another allosteric binding site on the enzyme.
Figure 2.

TEAS-AGPP/geraniline cocrystal structure. Full view of TEAS structure with a Sigma A-weighted Fo-Fc electron density map calculated based on data refined without the J/K loop (residues 521–530) (a). Strong density for entire loop demonstrates closed conformation of the active site. Active site view with 2Fo-Fc map calculated based on the final structure absent the geraniline, AGPP, magnesiums, and pyrophosphate (b). Map contoured at 1σ. Superposition of the TEAS-AGPP structure with the structure of TEAS bound to 2F-FPP (PDB code: 3M01) (c) or FHP (PDB code: 5EAT) (d).
Surprisingly, the 2Fo-Fc electron density map of the model with the ligand exhibited a donut-shaped region of electron density in the TEAS active site (Figure 2b). This unexpected electron density suggested that ionization and macrocyclization of the AGPP substrate within the TEAS active site had occurred. Assuming TEAS cyclizes AGPP analogously to FPP (Scheme 1), then the enzyme may promote the ionization of the pyrophosphate from AGPP to form an allylic carbocation such as shown in Scheme 2. Since the amino substituent of the aniline ring activates the ortho and para positions by donating electron density to the ring, electrophilic attack by the allylic carbocation would most likely occur at either of these positions. Model building into the electron density map suggested that cyclization indeed took place onto the para position of the aniline ring. Loss of a proton to the enzyme or solvent would then yield a macrocyclic [9]paracyclophane alkaloid (5). An energy-minimized (Chem3D) model of this macrocyclic C16H21N (5) structure was generated and easily placed into the TEAS active site with features in the ring of the electron density map accommodating the phenyl and methyl groups.
Scheme 2. A Proposed Mechanism for the Cyclization of Anilino-Geranyl Diphosphate (4) to “Geraniline” (5, 5b) by TEAS, a Tobacco Sesquiterpene Synthasea.

aIonization of the diphosphate moiety from AGPP is followed by attack of the para-aniline carbon on the newly created carbocation at C1 and subsequent loss of a proton from the benzene ring system would generate “geraniline”. 5 and 5b represent conformers discussed in the text.
It appears that we caught the enzyme in the act of catalyzing the cyclization of a substrate, given that some electron density remains in the space spanning C1 of AGPP and its pyrophosphate (red arrow, Figure 2b). Given this mixture of states, we modeled both the macrocylic geraniline 5 and the unionized AGPP substrate. Occupancy refinement of the structure considering dual residence of these groups revealed that 4 and 5 are present in roughly equal proportions. In cocrystal structures of TEAS bound to FPP analog 2F-FPP (PDB code 3M01),24 the farnesyl chain curves around the active site in a direction that leaves the terminal dimethyl allyl group poised under the pyrophosphate leaving group. We noted that the AGPP hydrocarbon chain curves along the active site in the same direction as substrate analog 2F-FPP,24 rather than farnesyl hydroxyphosphate (FHP;25 compare Figure 2c and d). This is the first time a hydrolyzable substrate has been observed in the active site of TEAS, which suggests that 2F-FPP is the more faithful of the substrate analogs, binding the active site in the catalytically relevant orientation.
The structural determinations corroborate the inhibition by AGPP; overlays of TEAS-alkaloid 5 structure with either 2F-FPP or FHP suggest that the substrate should be completely precluded from binding when AGPP is bound (Figure 2c,d). In the initial orientation, the most polar group of 4, its nitrogen atom, is relatively buried in the hydrophobic active site pocket. It is likely stabilized, however, by an electrostatic interaction between the partially negative ring dipole of Trp273 at the back of the active site. The rest of the hydrophobic groups can interact favorably with the hydrophobic active site surface. After ionization and attack on C1 by the para- carbon of the anilino group, the macrocycle swivels around to face the active site entrance where it is able to hydrogen bond with water molecules as well as engage in electrostatically favorable interactions with the ionized pyrophosphate. In addition, this orientation allows the phenyl ring of 5 to pack against the aromatic face of Tyr527 in an edge–face interaction. This interaction seems to aid in stabilizing the rest of the J/K loop, locking the active site in a closed conformation. Thus, although the initial inhibitor binding orientations match in the TEAS-alkaloid 5 and the TEAS–2F-FPP complex structures, the J/K loop conformation is more in line with that of the TEAS–FHP complex structure. By contrast, this loop is in various extents of disorder in the complexes with 2F-FPP.24 Given the rotational freedom around its single bonds, we verified this binding mode of 5 by modeling other conformations and orientations of 5 in the active site electron density of the Fo-Fc map. Conformation 5b fit the electron density moderately well, and its atomic positions and temperature factors were refined against the diffraction data. The resulting model, however, had higher temperature factors for the cyclic product (Supporting Information Table S1), and subsequent electron density maps calculated using this model exhibited poorer quality electron density in the active site. This suggested that the initially modeled conformation 5 was correct.
Verification that TEAS Utilizes AGPP as a Substrate
To characterize the catalytic turnover of AGPP by TEAS, additional enzyme assays were performed. TEAS catalyzes formation of an initial germacrenyl 10-membered ring intermediate between C1 and C10 of FPP in the biosynthesis of 5-epi-aristolochene. However, cyclization of AGPP to a 10-membered ring would require bond formation between C1 of AGPP and the aromatic ring carbon bonded to the nitrogen. GC profiling of hexane extracts from incubations of AGPP with TEAS indicated the formation of a product with a retention time of 18.05 min (Figure 3a). The retention time of this reaction product was distinct from 5-epi-aristolochene, the reaction product of incubations of TEAS with FPP, which migrated with a retention time of 12.68 min under these GC conditions (Figure 3b). The AGPP reaction product did not appear to arise from simple hydrolysis of the diphosphate substituent generating the parent alcohol anilinogeraniol (AGOH) because no reaction product with the retention time of authentic AGOH (20.73 min) was observed under these conditions (Supporting Information Figure S2). Biochemical and crystallographic evidence indicate that Mg2+ is a required cofactor in TEAS catalysis due to its role in coordinating binding of the diphosphate moiety of FPP, thereby facilitating both the formation of a catalytically competent enzyme–substrate complex as well as the ionization of the diphosphate group from the substrate.25,29 If formation of a putative cyclized product from AGPP is the result of binding and catalysis within the TEAS active site, then Mg2+ would be expected to function in a similar capacity in this reaction. Consistent with this notion, no reaction products were formed in incubations of TEAS and AGPP with 10 mM EDTA substituted for 40 mM MgCl2. Lastly, if AGPP could serve as a bona fide substrate, then a substrate dependent accumulation of product would be expected (Figure 4).
Figure 3.
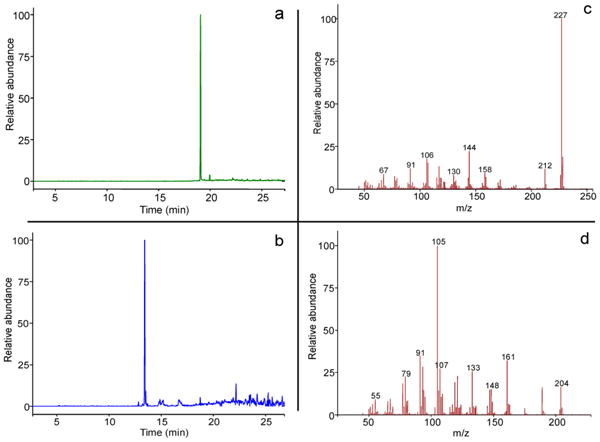
GC-MS comparison of the reaction products generated by TEAS incubated with AGPP (a) and FPP (b) and the mass spectrum for the dominant peaks at 18:05 min (c) and 12.68 (d).
Figure 4.

Reaction kinetics of 5-epi-aristolochene synthase for the non-native substrate AGPP. The TEAS enzyme (1 μM) was incubated with the indicated concentrations of AGPP at 37 °C for 0.5 h, the reaction products extracted into hexanes, then qualified and quantified by GC-MS. Error bars are standard errors of the mean of five independent experiments.
The slow reaction rates for TEAS catalyzed cyclization of AGPP; high concentrations of enzyme relative to substrate and difficulty in measuring product formed or substrate consumed within the first 10% of substrate consumption precluded analysis of the enzyme kinetics by Michaelis–Menten formalism. Nevertheless, we derived the AGPP product formation rates employing the standard Michaelis–Menten formalism in order to compare determinations with those for FPP. The average Km determined for AGPP in five independent experiments was approximately 8.2 μm, albeit exhibiting a turnover rate (kcat) of 0.00012 s−1, a rate approximately 100-times slower than for the native FPP substrate (Supporting Information Table S3).
When 1 μM TEAS was incubated with either 20 μM FPP or an equal amount of AGPP at 37 °C for 0.5 h, the TEAS enzyme was able to generate 27.2 nmol of 5-epi-aristolochene (2) per 50 μL reaction in comparison to 12.8 nmol of the AGPP reaction product 5 (Supporting Information Figure S3). However, when incubated with both FPP and AGPP, the TEAS enzyme yielded 7.3 nmol of the AGPP reaction product but only 1.5 nmol of 5-epi-aristolochene, consistent with our observations that AGPP is a suitable substrate analog (Km = 8.2 μM) and a potent noncompetitive inhibitor (Ki = 0.41 μM).
The exact mass and corresponding molecular formula for the product of TEAS catalyzed conversion of AGPP were deduced from high-resolution GC-MS experiments in comparison to the natural FPP substrate. Importantly, the reaction products of either incubation were dominated by a single product (Figure 3). The mass spectrum for the 5-epi-aristolochene standard exhibited a parent molecular ion at 204 Da as well as a fragmentation pattern consistent with that previously pub-lished.18,30 The mass spectrum for the hexane-extractable product generated by incubation of AGPP with TEAS was characterized by a parent molecular ion of 227 Da and a fragmentation pattern distinctly different from that of 5-epi-aristolochene (Figure 3c,d). The observed mass of 227 Da was consistent with the formula C16H21N. The monoisotopic mass calculated for C16H21N is 227.1675 Da, and the experimentally determined high resolution mass for the AGPP reaction product 5 was 227.1676 Da. This mass is consistent with either TEAS catalyzed elimination of diphosphate to form a linear triene or cyclization of AGPP after ionization and cleavage of the diphosphate group.
NMR of the AGPP Reaction Product
To confirm the structure, especially the geometry and stereochemistry, of the cyclized product, we employed NMR analysis summarized in Figure 5 (see Supporting Information Figures S4 and S5 for spectra). For these experiments, 200 μg of the reaction product was prepared by hexane extraction and chromatographic purification of preparative-scale incubations of TEAS with AGPP. 1H NMR 1D and COSY spectra in CDCl3 revealed resonances for four aromatic protons and only two olefinic protons, which is inconsistent with a linear triene structure. Many of these resonances were broadened by intermediate exchange at 24 °C. These resonances sharpened, shifted, and developed a J-coupling pattern consistent with a para-substituted aromatic ring when cooled to –50 °C. Analysis of 1H COSY, 1D NOE, and 1D selective decoupling experiments at 24 °C and −50 °C clearly demonstrated macrocycle closure onto the para- position of the aniline ring as well as retention of the E-configuration for both the 2,3 and 6,7 double bonds. Although the four aromatic resonances had different chemical shifts, the COSY correlations established a para-substitution pattern. The absence of an NOE in 1H NMR NOE experiments at −50 °C between the H1 and H8 protons excludes both ortho-ring closure and alkylation of the aniline nitrogen. Analysis of the 1H COSY spectrum at −50 °C revealed that each olefinic resonance was part of an isolated spin system. The E-configuration of both double bonds was confirmed by employing 1H NMR NOE experiments at −50 °C (Figure 5). Irradiation of the C9 methyl group (δ 1.31 ppm) leads to enhancement of the C1–H (δ 3.02–3.13 ppm) and C4–H (δ 1.92–2.02 ppm) resonances, and irradiation of the C10 methyl group (δ 1.44 ppm) leads to enhancement of the C5–H (δ 1.75–1.82 ppm) and C8–H (δ 3.26–3.36 ppm) resonances. A molecular model of the three-dimensional structure of 5 made using ChemBio3D gave a lowest energy conformation with a relatively high level of strain (40.1 kcal/mol) due to the distortion of the aromatic amine geometry. This strain was calculated to be reduced to 21.7 kcal/mol in models of the protonated molecule. Accordingly, 1H NMR 1D spectra recorded in 1 N HCl at 24 °C were simplified and showed a classic para-substituted AA′MM′ aromatic spin system. These observations suggest that a single, enantiomeric conformation of geraniline 5 is formed by TEAS cyclization of AGPP 3, that is racemized upon protonation.
Figure 5.

Key COSY spin system correlations and NOEs for geraniline, the reaction product generated when the TEAS enzyme is incubated with AGPP.
Mechanistic Considerations
The suitability of AGPP as a substrate for TEAS is intriguing with regard to the reaction mechanism proposed for this enzyme. Based largely on chemical rationalization, Whitehead et al.30 and Cane31 suggested earlier that the cyclization of FPP by TEAS proceeded via sequential partial reactions (Scheme 1). These partial reactions included (1) initial cleavage of the strong electron withdrawing diphosphate leaving group resulting in the generation of a carbocation at C1; (2) ring closure to generate the 10-membered germacrene A skeleton by nucleophilic attack of C10 on C1; (3) reprotonation at C6 followed by attack of C2 on C7 generating a new bicyclo eudesmane carbocation centered at C3; and finally, (4) a hydride shift from C2 to the new C3 carbocation followed by a methyl migration from C7 and final proton abstraction from C8 resulting in a double bond between C7 and C8 and formation of 5-epi-aristolochene 2. Structural and functional studies have supported this model of sequential partial steps. For example, tyrosine 520 is spatially positioned within the active site of TEAS such that it was proposed to donate a proton to the germacrene A intermediate and initiate the second round of partial reactions. Based on the assumption that a mutation of tyrosine 520 to phenylalanine would eliminate reprotonation of the germacrene intermediate, Rising et al.18 demonstrated that such a mutant did only synthesize germacrene A.
With respect to the cyclization of AGPP by TEAS (Scheme 2), the initial ionization and formation of the carbon–carbon bond between C1 and the para- position of the phenyl ring are directly analogous to the initial steps within the TEAS catalyzed reaction of FPP leading up to germacrene A (Scheme 1). However, formation of the 13 membered aza-[9]-paracyclophane ring is somewhat surprising given that cyclization of FPP gives the 10 membered germacrene A intermediate. There are four nucleophilic centers on the aniline ring, the nitrogen, the two ortho, and the para ring carbons, and cyclization is only observed onto the para-position. Ring closure onto the nitrogen or ortho-position would give a nine-membered ring (N-phenyl,-3,7-dimethyl-trans,trans-3,7-azacy-clononadiene) or an 11-membered ring (3,7-dimethyl-trans,-trans-3,7-aza[9]orthocyclophane-diene), respectively. The observed geraniline product indicates that the steric constraints imposed by the active site preclude these alternative products. These constraints allow cyclization of C1 onto the para position of the anilinogeranyl cation despite the acyclic precursor adopting a conformation distinct from that for the cyclization onto the 10-position of the farnesyl cation. That one cyclization product is seen rather than a mixture suggests that the AGPP cannot adopt conformations that would lead to formation of the other two products.
Why the initial cyclized AGPP intermediate does not go on to form additional rings or other structural rearrangements may be related to an inability of the double bonds in the 13-membered ring to achieve sufficient orbital overlap to allow bicycle-ring formation. The carbocation generated by proto-nation of C6 in geraniline would be unable to interact with the C2 nucleophile due to the conformation of the strained ring in 5. The C2 and C6 geraniline atoms are approximately 4.5 Å apart. Another possibility is that the positioning of geraniline within the active site pocket may prevent further reaction. Previous structural studies with FHP and F3-FPP, non-hydrolyzable substrates, helped to place the diphosphate moiety of these analogs at the entrance of the active site pocket. Subsequent modeling of the FPP cyclization events relied on positioning of the various intermediates within the active site with little overall movement or reorientation of the intermediates. In the electron density map of Figure 2b, the cyclization product of AGPP appears to be positioning the C6–C7 double bond 5 Å from Y520, which perhaps limits the possibilities for proton donation at this electron rich center or at C2–C3 and terminates further chemical transformations.
The orientation of the cyclized AGPP product also has implications for a limiting step in the cyclase reaction mechanism. Presteady state kinetic analysis of TEAS implied a rapid equilibration of the enzyme with FPP, followed by a slightly slower conversion of FPP to hydrocarbon, and a subsequent rate-limiting step.30 Whether this rate-limiting step constituted a chemical transformation, for example protonation of germacrene A, or a nonchemical step such as product release could not be determined. However, it is reasonable to assume that release of an aliphatic reaction product from the hydrophobic active site pocket into an aqueous environment would be slow. Addition of the amine nitrogen and positioning of the AGPP cyclized product near the entrance with the nitrogen closest to the bulk phase solvent may stabilize the product complex and result in a slower product release rate, consistent with the experimental data. Determination of such must await additional presteady-state kinetic analysis of the TEAS enzyme with AGPP as a substrate.
Implications for Synthesis of Novel Organic Compounds
Closure of acyclic intermediates to medium-sized rings is more difficult than five- and six-membered rings because of unfavorable entropic factors and the development of unfavorable transannular interactions.16 A significant aspect of enzyme function is the preorganization of substrates that substantially reduces the entropic penalties associated with the subsequent catalyzed reaction. Because of the chemo-, stereo-, and regio-selectivity inherent in enzyme mediated transformations, highly functionalized acyclic precursor molecules can be employed in ring synthesis. The cyclization of AGPP by the tobacco 5-epi-aristolochene synthase involves regioselective formation of a single bond to construct the medium-sized ring.32 Medium sized rings are typically prepared by intra- and intermolecular cyclization reactions under high dilution conditions.32 Medium sized rings can also be made through ring closing metathesis and intramolecular lactonization reactions.12 However, macrocycles with multiple double bonds like geraniline are difficult to obtain through ring closing metathesis since the product becomes highly strained.17 The observation that tobacco 5-epi-aristolochene synthase catalyzes the cyclization of the linear AGPP 4 to a single 13-membered ring product 5 demonstrates that the enzymatic cycloaddition approach to the generation of medium sized ring systems represents an important complementary alternative to existing conventional synthetic approaches.
Methods
Materials
[1-3H] FPP (1) (15–30 Ci/mmol) was purchased from PerkinElmer or American Radiolabeled Chemicals. 8-Anilino-geranyl pyrophosphate (AGPP (4)) and 8-anilino-geraniol (AGOH) were synthesized according to the procedure of Chedhade et al.20 and were stored at −20 °C. AGPP for kinetic and GC-MS experiments was stored as a 2 mM solution in water. FPP was obtained from Sigma Chemical Company and Echelon Bioscience. A 5-epi-Aristolochene standard for GC-MS was kindly provided by Dr. Robert Coates, Department of Chemistry, University of Illinois, Urbana–Champaign.
Purification of Recombinant TEAS
Bacterial expression and purification of TEAS were based on previously published procedures.18,29 In summary, a full-length TEAS cDNA containing a 5′ terminal extension sequence encoding a hexameric His sequence was inserted into the pET28B expression vector (Novagen), and this recombinant vector transformed into BL21-D3 (Novagen) E. coli cells. Recombinant cells were incubated until they entered into an exponential growth phase, then expression of the TEAS cDNA was induced by the addition of 1 mM IPTG for an additional 5 h. Cells were collected by centrifugation and resuspended in a lysis buffer containing lysozyme. Complete disruption of the cells was achieved by sonication and the lysate clarified by centrifugation. The supernatant was filtered, then applied to His-bind Ni2+ column and eluted with a linear or step gradients of 20 to 250 mM imidazole. Further purification, which was necessary for the crystallographic work, included chromatography on a MonoQ HR10/10 column (Pharmacia) eluted with a linear salt gradient from 0 to 500 mM NaCl, and gel filtration on a Superdex-200 column (Pharmacia) in the presence of 100 mM NaCl. Fractions containing the highest TEAS enzyme activity were combined, dialyzed, and concentrated to 10–20 mg mL−1 using a Millipore Ultrafree-4 Biomax-30 centrifigal concentrator. Glycerol was added to 50%, and the purified TEAS protein was stored at −80 °C.
Kinetic Characterization of the Interactions of AGPP with TEAS
The assay for TEAS activity is based upon partitioning of the hydrophobic product (3H-5-epi-aristolochene) into an organic solvent while the hydrophilic substrate (3H-FPP) remains in the aqueous phase,29 an assay previously validated by GC-MS analysis of the reaction product profile.26,27 The Ki describing the interaction of AGPP with TEAS was determined by measuring initial reaction velocities. Reactions (50 μL) contained 200 mM Tris-HCl, pH 7.5, 40 mM MgCl2, 40–333 nM TEAS, and 0.2 μCi of 3H-FPP, carrier free or in combination with unlabeled FPP to give final substrate concentrations of 0.1 to 90 μM. Fixed concentrations of AGPP ranged from 0 to 150 μM. Reactions were initiated by the addition of enzyme and were incubated for 10 min at RT. Reactions were quenched by rapid addition of 0.5 M EDTA and 50 mM KOH. Samples were subsequently extracted with 300 μL of hexanes, briefly centrifuged, then 200 μL of the hexane phase was treated with approximately 10 mg of silica powder to remove any contaminating FPP or farnesol, the latter generated by any contaminating phosphatase activities. Following gentle mixing and a brief centrifugation to pellet the silica, 100 μL of the hexane phase was analyzed for radioactivity by scintillation counting. Determination of reaction rate was based on percent conversion of FPP to product, a value that was obtained by comparing the radioactivity in the hexane phase to that in an untreated aliquot of the assay mixture. Near background levels of radioactivity were observed in hexane extracts derived from control reactions lacking enzyme. In addition, silica treatment did not significantly alter the amount of radioactivity observed in hexane extracts, indicating insignificant phosphatase contamination. Determinations were performed in triplicate with data analyzed using the Enzyme Kinetics 1.3 suite within the Sigma Plot 11.0 program (Trinity Software).
Characterization of the TEAS Reaction with AGPP As Substrate
Control reactions contained 20 μM FPP (the natural substrate), 40 mM MgCl2, 200 mM Tris-HCl, pH 7.5, and 0.1 to 1 μM TEAS in a volume of 50–100 μL. Reactions with AGPP contained 0.1 to 100 μM AGPP, 40 mM MgCl2, 10% glycerol, 200 mM Tris-HCl, pH 7.5, and 1 μM TEAS in a volume of 100 μL. Reactions were incubated at 4 °C for a period of 30 min to overnight, or at 37 °C for 30 min prior to extraction with hexanes. For high-resolution MS determinations, the pooled hexane extracts were concentrated to a minimum volume under N2. Assays were performed in triplicate, and the concentration of hexane-extractable product (0.1–3 μg/mL) was estimated by GC based on a comparison to the detection of a hexadecane standard.
The hexane extracts and a 5-epi-aristolochene standard were examined by GC-MS analyses using a Varian 3400 gas chromatograph and a Finnigan INCOS-50 quadrapole mass selective detector. The GC was equipped with a capillary DB-5MS column (15 m × 0.25 mm, 0.25 μm phase thickness), with He as the carrier gas (10 psi). Samples of the hexane extracts were introduced by splitless injection at an injection port temperature of 110 °C. The column temperature was maintained at 60 °C for 1 min following injection and then increased to 280 °C with a gradient of 10 °C per min. GC-MS analysis of the 5-epi-aristolochene standard included splitless injection at a port temperature of 110 °C. The column temperature was maintained at 50 °C for 1 min, then increased to 250 °C with a 4 °C per min gradient. All samples were introduced directly to the electron impact ionization source for mass spectral analysis. Spectra were recorded at 70 eV, scanning from 20 to 420 atomic mass units. Mass spectral data were compared to those published for 5-epi-aristolochene.18,30
The exact molecular mass of the hexane-extractable product generated upon incubation of AGPP with TEAS was determined by high-resolution mass spectrometry using a Kratos CONCEPT IIIH magnetic sectro mass selective detector. Samples were introduced directly to the electron impact ionization source, and each measurement was recorded at 70 eV. For each spectrum, approximately 20–30 scans were collected in a slow scanning mode (10 s/decade). The column bleed was used as the internal standard and was calibrated separately against perfluorokerosene. Reported data are based on a composite of the early strong scans for which an optimal response from the molecular ion was obtained.
NMR Analysis of the TEAS Reaction Products
The double bond geometry and aromatic substitution pattern for the hexane-extractable product generated upon incubation of AGPP with TEAS was determined by 1H COSY, NOE, and partial decoupling experiments in CDCl3 at −50 °C using a Varian Inova spectrometer operating at 400 MHz. Chemical shifts are reported in parts per million from CDCl3 internal peak at 7.27 ppm.
Crystallization and Data Collection
TEAS crystallizes in hanging drops as described by Starks et al.25 For structure determination of a TEAS–AGPP complex, TEAS crystals were soaked in mother liquor containing 1–5 mM AGPP, then stabilized for freezing in a similar solution which also included 20% ethylene glycol. The crystal was frozen in a nitrogen stream (∼190 K), and a diffraction data set was collected at Stanford Synchrotron Radiation Laboratory beamline 7–1 (Supporting Information Table S1).
Structure Determination and Refinement
A starting model consisting of protein residues 17–522 and 533–548 of the TEAS– farnesyl hydroxyphosphonate structure (PDB code 5EAT) was positioned with respect to the TEAS–AGPP data using rigid body refinement in PHENIX.33 The initial Fo-Fc difference electron density map revealed additional density for protein residues 523–532 as well as a bound reaction product in the active site. The missing regions of the protein were built, water molecules were added, and several rounds of positional and temperature factor refinement and manual model adjustment were carried out. An energy-minimized model of 5 was then placed in the active site electron density,34 aided by the clear density for its phenyl and methyl groups. Additional refinement of the model, including 5 was carried out with the program Coot;35 refinement, map calculation, and water molecule location were carried out with PHENIX. Final coordinates of the TEAS–5 complex have been submitted to the Protein Data Bank (4RNQ).
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health GM66152 (to H.P.S.) and the Kentucky Lung Cancer Research Program (to H.P.S.), and the NMR instruments used in this work were obtained with support from NSF CRIF Grant CHE-9974810. This work was also supported by NIH grant GM054029 to J.P.N. and J.C., grant 1RC2GM092521 to J.C. J.P.N. is an Investigator of the Howard Hughes Medical Institute. C.M.C. was supported by a T32 Cancer Training Fellowship at The Salk Institute for Biological Studies.
Footnotes
Supporting Information: Tables listing the crystallographic statistics, kinetic constants for the inhibition of the TEAS enzyme by AGPP and for AGPP as a substrate; Lineweaver–Burk plot of AGPP inhibition of TEAS activity; GC profile of AGOH; GC profile of reactions products from TEAS incubated with FPP, AGPP and FPP +AGPP; NMR spectra for AGPP and geraniline. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.5b00145.
The authors declare no competing financial interest.
References
- 1.Christianson DW. Structural biology and chemistry of the terpenoid cyclases. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 2.Davis EM, Croteau R. Cyclization enzymes in the biosynthesis of monoterpenes, sesquiterpenes, and diterpenes. Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids. 2000;209:53–95. [Google Scholar]
- 3.Connolly JD, Hill RA. Dictionary of Terpenoids. Chapman & Hall; London: 1991. [Google Scholar]
- 4.Vogeli U, Freeman JW, Chappell J. Purification and characterization of an inducible sesquiterpene cyclase from elicitor-treated tobacco cell-suspension cultures. Plant Physiol. 1990;93:182–187. doi: 10.1104/pp.93.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi S, Zhao YX, O'Maille PE, Greenhagen BT, Noel JP, Coates RM, Chappell J. Kinetic and molecular analysis of 5-epiaristolochene 1,3-dihydroxylase, a cytochrome P450 enzyme catalyzing successive hydroxylations of sesquiterpenes. J Biol Chem. 2005;280:3686–3696. doi: 10.1074/jbc.M411870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 7.Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981-2002. J Nat Prod. 2003;66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 9.Ortholand JY, Ganesan A. Natural products and combinatorial chemistry: back to the future. Cur Opin Chem Biol. 2004;8:271–280. doi: 10.1016/j.cbpa.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 10.Driggers EM, Hale SP, Lee J, Terrett NK. The exploration of macrocycles for drug discovery - an underexploited structural class. Nature Rev Drug Discovery. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 11.Mallinson J, Collins I. Macrocycles in new drug discovery. Future Med Chem. 2012;4:1409–1438. doi: 10.4155/fmc.12.93. [DOI] [PubMed] [Google Scholar]
- 12.Parenty A, Moreau X, Campagne JM. Macrolactonizations in the total synthesis of natural products. Chem Rev. 2006;106:911–939. doi: 10.1021/cr0301402. [DOI] [PubMed] [Google Scholar]
- 13.Wessjohann LA, Ruijter E, Garcia-Rivera D, Brandt W. What can a chemist learn from nature's macrocycles? - A brief, conceptual view. Mol Diversity. 2005;9:171–186. doi: 10.1007/s11030-005-1314-x. [DOI] [PubMed] [Google Scholar]
- 14.Gradillas A, Perez-Castells J. Macrocyclization by ring-closing metathesis in the total synthesis of natural products: Reaction conditions and limitations. Angew Chem, Int Ed. 2006;45:6086–6101. doi: 10.1002/anie.200600641. [DOI] [PubMed] [Google Scholar]
- 15.March J. Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. 4th. Wiley; New York: 1992. [Google Scholar]
- 16.Marsault E, Peterson ML. Macrocycles are great cycles: Applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J Med Chem. 2011;54:1961–2004. doi: 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- 17.Monfette S, Fogg DE. Equilibrium ring-closing metathesis. Chem Rev. 2009;109:3783–3816. doi: 10.1021/cr800541y. [DOI] [PubMed] [Google Scholar]
- 18.Rising KA, Starks CM, Noel JP, Chappell J. Demonstration of germacrene A as an intermediate in 5-epi-aristolochene synthase catalysis. J Am Chem Soc. 2000;122:6526–6526. [Google Scholar]
- 19.Lee HY, Yerkes N, O'Connor SE. Aza-tryptamine substrates in monoterpene indole alkaloid biosynthesis. Chem Biol. 2009;16:1225–1229. doi: 10.1016/j.chembiol.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chehade KA, Andres DA, Morimoto H, Spielmann HP. Design and synthesis of a transferable farnesyl pyrophosphate analogue to Ras by protein farnesyltransferase. J Org Chem. 2000;65:3027–3033. doi: 10.1021/jo991735t. [DOI] [PubMed] [Google Scholar]
- 21.Subramanian T, Liu S, Troutman JM, Andres DA, Spielmann HP. Protein farnesyltransferase-catalyzed isoprenoid transfer to peptide depends on lipid size and shape, not hydrophobicity. Chembiochem. 2008;9:2872–2882. doi: 10.1002/cbic.200800248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Troutman JM, Subramanian T, Andres DA, Spielmann HP. Selective modification of CaaX peptides with ortho-substituted anilinogeranyl lipids by protein farnesyl transferase: Competitive substrates and potent inhibitors from a library of farnesyl diphosphate analogues. Biochemistry. 2007;46:11310–11321. doi: 10.1021/bi700516m. [DOI] [PubMed] [Google Scholar]
- 23.Chen M, Knifley T, Subramanian T, Spielmann HP, O'Connor KL. Use of synthetic isoprenoids to target protein prenylation and Rho GTPases in breast cancer invasion. PloS One. 2014;9:e89892. doi: 10.1371/journal.pone.0089892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noel JP, Dellas N, Faraldos JA, Zhao M, Hess BA, Smentek L, Coates RM, O'Maille PE. Structural elucidation of cisoid and transoid cyclization pathways of a sesquiterpene synthase using 2-fluorofamesyl diphosphates. ACS Chem Biol. 2010;5:377–392. doi: 10.1021/cb900295g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Starks CM, Back KW, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 26.Greenhagen BT, O'Maille PE, Noel JP, Chappell J. Identifying and manipulating structural determinates linking catalytic specificities in terpene synthases. Proc Natl Acad Sci U S A. 2006;103:9826–9831. doi: 10.1073/pnas.0601605103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Maille PE, Chappell J, Noel JP. A single-vial analytical and quantitative gas chromatography–mass spectrometry assay for terpene synthases. Anal Biochem. 2004;335:210–217. doi: 10.1016/j.ab.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 28.Jahnke W, Rondeau JM, Cotesta S, Marzinzik A, Pelle X, Geiser M, Strauss A, Gotte M, Bitsch F, Hemmig R, Henry C, Lehmann S, Glickman JF, Roddy TP, Stout SJ, Green JR. Allosteric non-bisphosphonate FPPS inhibitors identified by fragment-based discovery. Nat Chem Biol. 2010;6:660–666. doi: 10.1038/nchembio.421. [DOI] [PubMed] [Google Scholar]
- 29.Mathis JR, Back K, Starks C, Noel J, Poulter CD, Chappell J. Pre-steady-state study of recombinant sesquiter-pene cyclases. Biochemistry. 1997;36:8340–8348. doi: 10.1021/bi963019g. [DOI] [PubMed] [Google Scholar]
- 30.Whitehead IM, Threlfall DR, Ewing DF. 5-Epi-aristolochene is a common precursor of the sesquiterpenoid phytoalexins capsidiol and debneyol. Phytochemistry. 1989;28:775–779. [Google Scholar]
- 31.Cane DE. Enzymatic formation of sesquiterpenes. Chem Rev. 1990;90:1089–1103. [Google Scholar]
- 32.Grimme S, Pischel I, Laufenberg S, Vogtle F. Synthesis, structure, and chiroptical properties of the first 4-oxa[7] paracyclophane. Chirality. 1998;10:147–153. [Google Scholar]
- 33.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moriarty NW, Grosse-Kunstleve RW, Adams PD. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr, Sect D: Biol Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.