

Abstract
The promise of exploiting combinatorial synthesis for small molecule discovery remains unfulfilled due primarily to the “structure elucidation problem”: the back-end mass spectrometric analysis that significantly restricts one-bead-one-compound (OBOC) library complexity. The very molecular features that confer binding potency and specificity, such as stereochemistry, regiochemistry, and scaffold rigidity, are conspicuously absent from most libraries because isomerism introduces mass redundancy and diverse scaffolds yield uninterpretable MS fragmentation. Here we present DNA-encoded solid-phase synthesis (DESPS), comprising parallel compound synthesis in organic solvent and aqueous enzymatic ligation of unprotected encoding dsDNA oligonucleotides. Computational encoding language design yielded 148 thermodynamically optimized sequences with Hamming string distance ≥ 3 and total read length <100 bases for facile sequencing. Ligation is efficient (70% yield), specific, and directional over 6 encoding positions. A series of isomers served as a testbed for DESPS’s utility in split-and-pool diversification. Single-bead quantitative PCR detected 9 × 104 molecules/bead and sequencing allowed for elucidation of each compound’s synthetic history. We applied DESPS to the combinatorial synthesis of a 75 645-member OBOC library containing scaffold, stereochemical and regiochemical diversity using mixed-scale resin (160-μm quality control beads and 10-μm screening beads). Tandem DNA sequencing/MALDI-TOF MS analysis of 19 quality control beads showed excellent agreement (<1 ppt) between DNA sequence-predicted mass and the observed mass. DESPS synergistically unites the advantages of solid-phase synthesis and DNA encoding, enabling single-bead structural elucidation of complex compounds and synthesis using reactions normally considered incompatible with unprotected DNA. The widespread availability of inexpensive oligonucleotide synthesis, enzymes, DNA sequencing, and PCR make implementation of DESPS straightforward, and may prompt the chemistry community to revisit the synthesis of more complex and diverse libraries.
Keywords: DNA-encoded libraries, combinatorial synthesis, split-and-pool, one-bead-one-compound
The NIH Molecular Libraries Program (MLP) was founded to translate the discoveries of the Human Genome Project into therapeutics. With gene sequences (and thereby target identities) in hand, the only obstacle to discovery was access to high-throughout screening (HTS) technology, which the MLP eliminated through its network of HTS centers.1 Despite major public investment, however, drug discovery remains a costly and specialized pursuit limited to a few major facilities, reminiscent of early DNA sequencing platforms that lead to the completion of the Human Genome Project,2 rather than the highly distributed and economical genome sequencing technology of today.3,4 At the heart of the problem is the compound library, a collection of molecular entities each inhabiting a single microtiter plate well and ranging in size from several thousand to several million different species. The management of these collections comes at enormous cost in terms of automation,5 analysis, and manpower, as does generation of molecular diversity by way of serial synthesis.6 These constraints constitute key technological barriers to transforming HTS-based small molecule discovery into a distributable and thereby economical enterprise.
Combinatorial synthesis potentially addresses these barriers by introducing enormous scaling advantages, both in its capability to generate large libraries of molecules and in storing the resulting libraries in a portable format. Combinatorial diversification7−10 mimics biological diversification in that randomly permuting chemically distinct monomers using one or several highly efficient bond-forming strategies generates exponentially increasing molecular diversity. When implemented in solid-phase synthesis, the resulting molecular libraries can be spectacularly large. These “one-bead-one-compound” (OBOC) libraries,9 which can contain thousands to millions of different members, are trivial to prepare by parallel synthesis involving a very modest 20–40 different chemical diversity elements (e.g., alkyl amines in a peptoid library11,12) and sometimes just one bond construction strategy, such as amide bond formation. Additionally, OBOC libraries exist simply as collections of beads in a tube. As such, they are consumable and thereby distributable, unlike conventional compound libraries, which are constantly curated resources.
Payment for exploiting the scaling and format advantages of combinatorial synthesis is due upon library screening. Screening a combinatorial solid-phase library entails incubating the library with a labeled target, isolating individual labeled “hit” beads for analysis, and then determining the compound structure on each bead. Hit structure elucidation almost always occurs via tandem mass spectrometric (MS/MS) fragmentation analysis, which imposes several significant restrictions on OBOC library design. First, most OBOC libraries lack stereochemical or regiochemical diversity because these features introduce mass redundancy, which generates ambiguous mass spectral signatures. Further, library design also must exclude any isomeric submonomer combinations if appropriate. Second, MS/MS fragmentation will only yield an interpretable ion series for sequencing if the general scaffold features multiple isoenergetic bonds, which favors limiting library design to polypeptides, peptoids, or other homogeneous oligomeric scaffolds. Third, synthesis steps must be highly efficient to minimize the formation of side products that could obscure the parent ion, and MS sensitivity demands relatively high compound loading per bead (>10 pmol), constraining synthesis to larger format resin. Finally, while highly sophisticated automation exists for analysis by scanning confocal fluorescence microscopy, robotic hit bead selection, and single-bead hit validation prior to resynthesis,13 there is sparingly little automation for single-bead chemical cleavage and de novo deconvolution of mass spectral data, severely limiting analysis throughput. These issues collectively constitute the “structure elucidation problem,” which has dogged combinatorial synthesis since its inception.
Synthesis encoding potentially circumvents the fundamental analytical limitations of MS-based compound structure elucidation by storing compound structure information in a more easily analyzed encoding molecule. Thus, encoded synthesis entails parallel synthesis of the target compound and installation of information encoding the synthetic steps executed in order to reach the target.14 Encoding molecules can be grown stepwise as a polymer to be sequenced, such as polypeptides15 and polynucleotides,16 or distinct encoding tags can be separately installed, such as electrophoric tags,17,18 alkylamines,19,20 or differential mass tags.21,22 In principle, any type of chemical complexity (e.g., stereochemical configuration) can be encoded with the caveat that the chemistry used to prepare the target molecule and the encoding molecule are orthogonal. The ultimate objective, then, is detecting and decoding the encoding molecule, highlighting detection sensitivity and throughput as additional considerations.
Nucleic acids are particularly attractive as encoding molecules for precisely the above considerations. PCR-based DNA amplification is possible from single template molecules and high-throughput sequencing can generate tens of millions of sequence reads in just over a day. Nucleic acid tags for encoding can be installed either as a parallel synthesis of the encoding oligonucleotide,16 as discrete sequence tags attached to library building blocks,23−27 or enzymatic ligation of oligonucleotides.28,29 Furthermore, the highly predictable secondary structure of nucleic acids can direct the assembly of combinatorial library building blocks, evoking the mRNA-templated polypeptide synthesis of biological translation. However, nucleic acids as encoding molecules are no panacea. The bases display a variety of nucleophiles that, upon modification, can inhibit enzymatic sequence replication or promote depurination.30 DNA contains acid-labile glycosidic bonds that, upon scission, result in phosphodiester backbone cleavage and concomitant genetic information loss,31 and their polyanionic character limits utility in nonaqueous solvents where solubility is an issue.
We set out to combine the best attributes of OBOC solid-phase combinatorial synthesis and nucleic acid encoding in order to overcome the disadvantages of each. We describe here DNA-encoded solid-phase synthesis (DESPS), which integrates solid-phase chemical synthesis of popular combinatorial library scaffold types (e.g., peptoids) as well as those derived from more specialized submonomers displaying stereochemical and regiochemical diversity.32 Accompanying the DESPS approach is a rationally designed encoding language that addresses constraints based on oligonucleotide secondary structure thermodynamics and compatibility with next-generation sequencing read lengths. Error correction informatics33 make the language almost resistant to the typical single-base errors of DNA sequencing analysis, which we demonstrate using PCR products obtained from single synthesis resin particles displaying chimeric oligomers that would otherwise prove to be analytically intractable by mass spectrometry. We finally apply DESPS to OBOC library synthesis, incorporating scaffold, regiochemical and stereochemical diversification, and we present a mixed-scale strategy that allows library quality control.
Results and Discussion
DESPS requires a bifunctional linker for parallel chemical synthesis and enzymatic DNA-based encoding. The DESPS bifunctional linker (Figure 1A) contains a coumarin chromophore for quantitative chromatographic analysis of synthesis yields, arginine to enhance mass spectrometric ionization efficiency, an alkyne for copper-catalyzed azide–alkyne cycloaddition (CuAAC)34,35 click chemistry, and protected terminal primary amine for compound synthesis. Sites for enzymatic DNA-based encoding are installed by CuAAC under conditions of substoichiometric azide-modified DNA “headpiece” (HDNA).28 The HDNA, two covalently tethered complementary sequences, presents a 5′-phosphate and 3′-dinucleotide overhang substrate for DNA ligase-catalyzed cohesive end ligation. Just as Fmoc quantitation measures resin loading capacity for solid-phase synthesis, quantitative PCR (qPCR) measures the number of HDNA sites that are accessible for enzymatic ligation and amplification (typically >1 × 106 sites per bead, see Supporting Information). The final DESPS product (Figure 1B) is a resin-bound oligomeric compound (synthesized via standard amide bond formation and nucleophilic displacement reactions conducted in organic solvent) and an encoding DNA (via directional cohesive end ligation of double-stranded oligonucleotides). The HDNA tether ensures that both strands remain associated with the bead throughout all chemical synthesis steps in denaturing organic solvent. PCR primer binding sites flank the DNA sequence that encodes the oligomer. DNA sequencing analysis of the PCR product reveals the synthesis history of each individual bead (Figure 1C), yielding the series of reaction conditions and monomers used to generate the oligomer.
Figure 1.
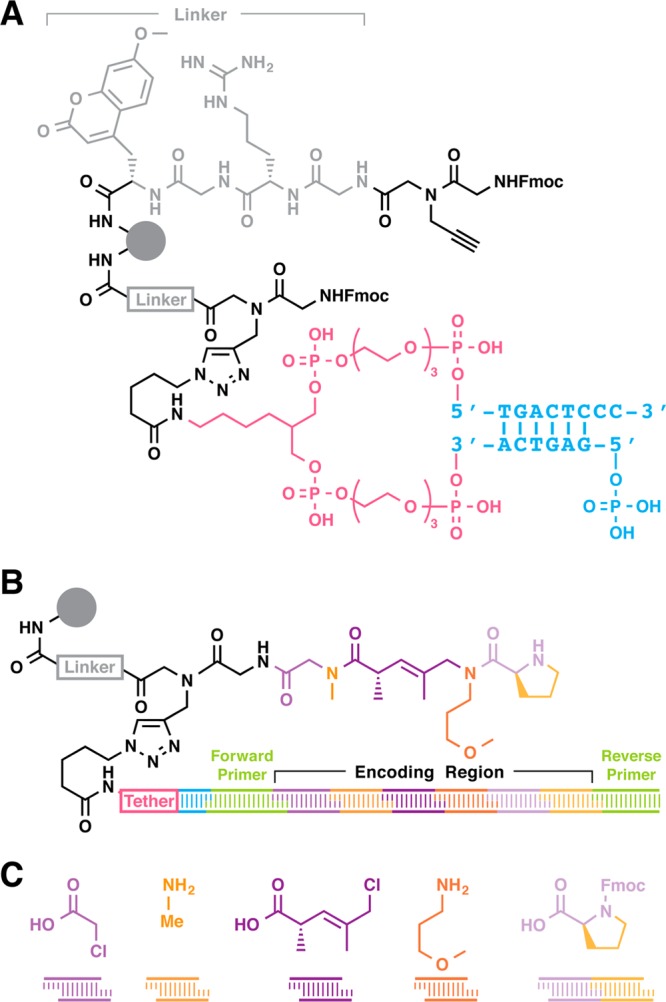
DNA-encoded solid-phase synthesis. (A) TentaGel Rink-amide resin (160-μm diameter) is first elaborated with a common linker (gray) containing a coumarin chromophore and arginine. Linker resin is further functionalized with an alkyne and Fmoc-protected glycine. Azide-functionalized DNA headpiece (HDNA), consisting of two complementary strands of DNA (cyan) covalently joined via two PEG tethers (magenta), is coupled substoichiometrically (0.004 equiv) to alkyne sites via CuAAC, yielding bifunctional-HDNA resin (Fmoc-protected amine for chemical coupling and 5′-phosphoryl-CC-3′ overhang for enzymatic cohesive end ligation). (B) A forward primer module (green) is first enzymatically ligated to resin. Encoded synthesis proceeds as alternating steps of monomer coupling (scaffold elements shown in purple hues, side chain elements shown in orange hues) and coding module ligation (correspondingly in purple or orange hues). After the last encoding step, a reverse primer module (green) is ligated. The finished resin displays oligomer and a structure-encoding DNA message flanked by primer binding sequences for PCR amplification. (C) The DNA sequence encodes the series of reaction conditions that the bead experienced. Here, the DNA sequence encodes acylation with chloroacetic acid, treatment with methylamine, acylation with (2S,3E)-5-chloro-2,4-dimethyl-3-pentenoic acid, treatment with 3-methoxypropylamine, and acylation with N-Fmoc-l-proline followed by Fmoc removal.
Parallel solid-phase synthesis of compound oligomer and encoding DNA proceeds through a series of alternating organic phase chemical reactions and aqueous phase enzymatic ligation steps. An example synthesis (Scheme 1) illustrates the intermediate compound structures explicitly while representing the growing DNA sequence as a string of 4-digit numeric identifiers. Each identifier uniquely specifies a sequence module. From left to right the digits indicate sequence set (0 = primer, 1 = set 1 coding sequences, 2 = set 2 coding sequences), the module’s position in the encoding DNA (0 = position 0, 1 = position 1, etc.), and the last two digits index unique sequences for encoding. The identifiers visually assist in correlating low-bit-depth DNA sequence with the higher complexity of molecular structure and appear bold in text. DESPS begins with removal of the Fmoc protecting group from the terminal primary amine, exchange of organic solvent to aqueous phase followed by enzymatic ligation of a forward PCR primer module 0001 (a DNA heteroduplex that contains an overhang complementary to the HDNA, a second overhang that is noncomplementary to HDNA, and intervening PCR primer sequence) to the resin-bound HDNA (cyan). Encoding continues with the resin-bound new overhang of 0001 serving as a substrate for enzymatic ligation of scaffold diversity-encoding module 11XX (a DNA heteroduplex that contains an overhang complementary to 0001, a second overhang that is noncomplementary to 0001, and intervening diversity-encoding sequence), followed by exchange of solvent to organic phase and acylation of the terminal primary amine with the encoded scaffold diversity element. Nucleophilic displacement of the terminal allylic halide with an alkylamine, solvent exchange to aqueous, and enzymatic ligation of side chain diversity-encoding module 22XX (a DNA heteroduplex that contains an overhang complementary to all 11XX sequences, a second overhang that is noncomplementary to all previous overhangs, and intervening diversity-encoding sequence) completes one cycle of an encoded submonomer-type synthesis. Alternating cycles of chemical synthesis (N-Fmoc-protected amino acid with defined stereochemistry and final submonomer-type scaffold and side chain diversification) and directional enzymatic oligonucleotide ligation terminating with ligation of the reverse primer module 0701 yields resin displaying the product oligomer and DNA encoding the reaction sequence. It is worth noting that each reaction arrow after the first represents a point at which resin could be pooled and split in an encoded combinatorial library synthesis. Single-bead analysis of the resin-bound product is accomplished by cleavage of oligomer under acidic conditions followed by mass spectrometric analysis, or by PCR amplification followed by sequencing. The DNA sequence, decoded for display as a string of numeric identifiers, reveals the order and conditions of synthetic transformations that each bead experienced.
Scheme 1. DNA-Encoded Solid-Phase Synthesis Reaction Sequence.

The sequence modules that are used to assemble the encoding DNA are composed of two hybridized, partially complementary synthetic oligonucleotides. The slipped heteroduplex structure (Figure 2A) features a central 8-base-pair complementary coding region and each strand displays a 5′-phosphorylated overhang. Use of distinct overhangs enables directional ligation based on sequence complementarity of one overhang with another, and their sequence is constant based on their position in the order of ligated modules. For example, after ligation of the first diversity-encoding module, the resin displays a 5′-phosphoryl-TGA-3′ overhang. All diversity-encoding modules for the second module ligation will display a 5′-phosphoryl-TCA-3′ complementary overhang, a 5′-phosphoryl-AAC-3′ noncomplementary overhang, and one of a set of different coding region sequences. Module coding region sequences conform to one of two sequence degeneracy patterns: 5′-NNRRRRNN-3′ (set 1 1XXX) or 5′-NNYYYYNN-3′ (set 2 2XXX). “N” is any DNA nucleobase, “R” is any purine DNA nucleobase, and “Y” is any pyrimidine DNA nucleobase. Constraining the sets to these sequence motifs eliminates the possibility of selecting palindromic sequences that could form a stable homoduplex and ligate, which terminates the encoding DNA by virtue of displaying the incorrect overhang for subsequent ligation (Figure 2B). Even after constraining the coding region sequences to these motifs, appending overhangs for directional ligations may result in oligonucleotides that are thermodynamically prone to formation of undesired heteroduplexes (Figure 2C) or intramolecular secondary structures (hairpins, Figure 2D).
Figure 2.
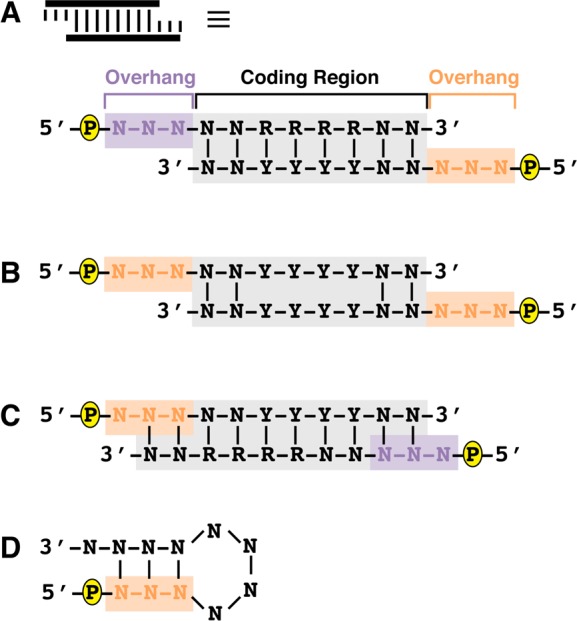
Encoding language design and optimization. (A) Each target heteroduplex coding module (schematic at top) is composed of two hybridized oligonucleotide strands. Each strand is 5′-phosphorylated (yellow “P”), displays a strand-specific overhang sequence (orange or purple), and coding region that is complementary (gray background). (B) Sufficiently self-complementary sequences may form undesired homoduplexes. Enforcing a coding region sequence structure of either 5′-NNRRRRNN-3′ or 5′-NNYYYYNN-3′ decreases the stability of potential homoduplexes relative to the target heteroduplex. (C) Some sequences (e.g., homopolymers) can form stable off-target heteroduplexes with occluded, unreactive overhangs. (D) Self-complementary sequences can form intramolecular secondary structures (hairpins) that prevent target heteroduplex formation.
To avoid selecting sequences that exhibit these potentially problematic features, we generated code to apply extant computational resources for biophysical oligonucleotide secondary structure prediction to large initial candidate sequence sets. The set 1 and set 2 motif each in combination with 3 different overhang sequences resulted in a total of 24,576 top ([+]) strand sequences and the same number of partially complementary overhang-appended bottom ([−]) strand sequences. For each sequence, mfold queries36 returned a series of Gibbs energies and melting temperatures (TM) that describe the thermodynamic stabilities of the desired heteroduplex module and all potential off-target structures. We discarded sequence candidates with predicted module TM < 30 °C or predicted hairpins within 15 °C of the module TM (ΔTM). Furthermore, we discarded all sequences where mfold predicted off-target heteroduplex (e.g., the structure of Figure 2C where [+] and [−] strand form a stable duplex that displays only single nucleotide overhangs) or homoduplex ΔG within 5.5 kcal/mol of the desired heteroduplex ΔG. For simplicity, we rejected any coding region sequence even if only 1 of the 3 overhang-appended candidates violated the above constraints. This rule ensured that any coding sequence would be well-behaved with all overhangs for its set. Finally, we culled the remaining sequences of homopolymeric coding regions (runs of >3) and recursively pruned set 1 and 2 such that each coding region sequence was ≥ 3 Hamming string distance from all other set members.37
The resulting computationally optimized encoding language contained 72 set 1 and 76 set 2 coding region sequences. Ten example coding sequences from each set (Table 1) illustrate the thermodynamic favorability of module formation over that of predicted off-target complexes. For a given coding sequence, the tabulated ΔG for an off-target complex is the most stable predicted of the 3 possible overhangs for that coding sequence. The average coding module ΔG is −13.6 kcal/mol, which is 9.9 kcal/mol more stable than the average predicted [+]-strand homoduplex, 9.1 kcal/mol more stable than the average predicted [−]-strand homoduplex, and 8.5 kcal/mol more stable than the average predicted off-target heteroduplex. Calculations predicted no stable hairpins for the selected example [+]-strand coding sequences. mfold predicted that four of the [−]-strand coding sequences would form stable hairpins, however the melting temperatures are far below that of the target heteroduplex and not a concern according to the above constraints.
Table 1. Encoding Sequence Thermodynamic Parameters and Ligation Yield.
| set 1 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| coding [+]a | TM,hetb (°C) | ΔGhetc (kcal/mol) | ΔGhomo[+]a,d (kcal/mol) | ΔGhet,2°e (kcal/mol) | ΔΔGhet/homog (kcal/mol) | ΔΔGhet/het,2°h (kcal/mol) | TM,hp[−]f (°C) | ΔTM[−]i (°C) | ΔGhomo[−]a,d (kcal/mol) | ΔΔGhet/homog (kcal/mol) | OH1 yieldj (%) | OH3 yieldj (%) | OH5 yieldj (%) | |
| 1X01 | TGGAAAGT | 37.1 | –13.4 | –3.9 | –5.0 | –9.5 | –8.4 | – | – | –3.6 | –9.8 | 70 | 71 | 68 |
| 1X02 | ACGGAGCA | 49.9 | –16.3 | –3.6 | –6.9 | –12.7 | –9.4 | – | – | –3.6 | –12.7 | 70 | 70 | 63 |
| 1X03 | TTGGAGTT | 37.1 | –13.4 | –1.6 | –5.0 | –11.8 | –8.4 | 1.9 | 35.2 | –3.6 | –9.8 | 72 | 73 | 69 |
| 1X04 | AAGGAGGT | 40.7 | –14.2 | –4.9 | –4.7 | –9.3 | –9.5 | – | – | –3.6 | –10.6 | 75 | 74 | 66 |
| 1X05 | AGAAAGCA | 38.5 | –13.8 | –3.5 | –3.1 | –10.2 | –10.6 | 20.6 | 17.9 | –3.6 | –10.1 | 74 | 74 | 67 |
| 1X06 | ACAGAACT | 36.5 | –11.4 | –3.5 | –2.0 | –7.8 | –9.4 | – | – | –3.6 | –7.7 | 72 | 72 | 61 |
| 1X07 | TAAGGAGT | 33.5 | –12.1 | –4.9 | –3.1 | –7.2 | –9.0 | – | – | –3.6 | –8.5 | 72 | 74 | 68 |
| 1X08 | ATGGGAGT | 40.9 | –14.1 | –5.4 | –6.5 | –8.7 | –7.6 | – | – | –3.6 | –10.5 | 74 | 75 | 65 |
| 1X09 | TGAAGGAA | 36.3 | –13.7 | –3.5 | –3.5 | –10.1 | –10.1 | – | – | –3.6 | –10.0 | 71 | 73 | 66 |
| 1X10 | TTGAGGAT | 35.4 | –13.2 | –1.6 | –3.1 | –11.6 | –10.1 | 20.0 | 15.4 | –4.6 | –8.6 | 75 | 75 | 70 |
| set 2 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| coding [+]a | TM,hetb (°C) | ΔGhetc (kcal/mol) | ΔGhomo[+]a,d (kcal/mol) | ΔGhet,2°e (kcal/mol) | ΔΔGhet/homog (kcal/mol) | ΔΔGhet/het,2°h (kcal/mol) | TM,hp[−]f (°C) | ΔTM[−]i (°C) | ΔGhomo[−]a,d (kcal/mol) | ΔΔGhet/homog (kcal/mol) | OH2 yieldj (%) | OH4 yieldj (%) | OH6 yieldj (%) | |
| 2X01 | CCTCCTAA | 35.9 | –13.8 | –3.6 | –6.2 | –10.2 | –7.6 | – | – | –5.4 | –8.5 | 52 | 69 | 70 |
| 2X02 | AACCTCAA | 37.1 | –13.4 | –3.6 | –5.5 | –9.8 | –8.0 | – | – | –5.4 | –8.1 | 61 | 73 | 70 |
| 2X03 | AATCCCAT | 36 | –14.6 | –3.6 | –3.1 | –10.9 | –11.4 | – | – | –5.4 | –9.2 | 65 | 74 | 69 |
| 2X04 | AACCCTAC | 37.6 | –13.3 | –3.6 | –6.8 | –9.7 | –6.6 | – | – | –6.3 | –7.0 | 63 | 74 | 70 |
| 2X05 | ATCCTCTC | 36.2 | –12.5 | –3.6 | –6.8 | –8.9 | –5.7 | – | – | –3.6 | –8.9 | 58 | 72 | 70 |
| 2X06 | CATTTCAA | 30.8 | –12.8 | –3.6 | –6.2 | –9.2 | –6.6 | – | – | –5.4 | –7.4 | 59 | 70 | 71 |
| 2X07 | CGCCTTCA | 47.3 | –16.9 | –3.6 | –9.8 | –13.3 | –7.1 | – | – | –3.6 | –13.3 | 66 | 75 | 72 |
| 2X08 | CGTTCCTG | 43.1 | –15.1 | –6.3 | –6.2 | –8.8 | –8.9 | – | – | –9.3 | –5.8 | 69 | 74 | 72 |
| 2X09 | TTCTTCAT | 30.9 | –12.1 | –3.6 | –5.1 | –8.5 | –6.9 | – | – | –5.4 | –6.7 | 61 | 71 | 71 |
| 2X10 | TCCTCTTA | 32.7 | –12.3 | –3.6 | –4.7 | –8.7 | –7.7 | 14.9 | 17.8 | –3.1 | –9.2 | 69 | 76 | 69 |
Strand designations are [+] for top strand and [−] for bottom strand.
TM,het = melting temperature of the target heteroduplex (50 mM Na+, 10 mM Mg2+, 1 mM nucleotide triphosphates, 10 μM each oligonucleotide).
ΔGhet = target heteroduplex free energy of formation.
ΔGhomo = most stable homoduplex (of all overhang-appended parents) free energy of formation.
ΔGhet,2° = most stable off-target heteroduplex (of all overhang-appended parents) free energy of formation.
TM,hp = highest hairpin melting temperature (of all overhang-appended parents); entries marked “–” yielded no predicted hairpin formation; no hairpins predicted for any overhang-appended [+] parent.
ΔΔGhet/homo = ΔGhet – ΔGhomo.
ΔΔGhet/het,2° = ΔGhet – ΔGhet,2°.
ΔTM,hp = TM,het – TM,hp.
OHX yield = experimentally measured ligation yield of overhang-appended parent for each set’s overhangs (OH1, OH3, and OH5 for set 1; OH2, OH4, and OH6 for set 2). OH1 = 5′-ATGG-3′; OH2 = 5′-TCA-3′; OH3 = 5′-GTT-3′; OH4 = 5′-CTA-3′; OH5 = 5′-TTC-3′; OH6 = 5′-CGC-3′.
The biochemistry of enzymatic cohesive end ligation provided a significant source of encoding language design constraints, both in ensuring the formation of heteroduplex module substrates for ligase and for enforcing directional, specific serial ligation of multiple coding sequence modules. Sequences that exhibit a heteroduplex TM (under experimental conditions of enzymatic ligation) > 30 °C should hybridize at room temperature. Discarding sequences where mfold predicts a hairpin ΔTM < 15 °C eliminates the possibility that a single strand could become trapped in intramolecular secondary structure and inhibit heteroduplex module formation. The ΔG-based considerations derived from an empirical desire to select sequences that thermodynamically favor heteroduplex module formation 10,000-fold (5.5 kcal/mol at RT) over all other predicted off-target structures. The above constraints served as a point of entry into experimental measurements of ligation yields and directionality.
We verified that the designed encoding sequence modules would serve as T4 DNA ligase substrates in an enzymatic cohesive end ligation reaction as predicted. To assess this, we performed a standard assay to measure ligation yield of a solution-phase module displaying a 5′-phosphorylated overhang to a resin-bound duplex displaying the complementary 5′-phosphorylated overhang. Reaction buffer, catalyst loading, time and temperature conditions approximated those of DESPS. The 20 example coding sequences were appended with overhangs according to set (OH1, OH3, OH5 for set 1; OH2, OH4, OH6 for set 2; Table 1 footnotes) and hybridized prior to ligation with appropriate partially complementary [−]-strand sequence to form modules for ligation. Module ligation yields with respective complementary solid-phase ligation partner averaged 70% for all overhangs investigated. Ligation yields were highly reproducible for any given overhang and were independent of the coding sequence. For example the average ligation yield of all 11XX (set 1 sequences displaying OH1) was 73 ± 2%. The 22XX (set 2 sequences displaying OH2) ligation efficiencies were similarly reproducible, though lower (62 ± 5%).
Ligation yields clustered independent of coding sequence for the 10 example sequences of each set, suggesting that the remaining 62 set 1 and 66 set 2 members will behave similarly. Furthermore, electrophoretic analysis of the ligation reaction products verified that ligase catalyzed at most one module ligation per site on solid supports. Some overhangs, while meeting the thermodynamic constraints, proved problematic in ligation assays; for example, the trinucleotide overhang 5′-CTG-3′ resulted in overhang homoligation and concomitant addition of two modules, highlighting the need to validate overhangs experimentally.
In addition to ligase biochemistry, a second source of encoding language design constraints derived from the downstream analytical limitations of current high-throughput DNA sequencing. While the read lengths of these instruments are relatively short (100–200 bases), a single analysis can yield >107 of such reads in hours and for ∼$1000. The 6-position code design meets this constraint in producing encoded messages that require a maximum read length of 98 bases: a 27-base forward primer, 12 bases for the first and sixth positions, 11 bases each for positions 2–5, and 4 bases downstream of the message for alignment. Discarding coding sequences that contain >3-base homopolymeric runs eliminates the highest source of error for pyrosequencing-based platforms.3,38 Finally, the minimum Hamming string distance of 3 between any two coding regions drastically diminishes the probability of random sequencing errors obfuscating the actual coding region identity. At least two sequencing errors within any coding region must occur to render the sequence unintelligible for decoding. The probability of such errors is platform dependent, but an average substitution error rate of 0.1% translates to a probability of 6 × 10–5, an insignificant fraction of total reads.
To demonstrate DESPS using the newly designed encoding language, we prepared a series of compounds that exhibit diversity in both scaffold configuration and isomerism. The synthesis of each compound (1–8, Chart 1) occurred in a filtration microtiter plate using bifunctional-HDNA resin (1 mg each compound) and the protocol outlined in Scheme 1. In parallel with the DESPS of each compound, we conducted DNA encoding ligation control experiments (DE+, also using bifunctional-HDNA resin) in which the complete unique encoding sequence of the compound was prepared by serial ligation of primer and appropriate encoding modules absent compound synthesis steps. DE+ beads simultaneously served as a measure of the maximum viable templates per bead for PCR amplification and as a benchmark for determining the impact of chemical synthesis conditions on the integrity of the encoding DNA. Single-bead qPCR analysis of DESPS and DE+ samples (N = 10 each analysis) measured an average of 4 × 106 DNA molecules/bead for single DE+ beads. Beads from the DESPS of 1–5 harbored an average of 2 × 105 DNA molecules/bead while DESPS beads of 6–8 harbored 4 × 103 DNA molecules/bead. Sequencing analysis of DESPS single-bead PCR products was consistent with the structure predicted using the sequence-structure database (see Supporting Information).
Chart 1. DNA-Encoded Oligomer Synthesis and Single-Bead Quantitation.
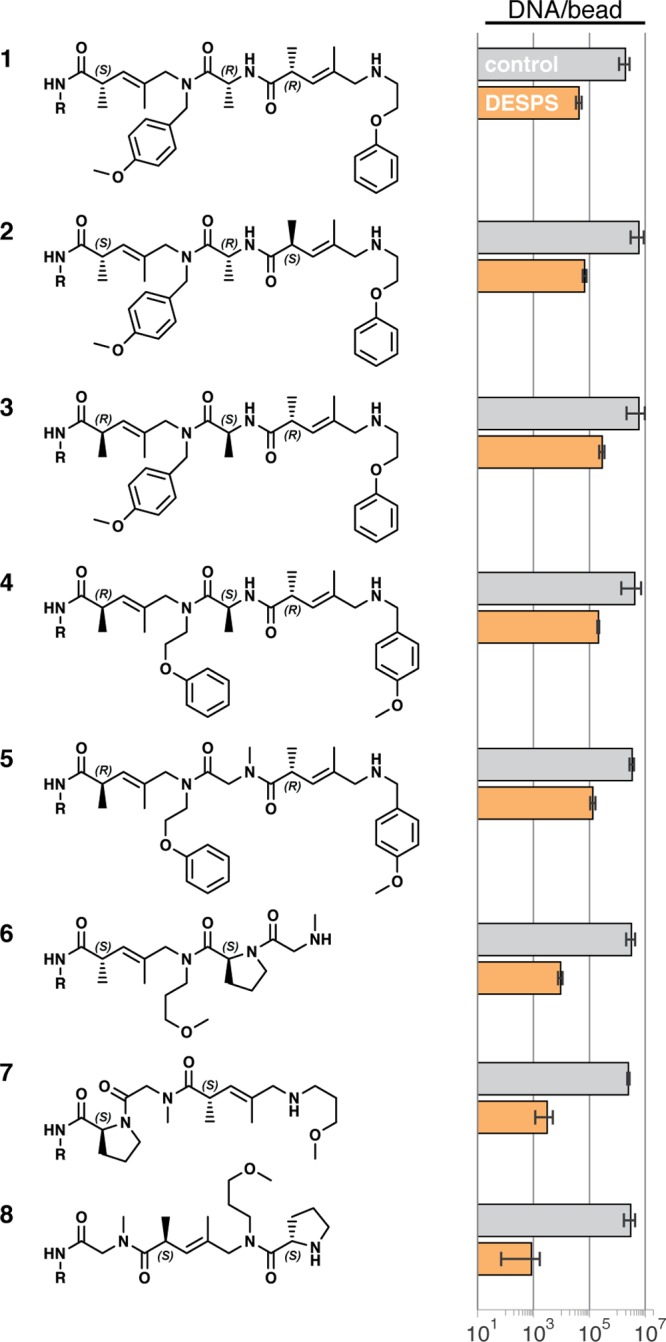
The single-bead qPCR and sequencing analysis of DESPS support the utility and practicality of employing DNA to encode the solid-phase synthesis of complex oligomers. Isomeric oligomers 1–5 feature not only a mixed scaffold of amino and pentenoic acids, but also stereochemical diversity at the three α-carbon centers and regioisomerism. Isomers 6–8 exhibit scaffold diversity by permuting the order of three scaffold types (proline, pentenoic acid, glycine). While both MALDI-TOF MS analysis of single beads and MS/MS fragmentation analysis of all purified compounds afforded unambiguous parent and fragment ions (see Supporting Information), the spectra were predictably insufficient to differentiate 1–5. The DNA sequence, on the other hand, clearly defined the synthesis history of each bead-bound oligomer, revealing the complete expected structure. Isomers 6–8 exhibited similar quality of MS and DNA sequence data, but MS/MS-ETD-based analysis did not yield interpretable fragmentation due to the internal proline. DESPS, however, is amenable to encoding and decoding this type of complex diversification, which one could encounter in a complex combinatorial library.
The large dynamic range and single-molecule sensitivity of PCR can tolerate high degrees of DNA loss, both as a result of chemical synthesis and ligation inefficiency. While different orders of chemical steps can result in a loss of 95–99.97% of maximum viable PCR templates, these losses sustained on an initial 4 × 106 viable templates per bead still provide ample remaining material for decoding. Furthermore, generation of enzymatic ligation sites via click coupling and subsequent enzymatic ligation steps is apparently quite inefficient. We expected to generate ∼1011 possible templates (calculated based on 0.004 equiv of HDNA coupling, ∼800 pmol/bead resin loading capacity, and 6% overall ligation yield over 8 steps). Despite these encoding inefficiencies and others, such as protein and substrate accessibility to bead interior sites,39 single-bead qPCR afforded structural decoding of compounds synthesized under reaction conditions with wide-ranging impact on DNA integrity.
Just as qPCR and sequencing studies verified the integrity of the encoding DNA, we next sought to demonstrate that the multiple exposures to aqueous-phase encoding conditions did not compromise either purity or yield of the synthesized compounds. LC-MS analysis of 1–8 cleaved from DESPS samples (Chart 2) revealed a predominant product peak by absorbance detection of the coumarin chromophore (λ = 330 nm) installed in the linker (R, the top branch of Figure 1A) and mass analysis of each predominant peak yielded parent ion m/z in agreement with the predicted mass of the compound. Trace side products included unreacted resin linker (a), hydrolyzed haloacid (b, d), and truncations (c, e, f). These side products appear with equal abundance in the control solid-phase syntheses (SPS+) of each compound that was performed (omitting intervening aqueous encoding conditions). DESPS of 1–5 yielded average compound purity of 48%. DESPS of 6–8 yielded average compound purity of 67%. Mass spectrometric analysis of HPLC purified 1–8 using ETD-based fragmentation (MS/MS-ETD)40 yielded z ion series sequencing data that agreed with the proposed oligomer sequence, and high-resolution MS analysis of 1–8 agreed within 3.2 ppm of the predicted exact mass.
Chart 2. DNA-Encoded Compound Purity and Side Product Identification.

Mass spectrometric and chromatographic results indicated that DESPS predominantly yields the desired product predicted by each encoded synthesis history. One might expect that exposure to aqueous conditions would compromise compound purity, however, the chromatographic analyses of SPS+ and DESPS samples of the same compound are indistinguishable (see Supporting Information), supporting the conclusion that compound impurity results solely from inefficiency of compound coupling or monomer impurity. While these side products did not obscure the product parent ion in the single-bead MALDI-TOF MS analysis of these samples, conventional combinatorial synthesis strictly relies on this analytical strategy for structure elucidation, engendering the need to prescreen monomers for minimal side product formation by using only the most efficient reactions. Additionally, de novo sequencing based on single-bead MALDI-TOF-MS/MS fragmentation data is only analytically tractable for libraries constructed from homogeneous scaffolds (e.g., polypeptides, peptoids) and devoid of mass redundancy. DESPS eliminates these concerns because structure elucidation occurs via decoding the synthesis history rather than direct mass analysis. Heterogeneous scaffolds (6–8) that would fragment unpredictably are no more difficult to decode than compounds displaying high degrees of mass redundancy (1–5). And, as long as synthesis on a single bead passaging through multiple splits of a combinatorial synthesis faithfully reproduces bulk-scale synthesis of a potential hit compound, side products could even become a source of diversity in a DESPS library.
High test compound yields and purities coupled with only modest losses of amplifiable DNA encoding tags suggested that DESPS would translate well to a combinatorial library synthesis. However, given that DESPS libraries using our language and reaction suite could easily contain one million distinct members, choice of resin scale became a critical consideration prior to moving forward. Using 160-μm resin to prepare a one-million-member library with an average redundancy of 1 would require 2 g of resin; the same synthesis using 10-μm resin would require only 0.5 mg. Miniaturization of DESPS to this scale should be limited only by our ability to generate sufficient PCR-viable templates per bead. If enzymatic reactions (e.g., DNA ligation) on TentaGel resin-bound substrate occur only on the bead surface, the 10-μm particles should yield 256-fold lower PCR templates per bead, or an average of ∼400 molecules based on our single-bead qPCR studies. While low, this amount of PCR-viable template is adequate. The only challenge with such a synthesis would be quality control. Although each 10-μm particle would display sufficient DNA for PCR detection and sequencing analysis, the 100-fmol single-bead loading capacity would not generate sufficient material for mass analysis to correlate with DNA sequence as for test compounds 1–8 above.
To address quality control concerns, we developed a mixed-scale synthesis strategy and applied it to the preparation of a modest-scale combinatorial library. We selected a linear scaffold of mixed amino acid and peptoid monomers, each displaying side chain diversity (Figure 3A). The 75 645-member library was prepared on mixed-scale resin containing both 160- and 10-μm beads. The 160-μm “quality control” (QC) beads were mixed with 10-μm “screening” beads at a bead stoichiometry of ∼1:20 000 (QC:screening). After library synthesis, QC beads were separated from screening beads via filtration for tandem DNA sequencing and mass analysis (Figure 3B). Single QC beads were first distributed into individual qPCRs. Then, each bead was subjected to TFA cleavage and MALDI-TOF mass spectrometric analysis. The QC beads yielded 1.3 ± 0.7 × 106 amplifiable DNA templates/bead (N = 26). Tandem analysis results from two QC beads demonstrated excellent correlation between DNA sequence-predicted structures (9 and 10) and predominant observed [M + H]+. Although the two DNA encoding sequences were very different and easily distinguished, the QC beads displayed compounds with mass redundancy at Pos3, which would have confounded MS/MS-based sequencing. Predicted structures for 19 QC beads agreed within 1 ppt of observed [M + H]+ (see Supporting Information), although some spectra were not as clean as those of 9 and 10, demonstrating the utility and superiority of DNA encoding over direct mass-based structure elucidation. The 10-μm screening beads yielded 1.2 ± 0.6 × 104 amplifiable DNA templates/beads (N = 33) after library synthesis, which is still easily detectable by qPCR.
Figure 3.

DNA-encoded combinatorial library plan and quality control. (A) The library scaffold features a linear arrangement of three positions for diversification (Pos1, Pos2, Pos3). Each position displays either an amino acid or N-substituted glycine. Amino acids featured Cα diversity in side chain, side chain stereochemistry, and N-methylation. The central position, Pos2, uniquely featured 1 of 6 different “linker” amino acids in addition to the L- and D- complement. N-substitution of glycine was executed with 1 of 21 different amines (gray). (B) The mixed-scale combinatorial DESPS was conducted in wells of a filtration microplate that housed a mixture of 160- and 10-μm bifunctional-HDNA library resin. The 160-μm QC beads were harvested by filtration and single beads were placed into separate wells for qPCR analysis. The resultant amplicons were purified and sequenced. The single QC beads were retrieved from qPCR supernatant, transferred to individual trifluoroacetic acid cleavage reactions, and the cleavage products subjected to mass spectrometric analysis. (C) DNA sequence data (shown as numeric identifiers) were used to predict the compound structure on each QC bead. The predicted exact mass of [M + H]+ (green) agreed with the observed predominant ion (black) in the MALDI-TOF mass spectra.
Solid-phase combinatorial synthesis and DNA-encoded libraries (DELs) represent two of the most powerful strategies for generating large nonbiological molecular libraries. To date, DELs containing diversity ranging from 105–1010 are possible by uniquely encoding single compounds with DNA.28,41,42 The solubility and information content of the DNA are the only fundamental limiting factors for library size and complexity. Furthermore, library screening throughput via solution-phase binding assays is equally impressive because it is a selection, which interrogates all members of the library simultaneously. This approach has recently gained incredible momentum as solution-phase encoded libraries have yielded ligands of, for example, sirtuins,43 tankyrase 1,44 and PAD4.45 However, the reactions used to generate DELs must solubilize DNA since the encoding DNA is the compound carrier, and HPLC purification accompanies each encoded synthesis step (which does not guarantee compound fidelity). Solid-phase synthesis, on the other hand, is compatible with a broad range of solvents, there are numerous solid-phase chemical reactions available,46 sample purification is trivial (washing), and each bead harbors numerous copies of the compound for synthesis quality control. However, solid-phase strategies rely almost exclusively on mass analysis for structure elucidation, limiting diversity (as discussed above) and analytical throughput.47
DESPS unites many of the advantages of these two approaches resulting in synergistic benefits for both encoding and synthesis. Using solid support as the compound carrier instead of DNA enables synthesis using a much wider range of solvents and reagents (e.g., activators, organic bases, reactive electrophiles). In fact, DNA information storage capacity and “DNA compatibility” of solid-phase chemical reaction conditions appear to be the only factors limiting the scope of DNA-encoded solid-phase library diversity. Even reactions that promote DNA lesions, such as deglycosylation (e.g., alkylation, acidification), nucleobase acylation, or mutagenesis (e.g., cytosine deamination), may not necessarily be incompatible with DESPS. For example, our reaction suite included both acylating and potentially alkylating reaction conditions, yet ample encoding DNA remained on each bead for PCR analysis and sequencing. Although more aggressive acylation conditions or other routine synthetic transformations, such as deprotection in organic acid, may be problematic for DESPS, the redundancy of encoding DNA per bead and sensitivity of PCR jointly confer a high degree of robustness to synthesis conditions. Exploring the full reaction scope of DESPS will require a strategy for simultaneously measuring reaction product yield and DNA integrity. Given that DESPS-compatible reactions need not be quantitative because the compound is no longer the subject of analysis for structure elucidation, and DNA template loss up to ∼99.9% over an entire library synthesis reaction sequence is tolerable, the reaction scope should be broad.
Accordingly, the design of our encoding language accommodates a large number of DESPS-compatible reactions using few coding sequences. We encoded each monomer position using two modules with each module being 1 of 10 different coding sequences. This provides an encoding capacity of 100 monomer diversity elements per position and the capability of encoding a library of 1 × 106 trimers, using only 60 encoding module stock solutions. Increasing diversity-encoding capacity with the additional optimized coding sequences is nonlinear; 15 modules per position can encode 225 diversity elements, or 1 × 107 trimers, and the module collection is still amenable for use in a 96-well microtiter plate. Increasing capacity by adding encoding positions is also a possibility. While a language based on dinucleotide overhang ligations is inherently limited to 6 encoding positions (12 nonpalindromic dinculeotides; 2 primer modules, 4 chemical diversity-encoding positions), our trinucleotide overhang-based language minimally provides the capacity for 30 diversity-encoding positions, at which point serial enzymatic ligation yield is limiting.
The mixed-scale combinatorial library synthesis introduces numerous advantages in miniaturizing the scale of both library synthesis and screening. Reduced reagent consumption at this scale not only enables usage of more expensive or designer monomers, such as the chiral chloropentenoic acids of this study,32 but also automated high-throughput flow cytometry-based screening.16,48 Oligonucleotide-encoded peptide synthesis reached this degree of miniaturization more than two decades ago,16,49 but this line of research has remained dormant likely because parallel oligonucleotide synthesis has poor step economy relative to information yield (3 synthesis steps yields 2 bits; one ligation step yielding 12 bits) and introduces numerous aggressive reaction conditions and two orthogonal protection strategies. DESPS provides a more approachable strategy for accessing the benefits of DNA-based encoding, and combined with 10-μm-scale library preparation, raises the possibility of functional screening (e.g., in microfluidic droplets50) by virtue of the solid-phase synthesis bead colocalizing many copies of one compound library member. Direct functional screening would provide a powerful alternative to solution-phase library competition binding as a mode of discovery.
We have demonstrated structure elucidation of complex non-natural oligomers from single synthesis resin particles using PCR amplification and DNA sequencing. The computationally designed encoding language ensures efficient and directional enzymatic cohesive end ligation. Furthermore, enforcing a minimum Hamming string distance of 3 between all sequences of the language and a full encoding DNA sequence length of ∼100 nucleotides facilitates downstream high-throughput sequencing. Finally, a mixed-scale combinatorial library synthesis illustrates the compatibility of DESPS with conventional split-and-pool diversification and miniaturized resin scales while enabling gold-standard mass spectrometric library QC. The widespread availability of advanced quantitative DNA amplification and high-throughput sequencing technology in combination with economical oligonucleotide synthesis sets the stage for application of combinatorial DESPS libraries to small molecule discovery.
Experimental Procedures
Materials Sources
All reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise specified. 5-Azidopentanoic acid, N-hydroxysuccinimide (NHS), 1,3-bis[tris(hydroxymethyl)methylamino]propane (Bis-Tris), 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC), sodium dodecyl sulfate (SDS), tris(hydroxymethyl)aminomethane (Tris), N,N′-diisopropylcarbodiimide (DIC), ethyl 2-cyano-2-(hydroxyimino)acetate (Oxyma), 1-hydroxy-7-azabenzotriazole (HOAt), N,N′-diisopropylethylamine (DIEA), 2,4,6-trimethylpyridine (TMP), triisopropylsilane (TIPS), α-cyano-4-hydroxycinnamic acid (HCCA), trifluoroacetic acid (TFA), triethylammonium acetate (TEAA, Life Technologies, Carlsbad, CA), M-280 streptavidin-coated magnetic resin (Life Technologies), biotin N-hydroxysulfosuccinimidyl ester (biotin-sNHS, Pierce Biotechnologies, Rockford, IL), Taq DNA polymerase (Taq, New England Biolabs, Ipswich, MA), 2′-deoxyribonucleotide triphosphate (dNTP, set of dATP, dTTP, dGTP, dCTP, Promega Corp., Milwaukee, WI), N-α-Fmoc-l-Ala-OH, N-α-Fmoc-d-Ala-OH (Bachem, Torrance, CA), N-α-Fmoc-β-(7-methoxy-coumarin-4-yl)-Ala-OH (Bachem), all amines, and Fmoc-protected amino acids for combinatorial library synthesis were used as provided. Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) was recrystallized three times in t-BuOH/H2O (1:1).51 Substituted chloropentenoic acid monomers (2S,3E)-5-chloro-2,4-dimethyl-3-pentenoic acid (S-Me-COPA) and (2R,3E)-5-chloro-2,4-dimethyl-3-pentenoic acid (R-Me-COPA) were prepared according to previously published methods.32
Buffers
Bind and wash buffer (BWB, 1 mM EDTA, 2 M NaCl, 10 mM Tris, pH 7.5), bind and wash buffer with Tween (BWBT, 1 mM EDTA, 2 M NaCl, 10 mM Tris, 0.1% Tween 20, pH 7.5), 10× Bis-Tris propane ligation buffer (BTPLB, 500 mM NaCl, 100 mM MgCl2, 10 mM ATP, 0.2% Tween 20, 100 mM Bis-Tris, pH 7.6), Bis-Tris propane wash buffer (BTPWB, 50 mM NaCl, 0.04% Tween 20, 10 mM Bis-Tris, pH 7.6), Bis-Tris propane breaking buffer (BTPBB 100 mM NaCl, 10 mM EDTA, 1% SDS, 1% Tween 20, 10 mM Bis-Tris, pH 7.6), click reaction buffer (CRB, 50% DMSO, 30 mM TEAA, 0.04% Tween 20, pH 7.5), 10X PCR buffer (2 mM each dNTP, 15 mM MgCl2, 500 mM KCl, 100 mM Tris, pH 8.3), 1× GC-PCR buffer (1× PCR buffer, 8% DMSO, 1 M betaine), saline-sodium citrate hybridization buffer (SSC, 150 mM NaCl, 15 mM citrate, 1% SDS, pH 7.6), denaturing polyacrylamide gel electrophoresis loading buffer (GLB, 6 M urea, 0.5 mg/mL bromophenol blue, 12% w/v Ficoll 400, 1× TBE buffer, pH 8.5), and crush and soak buffer (C&S, 500 mM NaCl, 1 mM EDTA, 10 mM Tris pH 7.6, 200 μL) were prepared in DI H2O.
Oligonucleotides
Oligonucleotides (Integrated DNA Technologies, Inc. Coralville, IA) were purchased as desalted lyophilate and used without further purification. Oligonucleotide ligation substrates were 5′-phosphorylated (/5Phos/). Amino-modified headpiece DNA (NH2–HDNA, /5Phos/GAGTCA/iSp9//iUniAmM//iSp9/TGACTCCC) was HPLC purified at the manufacturer and used without further purification. Oligonucleotides are indicated by “≈” followed by a 4-digit numeric identifier and “[+]” or “[−]” strand designation. The first digit groups the oligonucleotides by set: set 0 contains PCR primer sequences and sets 1 and 2 contain synthesis encoding sequences. The second digit denotes position in the encoding region. The third and fourth digits index the different coding sequences in each set. Oligonucleotide paired (OP) stock solutions of complementary oligonucleotides (60 μM [+], 60 μM [−], 50 mM NaCl, 1 mM Bis-Tris, pH 7.6) were heated (5 min, 60 °C) and cooled to ambient (5 min, RT) before each usage. These reagents are indicated with a “[±]” double-stranded designation. Table T1 presents a concise look-up table for generating all oligonucleotide sequences. For example, ≈1302[+] is from set 1, and built by concatenating overhang X3XX[+] “/5Phos/GTT” with encoding sequence 1X02[+] “ACGGAGCA” to yield the sequence “/5Phos/GTTACGGAGCA.” The complement, ≈1302[−], is also from set 1 and built by concatenating overhang 1302[−] “/5Phos/TAG” with encoding sequence 1X02[−] “TGCTCCGT” to yield the sequence “/5Phos/TAGTGCTCCGT.” Combining ≈1302[−] and ≈1302[+] and thermally processing as above yields the double-stranded coding module ≈1302[±], a position 3 OP stock solution (OP3) of set 1 parent sequence 02 (1X02). All sequences are written in the 5′ to 3′ direction.
Azido Headpiece DNA Synthesis, Purification, and Characterization
NH2–HDNA (300 nmol) was dissolved in phosphate buffer (1M, pH 8.0, 240 μL). 5-azidopentanoic acid NHS ester was prepared by dissolving NHS (9.6 μmoles), EDC (9.6 μmoles), and 5-azidopentanoic acid (7.2 μmoles) in DMF (20 μL) and incubating (30 min, 60 °C). The NH2–HDNA acylation reaction was assembled by sparging (N2, 1 min) the phosphate-buffered NH2–HDNA, followed by addition of 5-azidopentanoic acid N-hydroxysuccinimidyl ester solution (22 μL), and incubation (2 h, RT). A fresh solution of 5-azidopentanoic acid N-hydroxysuccinimidyl ester was prepared as described above, added to the acylation reaction, and the reaction incubated (1 h, RT). The reaction was quenched (1 M Tris, pH 7.6, 100 μL) and incubated (5 min, 60 °C). Azido-HDNA (N3–HDNA) product was precipitated twice in ethanol. The pellet was dried under N2, resuspended (20 mM TEAA, pH 8.0), and purified at semipreparative scale using reversed-phase HPLC (X-Bridge BEH C18 column, 10 mm × 150 mm, 130 Å, 5 μm, Waters Corp., Milford, MA) with gradient elution (mobile phase A H2O, 20 mM TEAA, pH 8; mobile phase B ACN; 5—12% B, 24 min). A product fraction aliquot (1 μL) was spotted to a MALDI-TOF MS target plate, dried, covered with matrix solution (18 mg/mL trihydroxyacetophenone, 7 mg/mL ammonium citrate dibasic in 50:50 acetonitrile/H2O) and mass analyzed via MALDI-TOF MS (Microflex, Bruker Daltonics Inc., Billerica, MA, Figure S2).
Biotin–HDNA and Biotin–HDNA Magnetic Resin Preparation
NH2–HDNA (56 nmol) was dissolved in carbonate buffer (60 mM, pH 8.5, 300 μL). Biotin–sNHS (2.25) was dissolved in DI H2O (225 μL), combined with NH2–HDNA solution and incubated (16 h, 4 °C). The DNA was precipitated in ethanol, resuspended in buffer (10 mM Tris, pH 8, 200 μL), and used without further purification. Streptavidin-coated magnetic resin (0.5 mg, 100 pmoles of sites for oligonucleotide) was washed (BWB, 2 × 200 μL), resuspended (BWB, 100 μL), combined with crude biotin–HDNA (120 pmol) and incubated (15 min, RT). The biotin–HDNA magnetic resin was washed (BWB + 0.1% Tween 20, 3 × 200 μL) and resuspended in BTPWB (200 μL).
Bifunctional Resin Synthesis and Characterization
TentaGel Rink amide resin (160 μm, 0.41 mmol/g, 600 mg, Rapp-Polymere, Tuebingen, Germany) was transferred to a fritted syringe (10 mL, Torviq, Niles, MI) and swelled in DMF (1 h, RT). Linker construction proceeded via iterative cycles of manual solid-phase peptide synthesis. Each cycle included: (1) 9-fluorenylmethoxycarbonyl (Fmoc) deprotection (20% piperidine in DMF, 2 × 6 mL, 5 min first aliquot, 15 min second aliquot); (2) N-α-Fmoc-amino acid (1.23 mmol, 2 mL DMF) activation with DIC/Oxyma/DIEA (1.23 mmol/1.23 mmol/2.46 mmol), and incubation (2 min, RT); (3) N-α-Fmoc-amino acid coupling to resin by transferring activated acid (6 mL) to resin and incubating (1 h, 37 °C). After each deprotection and coupling step, reactants were expelled and the resin washed (DMF, 3 × 5 mL; DCM, 1 × 5 mL; DMF, 1 × 5 mL). N-α-Fmoc-β-(7-methoxy-coumarin-4-yl)-Ala-OH, N-α-Fmoc-Gly-OH, N-α-Fmoc-Arg(Pbf)-OH, and N-α-Fmoc-Gly-OH were coupled sequentially. The pendant Fmoc-protected amine was deprotected and the resin washed (see above). The deprotected N-terminus was acylated by preparing a solution of bromoacetic acid (9.6 mmol) and DIC (4.8 mmol) in DMF (6 mL) to activate the bromoacid (2 min, RT), transferring the activated bromoacid to the resin and incubating (15 min, 37 °C). The bromoacetylated resin was washed, propargylamine solution (1 M in DMF, 6 mL) was transferred to resin, and the resin was incubated (2 h, 37 °C). The resin was washed and the product secondary amine acylated with addition of N-α-Fmoc-Gly-OH (see above) in DMF (6 mL) and incubation (1 h, 37 °C). The resin was washed and an aliquot (0.5 mg) was transferred to a clean fritted syringe, washed (DCM, 2 × 1 mL), and dried in vacuo. Cleavage cocktail (90% TFA, 10% H2O, 1 mL) was added to the dried resin and incubated (1 h, RT). Cleaved linker was expelled, concentrated in vacuo, resuspended (40% ACN, 0.1% TFA in H2O, 200 μL), and analyzed using reversed-phase HPLC (X-Bridge BEH C18 column, 10 mm × 150 mm, 130 Å, 5 μm) with gradient elution (mobile phase A, ACN; mobile phase B, 0.1% TFA in H2O; 5–65% A, 30 min) and UV absorbance detection (λ = 330 nm). The product fraction was collected, diluted (10:1, 50% ACN in 0.1% formic acid H2O), and infused directly into the electrospray source of a tandem mass spectrometer equipped with an electron transfer dissociation (ETD) module (LTQ-XL, Thermo Fisher Scientific, Waltham, MA). The doubly charged [M + 2H]2+ precursor ion was isolated (width = 5 m/z) followed by ETD activation (200 ms), with supplemental activation mode enabled. Spectra (n = 100, 0.33 Hz) were averaged during continuous infusion (2 μL/min). See Figure S2.
Bifunctional–HDNA Resin Preparation
Bifunctional resin (40 mg, 17.2 μmol) was aliquoted, washed (CRB, 3 × 200 μL), and equilibrated (CRB, 2 mL, 1 h, RT). Cu(II) sulfate (19.8 μmol), TBTA (34.4 nmol), and ascorbic acid (98.9 μmol) were dissolved in DMSO (66% in H2O, 312 μL). N3–HDNA (68.8 nmol) and ascorbic acid (264 nmol) were dissolved in TEAA buffer (200 mM, pH 7.5, 132 μL). Bifunctional resin was washed (CRB, 2 mL), resuspended (CRB, 3.05 mL), combined with Cu(II) solution (312 μL), mixed, and incubated with rotation (5 min, 40 °C, 15 rpm). The resin was centrifuged (30 s, 1000 rcf), N3–HDNA solution added (132 μL, 0.004 eq. to bead sites), and the reaction immediately mixed with vortexing and incubated with rotation (4 h, 40 °C, 15 rpm). Resin was centrifuged (30 s, 1000 rcf), the supernatant removed, the resin washed (BTPBB, 3 × 2 mL) and incubated with rotation (12 h, RT, 15 rpm). Resin was washed (BTPWB, 3 × 1 mL) then transferred to a fritted syringe, washed (DI H2O, 3 × 1 mL; DMF, 3 × 1 mL), and stored (DMF, −20 °C).
Bifunctional–HDNA Resin Characterization
Bifunctional resin (0.4 mg) and bifunctional-HDNA resin (0.4 mg) were aliquoted into individual fritted spin columns (Mobicol Classic, MoBiTec GmbH, Goettingen, Germany) then washed (BTPWB, 2 × 400 μL; BTPLB, 1 × 100 μL). Biotin–HDNA magnetic resin (25 μg) was aliquoted to a separate tube and washed (BTPWB, 2 × 400 μL; BTPLB, 1 × 50 μL). A first enzymatic oligonucleotide ligation reaction, consisting of ≈0001[±] (1.4 nmol), ≈1103[±] (1.4 nmol), ≈2204[±] (1.4 nmol), ≈1304[±] (1.4 nmol), and T4 DNA ligase (334 U), was combined (BTPLB, 815 μL) and aliquoted to bifunctional resin (400 μL), bifunctional–HDNA resin (400 μL), and biotin–HDNA magnetic resin (15 μL). Resin samples were incubated with rotation (4 h, RT, 8 rpm), then washed (BTPBB, 3 × 400 μL) and incubated with rotation (12 h, RT, 8 rpm). The resin samples were washed (DI H2O, 2 × 400 μL, BTPWB, 2 × 400 μL; BTPLB, 1 × 150 μL). A second ligation reaction, consisting of ≈2402[±] (1.4 nmol), ≈1501[±] (1.4 nmol), ≈2601[±] (1.4 nmol), ≈0701[±] (1.4 nmol), and T4 DNA ligase (334 U), was combined (BTPLB, 815 μL) and aliquoted to bifunctional resin (400 μL), bifunctional–HDNA resin (400 μL), and biotin–HDNA magnetic resin (15 μL). Resin samples were incubated with rotation (4 h, RT, 8 rpm), then washed (BTPBB, 3 × 400 μL; MeOH, 1 × 200 μL; BTPWB, 3 × 400 μL). Quantitative PCR (qPCR) mixture contained Taq (0.05 U/μL), oligonucleotide primers 5′-GCCGCCCAGTCCTGCTCGCTTCGCTAC-3′ and 5′-GTGGCACAACAACTGGCGGGCAAAC-3′ (0.3 μM each), and SYBR Green (0.1×, Life Technologies) in PCR buffer (1×). Single resin particles (bifunctional or bifunctional–HDNA) in BTPWB (1 μL) were added to separate amplification reaction wells (20 μL, 5 replicates for bifunctional resin sample, 8 replicates for bifunctional–HDNA resin sample). Each resin supernatant (1 μL) was added to respective negative control reaction wells (20 μL). Biotin–HDNA magnetic resin (25 pg, 1 μL) was added to a positive control amplification reaction well (20 μL). Template standards (100 amol, 10 amol, 1 amol, 100 zmol, 10 zmol, 1 zmol, 100 ymol, and 10 ymol in BTPWB) were added to separate reaction wells (20 μL). The reaction plate was thermally cycled (96 °C, 10 s; [95 °C, 20 s; 72 °C, 35 s] × 35 cycles; C1000 Touch Thermal Cycler, Bio-Rad, Hercules, CA) with fluorescence monitoring (CFX-96 Real-Time System, Bio-Rad). Samples were quantitated (CFX Manager, version 3.1, Bio-Rad) using single-threshold Cq determination mode (100 RFU). Supernatant background was subtracted from respective single-particle measurements. Background-subtracted replicates were averaged and %RSD calculated.
DNA-Encoded Control Compound Synthesis
Wells of a filtration microtiter plate (MultiScreen Solvinert Filter Plate, Millipore, Billerica, MA) were each wetted (DCM, 100 μL) then filled with resin aliquots (1 mg) for synthesis. DNA encoding positive control (DE+) wells (A1-A8) and DNA encoded solid-phase synthesis (DESPS) wells (B1–B8) contained bifunctional-HDNA resin. Solid-phase synthesis positive control (SPS+) wells (C1–C8) contained bifunctional resin. All resin samples were washed (DMF, 2 × 150 μL). Fmoc was removed (20% piperidine in DMF, 2 × 150 μL), the plate sealed (adhesive aluminum foil, catalog no. 60941-126, VWR) and incubated with shaking (5 min first aliquot, 15 min second aliquot, RT, 700 rpm) on a vortex mixer (MS 3 digital, IKA, Wilmington, NC) then washed (DMF, 3 × 150 μL; DCM, 3 × 150 μL; DMF, 3 × 150 μL). DESPS and DE+ resin samples were washed (BTPWB, 3 × 150 μL), sealed and incubated with shaking (1 h, RT, 800 rpm), and then washed (BTPLB, 1 × 150 μL). SPS+ resin samples were resuspended (DMF, 150 μL).
An encoding solution consisting of ≈0001[±] (1.5 nmol), appropriate OP stocks ≈11XX[±] (1.5 nmol, Table T2) and T4 DNA ligase (180 U) was combined for each sample in BTPLB (150 μL), then added to resin, sealed and incubated with shaking (14 h, RT, 700 rpm). DESPS and DE+ resin samples were washed (BTPWB, 3 × 150 μL; MeOH, 1 × 150 μL; DMF, 3 × 150 μL) and incubated with shaking (2 h, 37 °C, 700 rpm). DE+ resin samples were resuspended (DMF, 150 μL). DESPS and SPS+ were acylated by preparing a solution of the appropriate acid monomer (80 mM), DIC (250 mM), Oxyma (80 mM), and TMP (80 mM) in DMF (150 μL), incubating (5 min, RT), then the solution was added to resin in wells, the plate sealed and incubated with shaking (3 h, 37 °C, 700 rpm). DESPS and SPS+ samples were washed (DMF, 3 × 150 μL) and samples with a terminal chloride (B01–B06, B08, C01–C06, C08) were suspended in a solution of the appropriate primary amine (1 M in DMF, 150 μL), the plate sealed and incubated with shaking (3 h, 37 °C, 700 rpm). DESPS and SPS+ samples were washed (DMF, 3 × 150 μL; DCM, 2 × 150 μL; DMF, 3 × 150 μL). To remove Fmoc from appropriate DESPS and SPS+ samples (B07, C07), deprotection solution was added, the plate sealed, and incubated with shaking as before. DESPS and SPS+ resin samples were washed (DMF, 3 × 150 μL; DCM, 2 × 150 μL; DMF, 3 × 150 μL). DESPS and DE+ resin samples were washed (BTPWB, 3 × 150 μL) and incubated with shaking (1 h, RT, 700 rpm). DESPS and DE+ resin samples were washed (BTPLB, 1 × 150 μL). SPS+ resin samples were resuspended (DMF, 150 μL). An encoding solution consisting of appropriate OP stock ≈22XX[±] (1.5 nmol, Table T2) and T4 DNA ligase (90 U) was combined for each DESPS and DE+ sample in BTPLB (150 μL), then added to resin, the plate sealed and incubated with shaking (3 h, RT, 700 rpm). DESPS and DE+ resin samples were washed (BTPWB, 3 × 150 μL; BTPLB, 1 × 150 μL). SPS+ resin samples were resuspended (DMF, 150 μL).
An encoding solution, consisting of appropriate OP stock ≈13XX[±] (1.5 nmol, Table T2) and T4 DNA ligase (90 U) was combined for each DESPS and DE+ sample in BTPLB (150 μL), then added to resin, the plate sealed and incubated with shaking (3 h, RT, 700 rpm). DESPS and DE+ resin samples were washed (BTPWB, 3 × 150 μL; MeOH, 1 × 150 μL; DMF, 3 × 150 μL). All resin samples were resuspended (DMF, 150 μL), and incubated (8 h, RT, 700 rpm). DESPS and SPS+ were washed (DMF, 1 × 150 μL) and incubated with shaking (1 h, RT, 700 rpm). DESPS and SPS+ were acylated by preparing a solution of the appropriate acid monomer (80 mM), DIC (250 mM), Oxyma (80 mM), and TMP (80 mM) in DMF (150 μL), incubating (5 min, RT), then the solution was added to resin in wells, the plate sealed and incubated with shaking (3 h, 37 °C, 700 rpm). DESPS and SPS+ samples were washed (DMF, 3 × 150 μL) and samples with a terminal chloride (B05, B07–B08, C05, C07–C08) were suspended in a solution of the appropriate primary amine (1 M in DMF, 150 μL), then plate sealed and incubated with shaking (3 h, 37 °C, 700 rpm). DESPS and SPS+ samples were washed (DMF, 3 × 150 μL; DCM, 2 × 150 μL; DMF, 3 × 150 μL). Fmoc was removed from appropriate DESPS and SPS+ samples (B01–B04, B06, C01–C04, C06) by adding deprotection solution, the plate was sealed and incubated with shaking as before. DESPS and SPS+ resin samples were washed (DMF, 3 × 150 μL; DCM, 2 × 150 μL; DMF, 3 × 150 μL). DESPS and DE+ resin samples were washed (BTPWB, 3 × 150 μL) and incubated with shaking (1 h, RT, 700 rpm). DESPS and DE+ resin samples were washed (BTPLB, 1 × 150 μL). SPS+ resin samples were resuspended (DMF, 150 μL). An encoding solution, consisting of appropriate OP stock ≈24XX[±] (1.5 nmol, Table T2) and T4 DNA ligase (90 U) was combined for each DESPS and DE+ sample in BTPLB (150 μL), then added to resin, the plate sealed and incubated with shaking (3 h, RT, 700 rpm). DESPS and DE+ resin samples were washed (BTPWB, 3 × 150 μL; BTPLB, 1 × 150 μL). SPS+ resin samples were resuspended (DMF, 150 μL).
An encoding solution, consisting of appropriate OP stock ≈15XX[±] (1.5 nmol, Table T2) and T4 DNA ligase (90 U) was combined for each DESPS and DE+ sample in BTPLB (150 μL), then added to resin, the plate sealed and incubated with shaking (3 h, RT, 700 rpm). DESPS and DE+ resin samples were washed (BTPWB, 3 × 150 μL; MeOH, 1 × 150 μL; DMF, 3 × 150 μL). All resin samples were resuspended (DMF, 150 μL), and incubated (8 h, RT, 700 rpm). DESPS and SPS+ were washed (DMF, 1 × 150 μL) and incubated with shaking (1 h, RT, 700 rpm). DESPS and SPS+ were acylated by preparing a solution of the appropriate acid monomer (80 mM), DIC (250 mM), Oxyma (80 mM), and TMP (80 mM) in DMF (150 μL), incubating (5 min, RT), then the solution was added to resin in wells, the plate sealed and incubated with shaking (3 h, 37 °C, 700 rpm). DESPS and SPS+ samples were washed (DMF, 3 × 150 μL) and samples with a terminal chloride (B01–B07, C01–C07) were suspended in a solution of the appropriate primary amine (1 M in DMF, 150 μL), then plate sealed and incubated with shaking (3 h, 37 °C, 700 rpm). DESPS and SPS+ samples were washed (DMF, 3 × 150 μL; DCM, 2 × 150 μL; DMF, 3 × 150 μL). Fmoc was removed from appropriate DESPS and SPS+ samples (B08, C08) by adding deprotection solution, the plate was sealed and incubated with shaking as before. DESPS and SPS+ resin samples were washed (DMF, 3 × 150 μL; DCM, 2 × 150 μL; DMF, 3 × 150 μL). DESPS and DE+ resin samples were washed (BTPWB, 3 × 150 μL) and incubated with shaking (1 h, RT, 700 rpm). DESPS and DE+ resin samples were washed (BTPLB, 1 × 150 μL). SPS+ resin samples were resuspended (DMF, 150 μL).
An encoding solution, consisting of appropriate OP stocks ≈26XX[±] (1.5 nmol, Table T2), ≈0701[±] (1.5 nmol), and T4 DNA ligase (180 U) was assembled for each sample in BTPLB (150 μL), then added to resin, sealed and incubated with shaking (3 h, RT, 700 rpm). DESPS and DE+ resin samples were washed (BTPWB, 3 × 150 μL; MeOH, 1 × 150 μL;DMF, 3 × 150 μL) and incubated with shaking (12 h, RT, 700 rpm).
DE+ resin samples were washed (BTPWB, 3 × 150 μL), then transferred to separate tubes, resuspended (BTPWB, 500 μL), and incubated with rotation (14 h, RT, 8 rpm). DESPS resin samples were resuspended in filter plate wells (DMF, 150 μL), then ∼20 beads from each sample were transferred to separate tubes and washed (BTPWB, 3 × 500 μL).
Single-Bead Mass Spectrometric Analysis
Single resin beads were aliquoted from DESPS wells (B01–B08) into separate tubes, washed (DCM, 2 × 100 μL), and dried in vacuo. Cleavage cocktail (90% TFA, 5% DCM, 5% TIPS, 50 μL) was added to the dried resin, incubated (1 h, RT), and the sample was evaporated to dryness in vacuo. Residue was resuspended (50% ACN, 0.1% TFA in H2O, 1 μL) and spotted onto a MALDI target plate, dried, covered with HCCA matrix solution (1.5 mg/mL in 2:1 ACN/0.1% TFA in DI H2O), dried, and analyzed via MALDI-TOF MS (Microflex, Figure S4).
LC-MS Analysis and MS/MS Fragmentation Analysis of Model Compounds 1–8
DESPS and SPS resin samples (0.5 mg each) were aliquoted to separate fritted spin columns, washed (DCM, 3 × 400 μL), and dried in vacuo. Cleavage cocktail (90% TFA, 5% DCM, 5% TIPS, 400 μL) was added to the dried resin and incubated (1 h, RT). Cleavage product was collected, evaporated to dryness in vacuo, and resuspended (40% ACN, 0.1% TFA in H2O, 200 μL). Resuspended cleavage product (10 μL) was analyzed using LC-MS (Zorbax SB-C18, 4.6 × 100 mm, 80 Å, 3.5 μm, Agilent, Santa Clara, CA) with gradient elution (mobile phase A 0.1% formic acid in 5% ACN, 95% H2O; mobile phase B 0.1% formic acid in 95% ACN, 5% H2O; 0–55% B, 25 min), absorbance detection (λ = 330 nm) and total ion count. Cleavage product aliquots were also purified using reverse-phase HPLC (X-Bridge BEH C18 column, 10 mm × 150 mm, 130 Å, 5 μm) with gradient elution (mobile phase A, ACN; mobile phase B, 0.1% TFA in H2O; 5—65% A, 30 min) and absorbance detection (λ = 330 nm). The product fraction was collected, diluted (10:1, 50% ACN in 0.1% formic acid H2O), and infused directly for ETD-MS/MS fragmentation analysis (see above, Figures S5–S14).
Side Product Standard Synthesis
Truncation side-product 4b was prepared using bifunctional resin (1 mg) aliquoted to a fritted spin column and washed (DMF, 2 × 150 μL). Fmoc was removed (see above), and the resin was washed (DMF, 3 × 400 μL; DCM, 3 × 400 μL; DMF, 3 × 400 μL). The resin was acylated with (2R,3E)-5-chloro-2,4-dimethyl-3-pentenoic acid (see above), washed (DMF, 3 × 400 μL; H2O, 2 × 400 μL), and the terminal alyllic chloride hydrolyzed (1 mM NaOH, 0.02% Tween-20 in H2O, 500 μL, 16 h, 37 °C). Resin was washed (H2O, 3 × 400 μL; DMF, 3 × 400 μL; DCM, 2 × 400 μL), then incubated with rotation (DCM, 400 μL, 30 min, RT, 8 rpm), and dried in vacuo. Cleavage cocktail (90% TFA, 5% DCM, 5% TIPS, 300 μL) was added to the dried resin, incubated (1 h, RT), and cleavage product was evaporated to dryness in vacuo. Dried cleavage product was resuspended (10% ACN, 0.1% formic acid in H2O, 400 μL) and analyzed using LC-MS (see above) yielding an extracted-ion chromatogram (811 m/z).
Control Compound Encoding DNA Quantitation and Sequencing
DESPS and DE+ resin samples were aliquoted to separate tubes, washed (BTPWB, 3 × 500 μL) and incubated with rotation (1 h, RT, 8 rpm). qPCR mixture contained Taq (0.05 U/μL), oligonucleotide primers 5′-GCCGCCCAGTCCTGCTCGCTTCGCTAC-3′ and 5′-GTGGCACAACAACTGGCGGGCAAAC-3′ (0.3 μM each), and SYBR Green (0.1×, Life Technologies) in PCR buffer (1×). Single resin beads (DESPS or DE+) in BTPWB (1 μL) were added to separate amplification reactions (20 μL, 10 replicates each resin type for each compound). Each compound sample supernatant (1 μL) was added to respective negative control reactions (20 μL). Template standard solutions in BTPWB (1 fmol, 10 amol, 100 zmol, 1 zmol, 10 ymol) were added to separate reactions (20 μL). Reactions were thermally cycled ([95 °C, 20 s; 72 °C 35 s] x 35 cycles) with fluorescence monitoring and quantitated as above. DESPS single resin PCR samples (6 μL) were purified by denaturing PAGE (8% 19:1 polyacrylamide:bis, 8 M Urea in 1X TBE, 6 W, 30 min). Polyacrylamide gel slices containing 123-nt DNA products were excised, eluted in C&S buffer (200 μL) and incubated with rotation (14 h, RT, 8 rpm). PCR mixture contained Taq (0.05 U/μL), oligonucleotide primers 5′-GTTTTCCCAGTCACGAC-3′ (0.3 μM) and 5′-GTGGCACAACAACTG-3′ (0.28 μM) and 5′-CGCCAGGGTTTTCCCAGTCACGACCAACCACCCAAACCACAAACCCAAACCCCAAACCCAACACACAACAACAGCCGCCCAGTCCTGCTCGCTTCGCTAC-3′ (0.02 μM, FOX primer), in GC-PCR buffer (1×). PAGE-purified DNA templates (see above) in C&S (2 μL) were added to separate amplification reaction wells (20 μL). Each compound sample supernatant (1 μL) was added to respective negative control reactions (20 μL). Reactions were thermally cycled ([95 °C, 20 s; 52 °C, 15 s; 72 °C, 20 s] × 30 cycles), PCR products were purified (QIAquick PCR purification kit, QIAGEN, Valencia, CA) and sequenced using the M13F(−41) primer (GeneWiz, South Plainfield, NJ). Sequencing reads were trimmed to remove all called bases prior to the opening primer sequence (5′-GCCGCCCAGTCCTGCTCGCTTCGCTAC-3′) of the encoding region. DESPS sequence trace quality scores were averaged within the compound encoding region (positions 28–98) for each sample. Sequences were aligned to a degenerate reference sequence (5′-GCCGCCCAGTCCTGCTCGCTTCGCTACATGGNNNNNNNNTCANNNNNNNNGTTNNNNNNNNCTANNNNNNNNTTCNNNNNNNNCGCNNNNNNNNGCCTGTTTGCCCGCCAGTTGTTGTGCCAC-3′) and the encoding regions (5′-NNNNNNNN-3′) were matched to the structure-identifier lookup table (Table T3) to assign the synthesis history for each compound (Figures S15 and S16).
Mixed-Scale Library Resin Preparation
TentaGel M NH2 resin (10 μm, 0.23 mmol/g, 30 mg, Rapp-Polymere) was mixed with TentaGel MB Rink amide resin (160 μm, 0.41 mmol/g, 5 mg, Rapp-Polymere) and transferred to fritted spin-column (Mobicol Classic, large filter, 10-μm pore size) and swelled in DMF (1 h, RT). Linker construction proceeded via iterative cycles of manual solid phase peptide synthesis. Each cycle included (1) Fmoc deprotection (20% piperidine in DMF, 2 × 6 mL, 5 min first aliquot, 15 min second aliquot); (2) N-α-Fmoc-amino acid (90 μmol, 500 μL DMF) activation with DIC/Oxyma/DIEA (90 μmol/90 μmol/180 μmol), and incubation (2 min, RT); (3) N-α-Fmoc-amino acid coupling to resin by transferring activated acid (500 μL) to resin and incubating while rotating (1 h, 37 °C, 8 rpm). After each deprotection and coupling step, reactants were expelled and the resin washed (DMF, 3 × 5 mL; DCM, 1 × 5 mL; DMF, 1 × 5 mL). N-α-Fmoc-Gly-OH, N-α-Fmoc-Tyr(tBu)-OH, N-α-Fmoc-Arg(Pbf)-OH, and N-α-Fmoc-Gly-OH, were coupled sequentially. The pendant Fmoc-protected amine was deprotected and the resin washed (see above). The deprotected N-terminus was acylated by preparing a solution of bromoacetic acid (90 μmol) and DIC (90 μmol) in DMF (500 μL) to activate the bromoacid (2 min, RT), transferring the activated bromoacid (500 μL) to the resin and incubating (15 min, 37 °C). The bromoacetylated resin was washed, propargylamine solution (1 M in DMF, 500 μL) was transferred to resin, and the resin was incubated with rotation (2 h, 37 °C, 8 rpm). The resin was washed, then N-α-Fmoc-Gly-OH and N-α-Fmoc-Glu(OAll)-OH were coupled sequentially as above. The resin was washed, briefly sonicated, then an aliquot was filtered (150-μm mesh, CellTrics 150 μm, Partec, Sysmex America Inc., Lincolnshire, IL). The 160-μm particles (1 mg) were collected into a filtered spin column, washed (DCM, 4 × 400 μL), and dried in vacuo. Cleavage cocktail (90% TFA, 5% DCM, 5% TIPS, 50 uL) was added to dried 160-μm library resin sample, and incubated (RT, 1 h). Cleaved linker was eluted into a tube, concentrated in vacuo, resuspended (40% ACN, 0.1% TFA in H2O, 200 μL), and analyzed for purity using reversed-phase HPLC (X-Bridge BEH C18 column, 4.6 mm × 100 mm, 130 Å, 5 μm) with gradient elution (mobile phase A, ACN; mobile phase B, 0.1% TFA in H2O; 10–65% A, 40 min) and fluorescence detection (λex = 274 nm, λem = 303 nm).
Mixed-Scale Bifunctional–HDNA Library Resin Preparation
The mixed-scale library resin (10 μm, 0.23 mmol/g, 30 mg; 160 μm, 0.41 mmol/g, 4 mg) was transferred to a 5 mL tube, washed (CRB, 3 × 200 μL), and equilibrated (CRB, 2 mL, 1 h, RT). Cu(II) sulfate (9.5 μmol), TBTA (17 nmol), and ascorbic acid (47.8 μmol) were dissolved in DMSO (66% in H2O, 150 μL). N3–HDNA (33.3 nmol) and ascorbic acid (97 nmol) were dissolved in TEAA buffer (300 mM, pH 7.5, 132 μL). Bifunctional library resin was washed (CRB, 2 mL), then resuspended in (CRB, 1.8 mL), combined with Cu(II) solution (150 μL), mixed, and incubated with rotation (5 min, 40 °C, 15 rpm). The resin was centrifuged (60 s, 2000 rcf), N3–HDNA solution added (49 μL, 0.004 equiv N3–HDNA to bead sites), and the reaction immediately mixed with vortexing and incubated with rotation (16 h, 40 °C, 15 rpm). The resin was centrifuged (60 s, 2000 rcf), the supernatant removed, the resin washed (BTPBB, 3 × 2 mL) and incubated with rotation (24 h, RT, 15 rpm). Resin was washed (BTPWB, 3 × 1 mL) then transferred to a fritted syringe, washed (50:50 DI H2O/DMF, 3 × 1 mL; DMF, 3 × 1 mL) and stored (DMF, −20 °C).
Mixed-Scale Bifunctional–HDNA Library Resin Characterization
Bifunctional-HDNA library resin (10 μm, 0.4 mg; 160 μm, 0.05 mg) and bifunctional library resin (10 μm, 0.4 mg; 160-μm, 0.05 mg) were aliquoted into separate fritted spin columns (Mobicol Classic, MoBiTec GmbH) then washed (BTPWB, 2 × 400 μL; BTPLB, 1 × 100 μL). Biotin–HDNA magnetic resin (25 μg) was aliquoted into a separate tube and washed (BTPWB, 2 × 400 μL; BTPLB, 1 × 50 μL). Enzymatic oligonucleotide ligation was performed on the three resin aliquots as described previously for bifunctional-HDNA resin characterization. The samples were washed (BTPBB, 3 × 400 μL; MeOH, 1 × 200 μL; BTPWB, 3 × 400 μL). The mixed-scale resin samples were filtered (150-μm mesh, CellTrics 150 μm, Partec) and the 160-μm resin particles collected and stored (BTPWB, 700 μL, 4 °C). The eluted 10-μm resin samples were sonicated, vortexed, filtered (20-μm mesh, CellTrics 20 μm) and stored (BTPWB, 700 μL, 4 °C). Dilutions of each filtered 10-μm resin sample were prepared in BTPWB, bead concentration was determined by hemocytometer and normalized (∼100 beads/μL, BTPWB). Conditions for qPCR analysis were as previously described. Single 160-μm resin particles (bifunctional library resin or bifunctional-HDNA library resin) in BTPWB (1 μL) were added to separate amplification reaction wells (20 μL, 5 replicates for each sample). Quantitated 10-μm resin particles (79 beads/μL, bifunctional library resin; 93 beads/μL, bifunctional-HDNA library resin) in BTPWB (1 μL) were added to separate amplification reaction wells (20 μL, 5 replicates for each sample). Each resin sample supernatant (1 μL) was added to respective negative control reaction wells (20 μL). Biotin-HDNA magnetic resin (25 pg, 1 μL) was added to a positive control amplification reaction well (20 μL). Thermal cycling conditions and analysis were performed as previously described.
DNA-Encoded Solid-Phase Combinatorial Library Synthesis
Mixed-scale bifunctional-HDNA library resin (10 μm, 0.22 mmol/g, 30 mg; 160 μm, 0.41 mmol/g, 4 mg) was aliquoted to a fritted spin column (Mobicol Classic, MoBiTec GmbH), washed (DMF, 1 × 500 μL), Fmoc was removed (20% piperidine in DMF, 2 × 500 μL, 5 min first aliquot, 15 min second aliquot), and the resin was washed (DMF, 3 × 500 μL). Wells of two filtration microtiter plates (MultiScreen Solvinert Filter Plate, Millipore) were each wetted (DCM, 100 μL) then bifunctional-HDNA library resin aliquots were transferred to the two plates. The first plate (PLATE 1) contained aliquots for acylation with chloroacetic acid (20 wells, 0.75 mg 10-μm resin, 0.1 mg 160-μm resin, 0.21 μmol total bead loading capacity). The second plate (PLATE 2) contained aliquots for acylation with Fmoc-protected amino acids (22 wells, 0.68 mg 10-μm resin, 0.09 mg 160-μm resin, 0.19 μmol total bead loading capacity). Resin was acylated by preparing a solution of the appropriate acid monomers (40 mM), DIC (57 mM), HOAt (40 mM), and DIEA (40 mM) in DMF (150 μL), incubating (5 min, RT), then adding the activated carboxylic acid solutions to the appropriate wells of PLATE 1 or PLATE 2. The plates were sealed and incubated with shaking (1 h, 37 °C, 700 rpm). Resin samples were washed in parallel in the plates (DMF, 3 × 150 μL; DCM, 1 × 150 μL; DMF, 3 × 150 μL). Samples in PLATE 1 were suspended in a solution of the appropriate primary amine (1 M in DMF, 150 μL), the plate was sealed and incubated with shaking (3 h, 37 °C, 700 rpm) then washed (DMF, 3 × 150 μL; DCM, 2 × 150 μL; DMF, 3 × 150 μL). Fmoc deprotection solution (20% piperidine in DMF, 2 × 150 μL) was added to samples in PLATE 2, the plate was sealed and incubated with shaking (7 min each aliquot, RT, 700 rpm) then washed (DMF, 3 × 150 μL; DCM, 2 × 150 μL; DMF, 3 × 150 μL). Both plates were washed (50:50 DMF/H2O, 1 × 150 μL; BTPWB, 4 × 150 μL), sealed, incubated with shaking (30 min, RT, 700 rpm), then washed (BTPLB, 1 × 150 μL).
An encoding solution consisting of ≈0001[±] (1.2 nmol), appropriate OP stocks ≈11XX[±] (1.2 nmol, Table T4 “Scaffold”) and ≈22XX[±] (1.2 nmol, Table T4 “Side Chain”), and T4 DNA ligase (57 U) was combined in BTPLB (150 μL) for each resin sample and added to the appropriate wells. The plates were sealed and incubated with shaking (15 h, RT, 700 rpm). Both plates were washed (BTPWB, 3 × 150 μL; H2O, 2 × 150 μL; 50:50 H2O:DMF, 1 × 150 μL; DMF, 4 × 150 μL) and incubated with shaking (30 min, 700 rpm). All resin samples from PLATE 1 and PLATE 2 were pooled by transferring each sample (DMF, 3 × 150 μL; DCM, 2 × 150 μL DCM; DMF 1 × 150 μL) into a fritted spin column.
Pooled resin was mixed and transferred back to either PLATE 1 aliquots for acylation with chloroacetic acid (20 wells, 0.75 mg 10-μm resin, 0.1 mg 160-μm resin) or PLATE 2 aliquots for acylation with Fmoc-protected amino acids (25 wells, 0.68 mg 10-μm resin, 0.09 mg 160-μm resin). The 3 Fmoc-l-N(me)-amino acids were replaced with 6 monomers specific for the second diversification position (Figure 3). Resin was acylated by preparing a solution of the appropriate acid monomers (40 mM), DIC (57 mM), HOAt (40 mM), and DIEA (40 mM) in DMF (150 μL), incubating (5 min, RT), then adding the activated carboxylic acid solutions to the appropriate wells of PLATE 1 or PLATE 2. The plates were sealed and incubated with shaking (1 h, 37 °C, 700 rpm). Resin samples were washed in parallel in the plates (DMF, 3 × 150 μL; DCM, 1 × 150 μL; DMF, 3 × 150 μL). Samples in PLATE 1 were suspended in a solution of the appropriate primary amine (1 M in DMF, 150 μL), the plate was sealed and incubated with shaking (3 h, 37 °C, 700 rpm) then washed (DMF, 3 × 150 μL; DCM, 2 × 150 μL; DMF, 3 × 150 μL). Fmoc deprotection solution (20% piperidine in DMF, 2 × 150 μL) was added to samples in PLATE 2, the plate was sealed and incubated with shaking (7 min each aliquot, RT, 700 rpm) then washed (DMF, 3 × 150 μL; DCM, 2 × 150 μL; DMF, 3 × 150 μL). Both plates were washed (50:50 DMF/H2O, 1 × 150 μL; BTPWB, 4 × 150 μL), sealed, incubated with shaking (30 min, RT, 700 rpm), then washed (BTPLB, 1 × 150 μL).
An encoding solution consisting of appropriate OP stocks ≈13XX[±] (1.2 nmol, Table T4 “Scaffold”) and ≈24XX[±] (1.2 nmol, Table T4 “Side Chain”), and T4 DNA ligase (57 U) was combined in BTPLB (150 μL) for each resin sample and added to the appropriate wells. The plates were sealed and incubated with shaking (15 h, RT, 700 rpm). Both plates were washed (BTPWB, 3 × 150 μL; H2O, 2 × 150 μL; 50:50 H2O/DMF, 1 × 150 μL; DMF, 4 × 150 μL) and incubated with shaking (30 min, 700 rpm). All resin samples from PLATE 1 and PLATE 2 were pooled by transferring each sample (DMF, 3 × 150 μL; DCM, 2 × 150 μL DCM; DMF 1 × 150 μL) into a fritted spin column.
Pooled resin was mixed and transferred back to either PLATE 1 aliquots for acylation with chloroacetic acid (20 wells, 0.75 mg 10-μm resin, 0.1 mg 160-μm resin) or PLATE 2 aliquots for acylation with Fmoc-protected amino acids (22 wells, 0.68 mg 10-μm resin, 0.09 mg 160-μm resin). Resin was acylated by preparing a solution of the appropriate acid monomers (40 mM), DIC (57 mM), HOAt (40 mM), and DIEA (40 mM) in DMF (150 μL), incubating (5 min, RT), then adding the activated carboxylic acid solutions to the appropriate wells of PLATE 1 or PLATE 2. The plates were sealed and incubated with shaking (1 h, 37 °C, 700 rpm). Resin samples were washed in parallel in the plates (DMF, 3 × 150 μL; DCM, 1 × 150 μL; DMF, 3 × 150 μL). Samples in PLATE 1 were suspended in a solution of the appropriate primary amine (1 M in DMF, 150 μL), the plate was sealed and incubated with shaking (3 h, 37 °C, 700 rpm) then washed (DMF, 3 × 150 μL; DCM, 2 × 150 μL; DMF, 3 × 150 μL). Fmoc deprotection solution (20% piperidine in DMF, 2 × 150 μL) was added to samples in PLATE 2, the plate was sealed and incubated with shaking (7 min each aliquot, RT, 700 rpm) then washed (DMF, 3 × 150 μL; DCM, 2 × 150 μL; DMF, 3 × 150 μL). Both plates were washed (50:50 DMF:H2O, 1 × 150 μL; BTPWB, 4 × 150 μL), sealed, incubated with shaking (30 min, RT, 700 rpm), then washed (BTPLB, 1 × 150 μL). All resin samples from PLATE 1 and PLATE 2 were pooled by transferring each sample (DMF, 3 × 150 μL; DCM, 2 × 150 μL DCM; DMF 1 × 150 μL) into a fritted spin column.
DNA-Encoded Solid-Phase Combinatorial Library Quality Control
Library resin was sonicated, filtered (150-μm mesh, CellTrics 150 μm, Partec), and the 160-μm particles collected and stored (DMF, 700 μL, 4 °C). The eluted 10-μm library resin was collected into a tube, resuspended (DMF, 450 μL), sonicated, mixed by vortexing, filtered (20-μm mesh, CellTrics 20 μm) and eluted library resin was stored in filter-spin column (DMF, 500 μL, 4 °C). An aliquot of 10-μm library resin (0.05 mg) was transferred into 1.5 mL tube, washed (BTPWB, 4 × 500 μL; centrifuge 6000 rcf), resuspended (BTPWB, 500 μL), bead concentration determined by hemocytometer and normalized (1.2 beads/μL, BTPWB). An aliquot of 160-μm library beads (∼50 beads) were transferred into 1.5 mL tube, washed (BTPWB, 5 × 500 μL) and incubated (1 h, RT). Conditions for qPCR analysis were as previously described. Single 160-μm library resin particles in BTPWB (1 μL) were added to separate amplification reaction wells (20 μL, 22 replicates). Quantitated 10-μm library beads in BTPWB (1 μL, 1.2 beads/μL) were added to separate amplification reaction wells (20 μL, 33 replicates). Each resin sample supernatant (1 μL) was added to respective negative control reaction wells (20 μL, 3 replicates). Thermal cycling conditions and analysis were performed as previously described. Stochastic distribution of 10-μm beads was confirmed using a stereo zoom microscope. Each 160-μm library bead PCR sample was purified by denaturing PAGE, PCR amplified using the FOX primer, and sequenced as described previously. Sequence data from the individual 160-μm library bead-derived PCR amplicons were aligned and decoded using the lookup table (Table T4) as described previously.
The 160-μm library resin beads were individually transferred by pipet (1 μL) from PCR wells into separate wells of a filtration microplate, washed (DI H2O, 3 × 150 μL; 100 mM triethylammonium carbonate pH 8.5, 2 × 150 μL; DMF, 4 × 150 μL), incubated (15 min, RT), washed (DMF, 3 × 150 μL; DCM, 3 × 150 μL), transferred in DMF (5 μL) into fresh microplate wells, and dried in centrifugal evaporator (15 min, 40 °C). Cleavage cocktail (90% TFA, 5% DCM, 5% TIPS, 50 uL) was added to dried single 160-μm library bead samples, incubated (RT, 1 h) then samples were dried in vacuo. Residue was resuspended (50% ACN, 0.1% TFA in H2O, 7 μL) and an aliquot (1 μL) cospotted onto a MALDI-TOF MS target plate with HCCA matrix solution (see above), dried, and analyzed via MALDI-TOF/TOF MS/MS (4800 Plus MALDI TOF/TOF Analyzer, Applied Biosystems, Foster City, CA, Figures S17–20).
Acknowledgments
We thank Prof. Glenn C. Micalizio and Dr. Claudio Aquino for invaluable discussions on implementing the chloropentenoic acid monomers in encoded synthesis. We thank Mr. Jacob Roush for experimental assistance with enzymatic ligation assays.
Glossary
Abbreviations
- BTPLB
Bis-Tris propane ligation buffer
- BTPBB
Bis-Tris propane breaking buffer
- BWB
binding and washing buffer
- BWBT
binding and washing buffer with TWEEN
- HDNA
headpiece DNA
- DESPS
DNA-encoded solid-phase synthesis
- SPS+
solid-phase synthesis positive control
- DE+
DNA encoding positive control
- COPA
constrained oligomer of pentenoic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscombsci.5b00106.
Detailed experimental procedures for computationally selecting optimal sequences for ligation, determining enzymatic oligonucleotide ligation yield, model and library compound characterization data, and sequence-structure lookup tables (PDF)
B.M.P. gratefully acknowledges the support of a NIH Director’s New Innovator Award (OD008535) and the DARPA Fold F(X) Program (N66001-14-2-4057).
The authors declare no competing financial interest.
Supplementary Material
References
- Austin C. P.; Brady L. S.; Insel T. R.; Collins F. S. NIH Molecular Libraries Initiative. Science 2004, 306, 1138–1139 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- Collins F. S.; Patrinos A.; Jordan E.; Chakravarti A.; Gesteland R.; Walters L. New Goals for the U.S. Human Genome Project: 1998–2003. Science 1998, 282, 682–689 10.1126/science.282.5389.682. [DOI] [PubMed] [Google Scholar]
- Margulies M.; Egholm M.; Altman W. E.; Attiya S.; Bader J. S.; Bemben L. A.; Berka J.; Braverman M. S.; Chen Y.-J.; Chen Z.; et al. Genome Sequencing in Microfabricated High-Density Picolitre Reactors. Nature 2005, 437, 376–380 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler D. A.; Srinivasan M.; Egholm M.; Shen Y.; Chen L.; McGuire A.; He W.; Chen Y.-J.; Makhijani V.; Roth G. T.; et al. The Complete Genome of an Individual by Massively Parallel DNA Sequencing. Nature 2008, 452, 872–876 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- Roy A.; McDonald P. R.; Sittampalam S.; Chaguturu R. Open Access High Throughput Drug Discovery in the Public Domain: a Mount Everest in the Making. Curr. Pharm. Biotechnol. 2010, 11, 764–778 10.2174/138920110792927757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L. Challenges for Academic Drug Discovery. Angew. Chem., Int. Ed. 2012, 51, 11680–11684 10.1002/anie.201204625. [DOI] [PubMed] [Google Scholar]
- Houghten R. A. General Method for the Rapid Solid-Phase Synthesis of Large Numbers of Peptides Specificity of Antigen-Antibody Interaction at the Level of Individual Amino Acids. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 5131–5135 10.1073/pnas.82.15.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furka A.; Sebestyen F.; Asgedom M.; Dibo G. General-Method for Rapid Synthesis of Multicomponent Peptide Mixtures. Int. J. Pept. Protein Res. 1991, 37, 487–493 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]
- Lam K. S.; Salmon S. E.; Hersh E. M.; Hruby V. J.; Kazmierski W. M.; Knapp R. J. A New Type of Synthetic Peptide Library for Identifying Ligand-Binding Activity. Nature 1991, 354, 82–84 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- Bunin B. A.; Ellman J. A. A General and Expedient Method for the Solid-Phase Synthesis of 1,4-Benzodiazepine Derivatives. J. Am. Chem. Soc. 1992, 114, 10997–10998 10.1021/ja00053a067. [DOI] [Google Scholar]
- Simon R. J.; Kania R. S.; Zuckermann R. N.; Huebner V. D.; Jewell D. A.; Banville S.; Ng S.; Wang L.; Rosenberg S.; Marlowe C. K. Peptoids: a Modular Approach to Drug Discovery. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 9367–9371 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam K. S.; Lebl M.; Krchnak V. The “One-Bead-One-Compound” Combinatorial Library Method. Chem. Rev. 1997, 97, 411–448 10.1021/cr9600114. [DOI] [PubMed] [Google Scholar]
- Hintersteiner M.; Kimmerlin T.; Kalthoff F.; Stoeckli M.; Garavel G.; Seifert J.-M.; Meisner N.-C.; Uhl V.; Buehler C.; Weidemann T.; et al. Single Bead Labeling Method for Combining Confocal Fluorescence on-Bead Screening and Solution Validation of Tagged One-Bead One-Compound Libraries. Chem. Biol. 2009, 16, 724–735 10.1016/j.chembiol.2009.06.011. [DOI] [PubMed] [Google Scholar]
- Brenner S.; Lerner R. A. Encoded Combinatorial Chemistry. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 5381–5383 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J. M.; Banville S. C.; Zuckermann R. N. Encoded Combinatorial Peptide Libraries Containing Nonnatural Amino-Acids. J. Am. Chem. Soc. 1993, 115, 2529–2531 10.1021/ja00059a070. [DOI] [Google Scholar]
- Needels M. C.; Jones D. G.; Tate E. H.; Heinkel G. L.; Kochersperger L. M.; Dower W. J.; Barrett R. W.; Gallop M. A. Gallop, M. A. Generation and Screening of an Oligonucleotide-Encoded Synthetic Peptide Library. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 10700–10704 10.1073/pnas.90.22.10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlmeyer M. H.; Swanson R. N.; Dillard L. W.; Reader J. C.; Asouline G.; Kobayashi R.; Wigler M.; Still W. C. Complex Synthetic Chemical Libraries Indexed with Molecular Tags. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 10922–10926 10.1073/pnas.90.23.10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler H. P.; Bartlett P. A.; Still W. C. A General Method for Molecular Tagging of Encoded Combinatorial Chemistry Libraries. J. Org. Chem. 1994, 59, 4723–4724 10.1021/jo00096a008. [DOI] [Google Scholar]
- Ni Z. J.; Maclean D.; Holmes C. P.; Murphy M. M.; Ruhland B.; Jacobs J. W.; Gordon E. M.; Gallop M. A. Gallop, M. A. Versatile Approach to Encoding Combinatorial Organic Syntheses Using Chemically Robust Secondary Amine Tags. J. Med. Chem. 1996, 39, 1601–1608 10.1021/jm960043j. [DOI] [PubMed] [Google Scholar]
- Blackwell H. E.; Pérez L.; Stavenger R. A.; Tallarico J. A.; Cope Eatough E.; Foley M. A.; Schreiber S. L. A One-Bead, One-Stock Solution Approach to Chemical Genetics: Part 1. Chem. Biol. 2001, 8, 1167–1182 10.1016/S1074-5521(01)00085-0. [DOI] [PubMed] [Google Scholar]
- Geysen H. M.; Wagner C. D.; Bodnar W. M.; Markworth C. J.; Parke G. J.; Schoenen F. J.; Wagner D. S.; Kinder D. S. Isotope or Mass Encoding of Combinatorial Libraries. Chem. Biol. 1996, 3, 679–688 10.1016/S1074-5521(96)90136-2. [DOI] [PubMed] [Google Scholar]
- Song A.; Zhang J.; Lebrilla C. B.; Lam K. S. A Novel and Rapid Encoding Method Based on Mass Spectrometry for “One-Bead-One-Compound” Small Molecule Combinatorial Libraries. J. Am. Chem. Soc. 2003, 125, 6180–6188 10.1021/ja034539j. [DOI] [PubMed] [Google Scholar]
- Gartner Z. J.; Liu D. R. The Generality of DNA-Templated Synthesis as a Basis for Evolving Non-Natural Small Molecules. J. Am. Chem. Soc. 2001, 123, 6961–6963 10.1021/ja015873n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner Z. J.; Tse B. N.; Grubina R.; Doyon J. B.; Snyder T. M.; Liu D. R. DNA-Templated Organic Synthesis and Selection of a Library of Macrocycles. Science 2004, 305, 1601–1605 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin D. R.; Harbury P. B. DNA Display I. Sequence-Encoded Routing of DNA Populations. PLoS Biol. 2004, 2, E173. 10.1371/journal.pbio.0020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkko S.; Scheuermann J.; Dumelin C.; Neri D. Encoded Self-Assembling Chemical Libraries. Nat. Biotechnol. 2004, 22, 568–574 10.1038/nbt961. [DOI] [PubMed] [Google Scholar]
- Wrenn S. J.; Weisinger R. M.; Halpin D. R.; Harbury P. B. Synthetic Ligands Discovered by in Vitro Selection. J. Am. Chem. Soc. 2007, 129, 13137–13143 10.1021/ja073993a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M. A.; Acharya R. A.; Arico-Muendel C. C.; Belyanskaya S. L.; Benjamin D. R.; Carlson N. R.; Centrella P. A.; Chiu C. H.; Creaser S. P.; Cuozzo J. W.; et al. Design, Synthesis and Selection of DNA-Encoded Small-Molecule Libraries. Nat. Chem. Biol. 2009, 5, 647–654 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- Hili R.; Niu J.; Liu D. R. DNA Ligase-Mediated Translation of DNA Into Densely Functionalized Nucleic Acid Polymers. J. Am. Chem. Soc. 2013, 135, 98–101 10.1021/ja311331m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates K. S.Chemical Reactions of DNA Damage and Degradation. In Reviews of Reactive Intermediate Chemistry; Platz M. S., Moss R. A., Jones M. Jr., Eds.; Wiley-Interscience: Hoboken, NJ, 2007; pp 333–378. [Google Scholar]
- Zoltewicz J. A.; Clark D. F.; Sharples T. W.; Grahe G. Kinetics and Mechanism of Acid-Catalyzed Hydrolysis of Some Purine Nucleosides. J. Am. Chem. Soc. 1970, 92, 1741–1750 10.1021/ja00709a055. [DOI] [PubMed] [Google Scholar]
- Aquino C.; Sarkar M.; Chalmers M. J.; Mendes K.; Kodadek T.; Micalizio G. C. A Biomimetic Polyketide-Inspired Approach to Small-Molecule Ligand Discovery. Nat. Chem. 2011, 4, 99–104 10.1038/nchem.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystrykh L. V. Generalized DNA Barcode Design Based on Hamming Codes. PLoS One 2012, 7, e36852. 10.1371/journal.pone.0036852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click Chemistry: Diverse Chemical Function From a Few Good Reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res. 2003, 31, 3406–3415 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamming R. W. Error Detecting and Error Correcting Codes. Bell Syst. Tech. J. 1950, 29, 147–160 10.1002/j.1538-7305.1950.tb00463.x. [DOI] [Google Scholar]
- Ronaghi M.; Karamohamed S.; Pettersson B.; Uhlen M.; Nyren P. Real-Time DNA Sequencing Using Detection of Pyrophosphate Release. Anal. Biochem. 1996, 242, 84–89 10.1006/abio.1996.0432. [DOI] [PubMed] [Google Scholar]
- Hintersteiner M.; Buehler C.; Auer M. On-Bead Screens Sample Narrower Affinity Ranges of Protein-Ligand Interactions Compared to Equivalent Solution Assays. ChemPhysChem 2012, 13, 3472–3480 10.1002/cphc.201200117. [DOI] [PubMed] [Google Scholar]
- Sarkar M.; Pascal B. D.; Steckler C.; Aquino C.; Micalizio G. C.; Kodadek T.; Chalmers M. J. Decoding Split and Pool Combinatorial Libraries with Electron-Transfer Dissociation Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2013, 24, 1026–1036 10.1007/s13361-013-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichert M.; Krall N.; Decurtins W.; Franzini R. M.; Pretto F.; Schneider P.; Neri D.; Scheuermann J. R. Dual-Display of Small Molecules Enables the Discovery of Ligand Pairs and Facilitates Affinity Maturation. Nat. Chem. 2015, 7, 241–249 10.1038/nchem.2158. [DOI] [PubMed] [Google Scholar]
- Franzini R. M.; Ekblad T.; Zhong N.; Wichert M.; Decurtins W.; Nauer A.; Zimmermann M.; Samain F.; Scheuermann J.; Brown P. J.; et al. Identification of Structure-Activity Relationships From Screening a Structurally Compact DNA-Encoded Chemical Library. Angew. Chem., Int. Ed. 2015, 54, 3927–3931 10.1002/anie.201410736. [DOI] [PubMed] [Google Scholar]
- Disch J. S.; Evindar G.; Chiu C. H.; Blum C. A.; Dai H.; Jin L.; Schuman E.; Lind K. E.; Belyanskaya S. L.; Deng J.; et al. Discovery of Thieno[3,2-d]Pyrimidine-6-Carboxamides as Potent Inhibitors of SIRT1, SIRT2, and SIRT3. J. Med. Chem. 2013, 56, 3666–3679 10.1021/jm400204k. [DOI] [PubMed] [Google Scholar]
- Samain F.; Ekblad T.; Mikutis G.; Zhong N.; Zimmermann M.; Nauer A.; Bajic D.; Decurtins W.; Scheuermann J.; Brown P. J.; et al. Tankyrase 1 Inhibitors with Drug-Like Properties Identified by Screening a DNA-Encoded Chemical Library. J. Med. Chem. 2015, 58, 5143–5149 10.1021/acs.jmedchem.5b00432. [DOI] [PubMed] [Google Scholar]
- Lewis H. D.; Liddle J.; Coote J. E.; Atkinson S. J.; Barker M. D.; Bax B. D.; Bicker K. L.; Bingham R. P.; Campbell M.; Chen Y. H.; et al. Inhibition of PAD4 Activity Is Sufficient to Disrupt Mouse and Human NET Formation. Nat. Chem. Biol. 2015, 11, 189–191 10.1038/nchembio.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seneci P.Solid-Phase Synthesis and Combinatorial Technologies; John Wiley & Sons, Inc.: New York, 2002; pp 91–135. [Google Scholar]
- Franz A. H.; Liu R. W.; Song A. M.; Lam K. S.; Lebrilla C. B. High-Throughput One-Bead-One-Compound Approach to Peptide-Encoded Combinatorial Libraries: MALDI-MS Analysis of Single TentaGel Beads. J. Comb. Chem. 2003, 5, 125–137 10.1021/cc020083a. [DOI] [PubMed] [Google Scholar]
- Doran T. M.; Kodadek T. A Liquid Array Platform for the Multiplexed Analysis of Synthetic Molecule Protein-Interactions. ACS Chem. Biol. 2014, 9, 339–346 10.1021/cb400806r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J.; Brenner S.; Janda K. D. Synthetic Methods for the Implementation of Encoded Combinatorial Chemistry. J. Am. Chem. Soc. 1993, 115, 9812–9813 10.1021/ja00074a063. [DOI] [Google Scholar]
- Price A. K.; MacConnell A. B.; Paegel B. M. Microfluidic Bead Suspension Hopper. Anal. Chem. 2014, 86, 5039–5044 10.1021/ac500693r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan T. R.; Hilgraf R.; Sharpless K. B.; Fokin V. V. Polytriazoles as Copper(I)-Stabilizing Ligands in Catalysis. Org. Lett. 2004, 6, 2853–2855 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.