

Abstract
The progressive loss of the nigrostriatal pathway is a distinguishing feature of Parkinson’s disease. As terminal field loss seems to precede cell body loss, we tested whether alterations of axonal transport motor proteins would be early features in Parkinson’s disease. There was a decline in axonal transport motor proteins in sporadic Parkinson’s disease that preceded other well-known nigral cell-related pathology such as phenotypic downregulation of dopamine. Reductions in conventional kinesin levels precede the alterations in dopaminergic phenotypic markers (tyrosine hydroxylase) in the early stages of Parkinson’s disease. This reduction was significantly greater in nigral neurons containing α-synuclein inclusions. Unlike conventional kinesin, reductions in the levels of the cytoplasmic dynein light chain Tctex type 3 subunit were only observed at late Parkinson’s disease stages. Reductions in levels of conventional kinesin and cytoplasmic dynein subunits were recapitulated in a rat genetic Parkinson’s disease model based on over-expression of human mutant α-synuclein (A30P). Together, our data suggest that α-synuclein aggregation is a key feature associated with reductions of axonal transport motor proteins in Parkinson’s disease and support the hypothesis that dopaminergic neurodegeneration following a ‘dying-back’ pattern involving axonal transport disruption.
Keywords: conventional kinesin, cytoplasmic dynein, axonal neuropathies, α-synuclein, Parkinson’s disease
Introduction
Disruptions in axonal transport represent early pathogenic features in neurodegeneration and may cause disease progression in a number of neurodegenerative diseases including Parkinson’s disease (Roy et al., 2005; Morfini et al., 2009). Accordingly, pathological studies identified axonal enlargements and aberrant accumulations of organelle cargos in Parkinson’s disease brains (Chung et al., 2009). Further, nigrostriatal dopaminergic neurons affected in Parkinson’s disease follow a ‘dying back’ pattern of degeneration that is consistent with alteration in axonal transport (Iseki et al., 2001; Raff et al., 2002; Morfini et al., 2009).
Alpha-synuclein (α-synuclein) is a synaptic protein of unknown function that mainly localizes to termini (Lykkebo and Jensen, 2002) and accumulates in Lewy bodies and Lewy neurites characteristic of cases with idiopathic, sporadic Parkinson’s disease. Significantly, several mutations in α-synuclein, as well as increased genetic doses of α-synuclein, lead to familial forms of Parkinson’s disease, suggesting a critical role of α-synuclein in sporadic Parkinson’s disease pathogenesis (Dauer and Przedborski, 2003). It is assumed that α-synuclein modulates various physiological functions in the presynaptic compartment, such as regulating the activity of the dopamine transporter or fine-tuning neurotransmitter release (Madine et al., 2006). Experimental evidence suggests that both mutations and over-expression of α-synuclein might lead to axonal transport defects. For example, viral over-expression of mutant A53T α-synuclein in rats induced dystrophic axons and alterations in axonal transport that preceded neuronal loss (Chung et al., 2009). Also, mild over-expression of α-synuclein in cultured hippocampal neurons promotes reductions in the levels of synaptic proteins at presynaptic terminals, a phenomenon termed ‘vacant synapses’ (Scott et al., 2010). These and other studies linked alterations in axonal transport to familial Parkinson’s disease pathogenesis, but the relevance of these findings to sporadic Parkinson’s disease remains unknown.
Our recent studies indicate that accumulation of perikaryal α-synuclein aggregates in sporadic Parkinson’s disease and familial Parkinson’s disease models is associated with a loss of dopamine phenotype (Chu et al., 2006), as well as disruptions in both lysosomal and proteosomal functions (Chu et al., 2009). Accumulation and aggregation of α-synuclein also correlates with reduced levels of myocyte enhancer factor 2D (MEF2D), a protein that plays an important role in synapse formation and maintenance (Flavell et al., 2006, 2008; Chu et al., 2011).
Whether disease-associated α-synuclein inclusions are associated with axonal transport alterations needs to be investigated in the Parkinson’s disease brain. To evaluate this possibility, our study determined the relative levels of critical motor protein subunits in early and late Parkinson’s disease, compared with age-matched controls. Immunohistochemical analysis helped establish (i) whether axonal transport proteins are altered in remaining neuromelanin-laden nigral neurons; (ii) whether reductions in axonal transport proteins precede and exceed reductions in dopaminergic markers and (iii) whether reductions of axonal transport proteins are associated with α-synuclein inclusion formation in nigral neurons. In addition, we also over-expressed α-synuclein in the rodent substantia nigra and performed similar assessments in this rodent model of Parkinson’s disease. Results from these studies suggest that abnormalities in axonal transport motor proteins represent an early degenerative event in both familial and sporadic Parkinson’s disease.
Materials and methods
Human tissue acquisition and processing
To first establish the quality of the human tissue (both control and Parkinson’s disease), we immunostained sections from each case using a monoclonal anti-tyrosine hydroxylase antibody that consistently yields well-established immunostaining patterns in hypothalamus. Cases in which brain tissue demonstrated weak tyrosine hydroxylase immunoreactivity in hypothalamic dopaminergic neurons in arcuate nucleus and periventricular nucleus were excluded from the study. We analysed tissues from 25 subjects with clinical and neuropathological diagnoses of Parkinson’s disease (n = 16) and age-matched controls (n = 9). According to Hoehn and Yahr (1967; OFF medication) scores, cases with Parkinson’s disease were divided into early (Hoehn and Yahr 1–2; n = 6) and late Parkinson’s disease (Hoehn and Yahr 3–5; n = 10) stages. There were no differences in age at the time of death (P > 0.05) or post-mortem interval (P > 0.05) among the three groups examined (Table 1). All patients with Parkinson’s disease were diagnosed by movement disorder specialists in the Section of Movement Disorders in the Department of Neurological Sciences at Rush University Medical Centre. Post-mortem, a board-certified neuropathologist at Rush University Medical Centre confirmed the clinical diagnosis. For Parkinson’s disease, inclusion criteria included a history compatible with idiopathic Parkinson’s disease and at least two of the four cardinal motor signs (rest tremor, rigidity, akinesia/bradykinesia and gait disturbance/postural reflex impairment). The Unified Parkinson’s Disease Rating Scale 3 (UPDRS3 ON and OFF medication) and Hoehn and Yahr (ON and OFF medication) were recorded. The pathological diagnosis was based on finding Lewy bodies in catecholamine nuclei such as the substantia nigra. Exclusion criteria included familial Parkinson’s disease, the Lewy body variant of Alzheimer’s disease or the combination of Parkinson’s disease and Alzheimer’s disease. Age-matched control subjects were all participants in the Rush University Religious Order Study, a longitudinal clinical–pathological study of ageing and Alzheimer’s disease, which comprised older Catholic nuns, priests and brothers. Each participant received a clinical evaluation that included an assessment for movement disorders. Details of the clinical evaluation were previously reported (Chu et al., 2006). Subjects without psychiatric illnesses during life and neurological abnormalities at post-mortem were included in the control group. The Human Investigation Committee at Rush University Medical Centre approved this study. At autopsy, the brains were removed from the calvarium and processed as described previously (Chu et al., 2006). Briefly, each brain was cut into 1 cm coronal slabs using a Plexiglas brain slice apparatus and then hemisected. The slabs were fixed in 4% paraformaldehyde for 48 h at 4°C. The left-side brain slabs were used for pathological diagnoses. The right-side brain slabs were cryoprotected in 0.1 M PBS pH 7.4 containing 2% dimethyl sulphoxide, 10% glycerol for 48 h followed by 2% dimethyl sulphoxide and 20% glycerol in PBS for at least 2 days before sectioning. The fixed slabs were cut into 18 adjacent series of 40 -μm-thick sections on a freezing sliding microtome. All sections were collected and stored in a cryoprotectant solution before processing.
Table 1.
Summary of case demographics
| Case no. | Age (years) | Gender | Post-mortem interval (h) | UPDRS III (OFF) | Hoehn and Yahr score (OFF) |
|---|---|---|---|---|---|
| Early stage of Parkinson’s disease | |||||
| 1 | 76 | F | 7.8 | 36 | 2 |
| 2 | 92 | M | 2 | 7 | 1 |
| 3 | 89 | F | 3.6 | 13 | 2 |
| 4 | 72 | M | 2 | 17 | 2 |
| 5 | 79 | M | 5.15 | 23 | 2 |
| 6 | 74 | M | 8.21 | 38 | 2 |
| Mean ± SE | 80.33 ± 3.37 | 4.79 ± 1.41 | 22.42 ± 5.1 | 1.91 ± 0.20 | |
| Late stage of Parkinson's disease | |||||
| 1 | 72 | F | 11.5 | 69 | 5 |
| 2 | 63 | F | 5 | 58 | 5 |
| 3 | 72 | F | 11 | 49 | 5 |
| 4 | 88 | F | n/a | 24 | 3 |
| 5 | 64 | M | 3.75 | 40 | 3 |
| 6 | 83 | F | 12 | 31 | 3 |
| 7 | 77 | F | n/a | 53 | 3 |
| 8 | 78 | M | 3 | 63 | 5 |
| 9 | 82 | F | 3 | 39 | 4 |
| 10 | 78 | M | 5 | 30 | 3 |
| Mean ± SE | 75.70 ± 2.54 | 6.78 ± 1.41 | 46.27 ± 4.0* | 3.75 ± 0.27** | |
| Age (years) | Gender | Post-mortem interval (h) | MMSE | ||
| Age-matched controls | |||||
| 1 | 74 | M | 5.49 | 28 | |
| 2 | 90 | F | 5.3 | 28 | |
| 3 | 71 | M | 5.3 | 28 | |
| 4 | 89 | M | 4.3 | 29 | |
| 5 | 79 | F | 5.8 | 29 | |
| 6 | 91 | F | 10.7 | 27 | |
| 7 | 92 | F | 3.1 | 29 | |
| 8 | 82 | F | 3.6 | 30 | |
| 9 | 85 | F | 11 | 28 | |
| Mean ± SE | 83.67 ± 2.56 | 6.06 ± 0.95 | 28.44 ± 0.29 |
F = female; M = male; MMSE = Mini-Mental State Examination; n/a = not available; UPDRS = Unified Parkinson’s Disease Rating Scale.
*P < 0.0042, **P < 0.0005 compared with early stage of Parkinson’s disease.
Injection of adeno-associated virus into rat brain
Young adult Sprague–Dawley rats (both male and female; Charles River Laboratories) were housed two to a cage with ad libitum access to food and water during a 12-h light/dark cycle; all animal experiments were approved by the Rush University Institutional Animal Care and Use Committee. Recombinant adeno-associated virus (rAAV) serotype 6 vector encoding human mutant (A30P) α-synuclein gene (rAAV-h-A30P) and green fluorescent protein gene (rAAV-GFP) were prepared and titred as described previously (Towne et al., 2008). Under xylazine/ketamine anaesthesia, 2 μl of the vector suspension was injected stereotaxically into the left nigral region (5.3 mm posterior and 2.3 mm lateral to bregma; 7.7 mm ventral to dura). The needle was kept in place for an additional 5 min before slowly being withdrawn. At 6 weeks after injection, animals (rAAV-h-A30P, n = 8; rAAV-GFP, n = 8) were perfused through the ascending aorta with physiological saline, followed by 4% ice-cold para-formaldehyde. The brains were post-fixed in the same solution for 2 h, transferred to 10, 20 and 30% sucrose and sectioned on a freezing microtome at 40 μm in the coronal plane. All sections were collected and stored in order in a cryoprotectant solution before processing.
Antibodies
Several antibodies that recognize axonal transport motor proteins, tyrosine hydroxylase and α-synuclein were used (Table 2). To control for antibody specificity, adsorption experiments were performed for kinesin heavy chain (KHC) and kinesin light chain 1 (KLC1) antibodies. Briefly, these antibodies were combined with a 5-fold volume (by weight) of blocking peptides (BP1575a for KHC, BP8637c for KLC1) separately in Tris-buffered saline and incubated overnight at 4°C. After centrifugation at 10 000 rpm for 20 min, adsorbed antibodies were used in lieu of the primary antibody. Staining specificity was further confirmed by omitting the primary antibody (which controls for the specificity of the staining procedure) and replacement of the primary antibody with an irrelevant IgG matched for protein concentration. All these control experiments resulted in a total absence of staining.
Table 2.
Antibodies
| Name | Catalogue number/company | Host | References |
|---|---|---|---|
| KHC | ab25715/Abcam | Rabbit | Tomishige et al. (2006) |
| KHC | MA1-19352/Thermo | Mouse | |
| KLC1 | AP8637c/ABGENT | Rabbit | Gyoeva et al. (2000) |
| DYNLT3 | ab121209/Abcam | Rabbit | Lo et al. (2007) |
| Tyrosine hydroxylase | 22941/ImmunoStar | Mouse | Chu et al. (2006) |
| α-Synuclein (α-syn, LB509) | 18-0215/Invitrogen | Mouse | Jakes et al. (1999) |
| Phospho S129 α-syn | ab51253/Abcam | Rabbit | Mbefo et al. (2010) |
Fluorescence intensity measurements
Fluorescence intensity measurements were performed according to our previously published procedures (Chu et al., 2006, 2009; Kanaan et al., 2007). All immunofluorescence double-labelled images were scanned with an Olympus Confocal Fluoroview microscope equipped with argon and krypton lasers. At low magnification (×4), a virtual slice was taken of each section and the substantia nigra pars compacta drawn. With a 20× magnification objective and a 488, 468, or 647 nm excitation source, this system is specifically designed to acquire images at each sampling site in substantia nigra pars compacta. The images were saved to a file; the stage automatically moves to the next sampling site to ensure a completely non-redundant evaluation. Once all images were acquired, optical density measurements were performed on individual nigral neurons at the nuclear level. To maintain consistency of the scanned image for each slide, the laser intensity, confocal aperture, photomultiplier voltage, offset, electronic gain, scan speed, image size, filter and zoom were set for the background level whereby autofluorescence was not visible with a control section. These settings were maintained throughout the entire experiment (Chu et al., 2011). The intensity mapping sliders ranged from 0 to 4095; 0 represented a maximum black image and 4095 represented a maximum bright image. The KHC-, KLC1- or DYNLT3-immunoreactive perikarya co-labelled with tyrosine hydroxylase were identified and outlined separately by an investigator blinded to the clinical and pathological data. Quantitative optical density of immunofluorescence intensity was performed on individual KHC- and KLC1- or DYNLT3-immunoreactive soma with tyrosine hydroxylase immunolabelling in different channels. The same methods were used for quantitating optical density of KLC1- or dynein-immunoreactive soma with or without α-synuclein-immunoreactive inclusions. For each marker, five equispaced sections across the entire length of the substantia nigra were sampled and evaluated. The number of cells per case was analysed as follows: >200 nigral cells in normal cases, 50–70 nigral cells per Parkinson’s disease case that contained inclusions and >200 nigral cells per Parkinson’s disease case that did not contain inclusions. To account for differences in background staining intensity, five background intensity measurements lacking immunofluorescent profiles were taken from each section. The mean of these five measurements constituted the background intensity that was then subtracted from the measured immunofluorescence intensity of each individual neuron to provide a final immunofluorescence intensity value. To confirm co-localization of the axonal transport marker and tyrosine hydroxylase or α-synuclein immunofluorescence, optical scanning through the neuron’s z-axis was performed at 1-µm thickness and neurons suspected of being double labelled were confirmed with confocal cross-section.
Data analyses
Demographic and clinical characteristics and optical density measurements were compared across groups with one-way ANOVA, Kruskal–Wallis test followed by Dunn’s post hoc test for multiple comparisons (Prism 4, GraphPad Software, Inc.). Descriptive statistical level of significance was set at 0.05 (two-tailed).
Digital illustrations
Confocal images were exported from the Olympus laser scanning microscope with Fluoview software and stored as .tif files. Conventional light microscopic images were acquired using a Nikon Microphoto-FXA microscope attached to a Nikon digital camera DXM1200 and stored as .tif files. All figures were prepared using Photoshop 8.0 graphics software. Only minor adjustments of brightness were made.
Results
Characteristics of tyrosine hydroxylase labelling in different stages of sporadic Parkinson’s disease
Immunohistochemistry revealed that the extent of tyrosine hydroxylase-immunoreactive loss in the putamen was much greater than in the substantia nigra at early sporadic Parkinson’s disease stages (Supplementary Fig. 1E and G). Cases with Hoehn and Yahr 1 Parkinson’s disease showed extensive and intense tyrosine hydroxylase labelling in the substantia nigra, which was similar to that seen in age-matched controls. A high density of tyrosine hydroxylase-immunoreactive soma and an intricate local plexus of tyrosine hydroxylase-immunoreactive processes were observed within the substantia nigra (Supplementary Fig. 1E and F). In contrast, tyrosine hydroxylase labelling in putamen (Supplementary Fig. 1G and H) was remarkably decreased in early stages of Parkinson’s disease compared with age-matched controls (Supplementary Fig. 1C and D). In age-matched controls, dense fine tyrosine hydroxylase-immunoreactive fibres (<0.25 µm) were distributed throughout the grey matter of putamen (Supplementary Fig. 1C), consisting of a fine mesh of fibres (Supplementary Fig. 1D). A small number of tyrosine hydroxylase-immunoreactive thick fibres (>0.5 µm) were also observed in the putamen. In early stage of Parkinson’s disease, the number of tyrosine hydroxylase-immunoreactive fine fibres was dramatically decreased throughout the putamen (Supplementary Fig. 1G). Optical density measurements demonstrated a range of reductions between 31.64 and 60.79% within putamen in the early stage of Parkinson’s disease, relative to age-matched controls. The few remaining thick fibres displayed an abnormal morphology characterized by swollen varicosities (Supplementary Fig. 1H). In severe Parkinson’s disease (Hoehn and Yahr: 3–5), there was a marked decrease of tyrosine hydroxylase labelling throughout the entire nigrostriatal system (Supplementary Fig. 1I–P). Both tyrosine hydroxylase-immunoreactive nigral somas and dendrites in substantia nigra (Supplementary Fig. 1J and N) were severely reduced compared with both cases with Hoehn and Yahr 1 Parkinson’s disease and age-matched controls. In addition, many neuromelanin-laden nigral neurons displayed no tyrosine hydroxylase labelling. Few fine tyrosine hydroxylase-immunoreactive fibres were observed in the more ventromedial putamen near globus pallidus (Supplementary Fig. 1K and O). Tyrosine hydroxylase-immunoreactive fine fibres were barely detected in the putamen, and the few remaining thick fibres displayed swollen varicosities and intervaricose segments (Supplementary Fig. 1L and P).
Collectively, these data indicated that loss of tyrosine hydroxylase in target (putamen) of nigral neurons long precedes and exceeds that of nigral cell bodies (substantia nigra) early in sporadic Parkinson’s disease, suggesting that terminals are first impaired during dopaminergic neurodegeneration in sporadic Parkinson’s disease.
Reduced levels of conventional kinesin in nigral neurons affected in sporadic Parkinson’s disease
Findings of axonal transport alterations in familial Parkinson’s disease (Chung et al., 2009; Scott et al., 2009) and toxin-based (Morfini et al., 2007; Serulle et al, 2007) models led us to hypothesize that the early decline of tyrosine hydroxylase-immunoreactive terminals in nigrostriatal system observed in sporadic Parkinson’s disease (Supplementary Fig. 1) was associated with alterations in axonal transport motor proteins. Conventional kinesin, a major microtubule-based motor protein responsible for anterograde axonal transport is a heterotetramer composed of two KHC and two KLC subunits. KHC-immunoreactive (Fig. 1A) and KLC1-immunoreactive (Fig. 1E) neurons were widely distributed throughout the substantia nigra of age-matched control brains. Strong KHC- and KLC1-immunoreactive profiles included both perikarya and fibres in substantia nigra, and virtually all neuromelanin-laden neurons were KHC- and KLC1 immunopositive. Intense KHC- and KLC1 immunoreactivity were also observed in scattered non-neuromelanin-laden small cells. In contrast, immunoreactivity for kinesin markers was markedly decreased in all Parkinson’s disease brains. In the early stage of Parkinson’s disease (Hoehn and Yahr: 1–2), the immunoreactivity of KHC (Fig. 1B) and KLC1 (Fig. 1F) was significantly lost in remaining neuromelanin-laden neurons. Some neuromelanin-laden neurons displayed KHC- and KLC-immunoreactive somas, but not processes, whereas staining others displayed none (Fig. 2B and F). At later Parkinson’s disease stages (Hoehn and Yahr: 3–5), the majority of remaining neuromelanin-laden neurons failed to display detectable KHC (Fig. 1C and D) and KLC1 (Fig. 1G and H), whereas a few neurons displayed a lightly stained soma. These reductions in KHC and KLC1 were specific and not due to Parkinson’s disease-related degeneration of the actual neurite in substantia nigra, as tyrosine hydroxylase-immunoreactive processes were observed in remaining nigral neurons (Supplementary Fig. 1). Non-neuromelanin-laden neurons in the nigra, which are not dopaminergic, displayed KHC and KLC1 staining in the both the soma and processes (Fig. 1C and D) demonstrating the specificity of this effect for dopaminergic nigral neurons.
Figure 1.
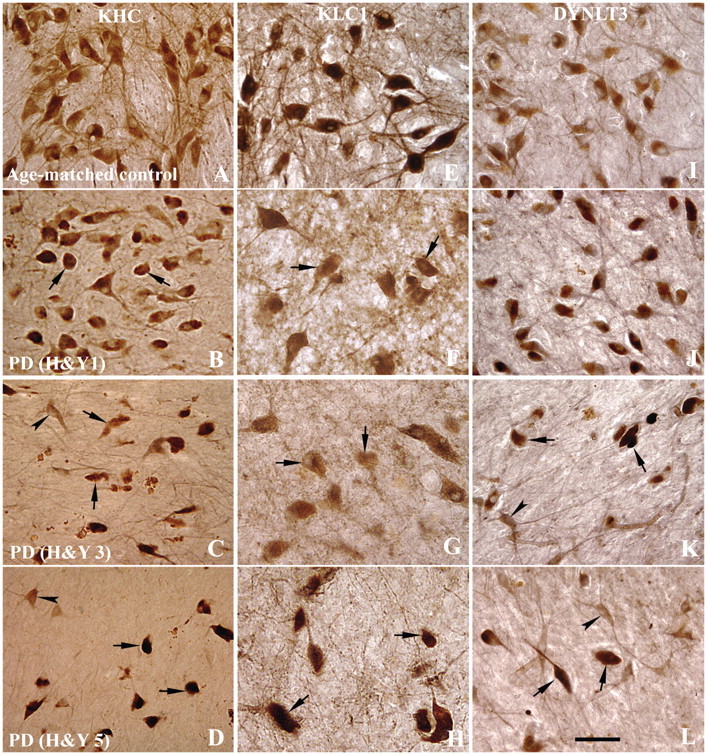
Sections through the mid-substantia nigra show immunoreactivity patterns of KHC (A–D), KLC1 (E–H) and DYNLT3 (I–L) in age-matched controls (A, E and I), Hoehn and Yahr (H&Y) stage 1 Parkinson’s disease (PD; B, F and J), Hoehn and Yahr stage 3 Parkinson’s disease (C, G and K) and Hoehn and Yahr stage 5 Parkinson’s disease (D, H and L) cases. In age-matched controls, every neuromelanin-laden nigral neuron displayed intense KHC and KLC1 immunoreactivities in the soma and processes (A and E). In contrast, nigral neurons of Hoehn and Yahr stage 1 Parkinson’s disease cases display much lighter KHC- and KLC1-immunoreactive levels in neuronal somas, being hardly detected in some cells (arrows; B and F). KHC-labelling processes were severely reduced even when many neuromelanin-laden neurons remain present (B and F), compared with age-matched control (A and E). In Hoehn and Yahr stages 3 and 5 Parkinson’s disease cases, most of remaining neuromelanin-laden neurons showed no detectable KHC and KLC1 labelling (arrows; C, D, G and H). However, non-neuromelanin-laden cells exhibited strong KHC immunoreactivity in the soma and processes (arrowheads; C and D). DYNLT3 immunoreactivity extent and intensity were not obviously decreased in Hoehn and Yahr stage 1 Parkinson’s disease (J) but severely declined in Hoehn and Yahr 3 (K) and Hoehn and Yahr 5 (L) compared with age-matched controls (I). Some remaining neuromelanin-laden neurons exhibited no detectable DYNLT3 (arrows; K and L) but non-neuromelanin-laden neuron appeared DYNLT3-labelling soma and processes (arrowheads; K and L). Scale bar = 70 µm (applies to all).
Figure 2.
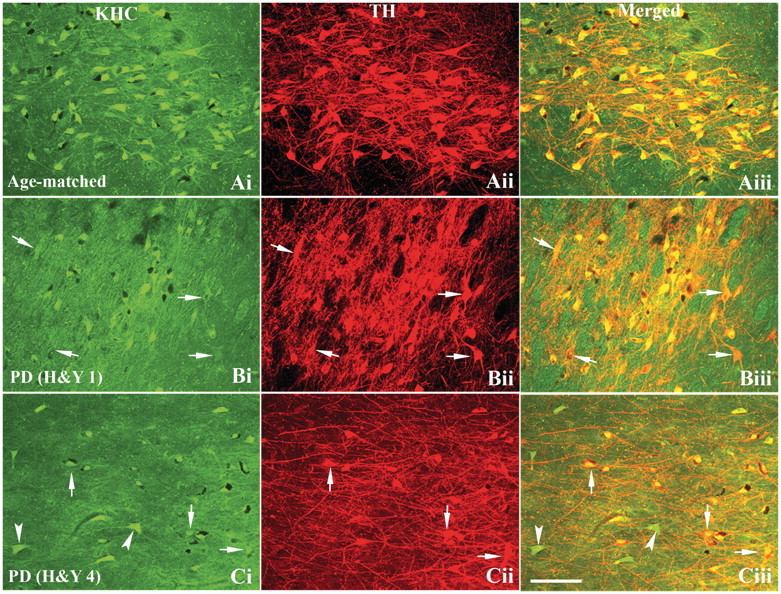
Confocal microscopy images of substantia nigra from age-matched control [A(i–iii)], Hoehn and Yahr (H&Y) stage 1 Parkinson’s disease (PD) [B(i–iii)] and H&Y stage 4 Parkinson’s disease [C(i–iii)] show immunostaining patterns for KHC [green; A(i), B(i) and C(i)], tyrosine hydroxylase [TH; red; A(ii), B(ii) and C(ii)] and co-localization of KHC and tyrosine hydroxylase [merged; A(iii), B(iii) and C(iii)]. The extent and intensity of tyrosine hydroxylase immunoreactivity in both nigral neuronal soma and processes were similar between Hoehn and Yahr stage 1 Parkinson’s disease [B(ii)] and age-matched control [A(ii)]. However, KHC immunofluorescence intensity [arrows; B(i and iii)] was markedly reduced in tyrosine hydroxylase-immunoreactive neurons [arrows; B(ii)] of Hoehn and Yahr stage 1 Parkinson’s disease cases. In Hoehn and Yahr stage 4 Parkinson’s disease, both tyrosine hydroxylase and KHC immunoreactivities were reduced in remaining neuromelanin-laden nigral neurons. Non-tyrosine hydroxylase-immunoreactive cells [arrowheads; C(i and iii)] exhibited intensive KHC-immunoreactive in neuronal soma and processes. Scale bar = 160 µm (applies to all).
Analysis of cytoplasmic dynein immunoreactivity in nigral neurons in Parkinson’s disease
We next evaluated whether levels of the retrograde axonal transport motor protein cytoplasmic dynein were affected in dopaminergic neurons of patients with sporadic Parkinson’s disease. Cytoplasmic dynein exists as a large, multisubunit protein complex in vivo, formed by at least two heavy chains, two intermediate chains, four light intermediate chains and several light chains (Pfister et al., 1998). Among these, dynein light chain Tctex type 3 (DYNLT3) was chosen to be examined in sporadic Parkinson’s disease brains, as this subunit has been shown to contribute to dynein cargo binding specificity (Lo et al., 2007). DYNLT3 antibody labelled soma and processes of neuromelanin-laden neurons in age-matched controls (Fig. 1I). The pattern of DYNLT3 immunostaining in Hoehn and Yahr 1 Parkinson’s disease (Fig. 1I) was similar to age-matched controls, as DYNLT3 was detected in almost all remaining neuromelanin-laden neurons. In Hoehn and Yahr 3–5 cases with Parkinson’s disease, many neuromelanin-laden neurons failed to display detectable DYNLT3, whereas non-neuromelanin-laden cells exhibited DYNLT3-labelling soma and processes (Fig. 1K and L). Taken together, these data indicate that levels of DYNLT3 are severely reduced in late stages of sporadic Parkinson’s disease, suggesting alterations in dynein functions in Parkinson’s disease.
Co-localization and quantitative analysis of axonal transport and dopaminergic markers in nigral neurons in Parkinson’s disease
To evaluate whether the decrease in axonal transport proteins documented in sporadic Parkinson’s disease precedes those seen for dopamine markers, double immunostaining for tyrosine hydroxylase and either conventional kinesin or cytoplasmic dynein markers was performed in nigral neurons of cases with sporadic Parkinson’s disease. Optical density measurements of these staining profiles were measured as described in the ‘Materials and methods’ section. In early Hoehn and Yahr 1 Parkinson’s disease cases, co-localization analyses revealed that remaining neuromelanin-laden neurons featuring intense tyrosine hydroxylase immunostaining [Fig. 2B(ii) and Supplementary Fig. 2B(2)] display light KHC-immunoreactive [Fig. 2B(i)] and KLC1-immunoreactive [Supplementary Fig. 2B(1)] staining, compared with age-matched controls [Fig. 2A(i) and Supplementary Fig. 2A(1)], with KHC- and KLC1-immunoreactive signals mainly detected in perikarya and less extensively in processes, both of which were intensely labelled by tyrosine hydroxylase. In late cases with Parkinson’s disease (Hoehn and Yahr 4), minimal KHC [Fig. 2C(i)] and KLC1 [Supplementary Fig. 2C(1)] staining was seen in neuronal cell bodies. Significantly, non-dopaminergic neurons displayed intense KHC-immunostained perikarya and processes [Fig. 2C(iii)] suggesting that alterations in conventional kinesin mainly affect nigral dopaminergic neurons in sporadic Parkinson’s disease.
The decline in intensity of DYNLT3 staining was observed in late sporadic Parkinson’s disease stages. In cases with early-stage sporadic Parkinson’s disease, most of the remaining tyrosine hydroxylase-immunoreactive neurons exhibited intensive DYNLT3-labelled soma and processes but a few of them appeared lightly DYNLT3 labelled [Fig. 3B(i–iii)] compared with age-matched controls [Fig. 3A(i–iii)]. In late sporadic Parkinson’s disease stages, minimal DYNLT3 immunoreactivity was detected in tyrosine hydroxylase-immunoreactive nigral neurons, whereas intensive DYNLT3 staining was observed in non-tyrosine hydroxylase-immunoreactive cells [Fig. 3C(i–iii)].
Figure 3.
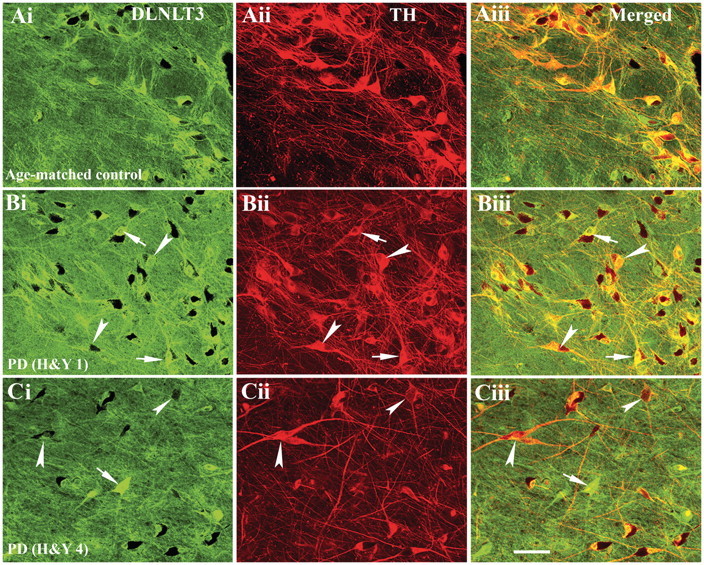
Confocal microscopy images of substantia nigra from age-matched control [A(i–iii)], Hoehn and Yahr (H&Y) stage 1 Parkinson’s disease (PD) [B(i–iii)] and Hoehn and Yahr stage 4 Parkinson’s disease [C(i–iii)] illustrating immunostaining for DYNLT3 [green; A(i), B(i) and C(i)], tyrosine hydroxylase [TH; red; A(ii), B(ii) and C(ii)] and co-localization of DYNLT3 and tyrosine hydroxylase [merged; A(iii), B(iii) and C(iii)]. Every tyrosine hydroxylase-immunoreactive neuron exhibited DYNLT3 immunostaining in neuronal soma and processes in age-matched control [A(iii)]. In Hoehn and Yahr stage 1 Parkinson’s disease cases, most tyrosine hydroxylase-immunoreactive nigral neurons exhibited DYNLT3 labelling in both soma and processes [arrows; B(i–iii)], whereas fewer neurons exhibited light DYNLT3 staining [arrowheads; B(i–iii)]. DYNLT3 immunofluorescence intensity was markedly reduced in tyrosine hydroxylase-immunoreactive neurons [arrowheads; C(i and iii)], but intensive DYNLT3 staining was observed in non-tyrosine hydroxylase-immunoreactive cells [arrow; C(i and iii)] in Hoehn and Yahr stage 4 Parkinson’s disease. Scale bar = 120 µm (applies to all).
Quantitative analyses of KHC-, KLC1- and DYNLT3-immunoreactivities were obtained for individual tyrosine hydroxylase-immunoreactive cell bodies in early Parkinson’s disease (Hoehn and Yahr 1–2), late Parkinson’s disease (Hoehn and Yahr 3–5) and age-matched control cases (Fig. 4). Kruskal–Wallis test revealed significant reductions in KHC levels across these experimental groups (Fig. 4A; P < 0.001). Post hoc analyses further revealed statistically significant decreases in optical densities of KHC immunofluorescence signals in early Parkinson’s disease (P < 0.05) and late Parkinson’s disease stages (P < 0.001), compared with age-matched controls. Similarly, significant differences in optical density of KLC1 immunofluorescence signals were observed among the three groups analysed (Fig. 4B; P < 0.0001). Post hoc analyses further revealed a significant decline in the optical density of the KLC1 immunofluorescence signal between early-stage Parkinson’s disease cases (P < 0.05) and age-matched controls, between late Parkinson’s disease stages (P < 0.001) and age-matched controls, and between early and late stages of Parkinson’s disease (P < 0.05). Kruskal–Wallis test demonstrated significant differences (P < 0.0001) in the optical density of the DYNLT3 immunofluorescence signals across all experimental groups (Fig. 4C; P < 0.001). Post hoc analyses demonstrated a significant decrease in the optical density of the DYNLT3 immunofluorescence signal only at late Parkinson’s disease stages (P < 0.001; P < 0.05), compared with both age-matched control and early Parkinson’s disease stages but not between early Parkinson’s disease stages and age-matched controls (P > 0.05). The optical density of the tyrosine hydroxylase immunofluorescence signal was also measured in individual cells that were KLC1 immunopositive from all Parkinson’s disease and age-matched control cases (Fig. 4D; P < 0.001). Post hoc analyses revealed a significant decline in the optical density of tyrosine hydroxylase immunofluorescence signal in the late stage of Parkinson’s disease (P < 0.05; P < 0.001), relative to both early Parkinson’s disease stages and age-matched controls but not between the early stage of Parkinson’s disease and the age-matched control (P > 0.05).
Figure 4.
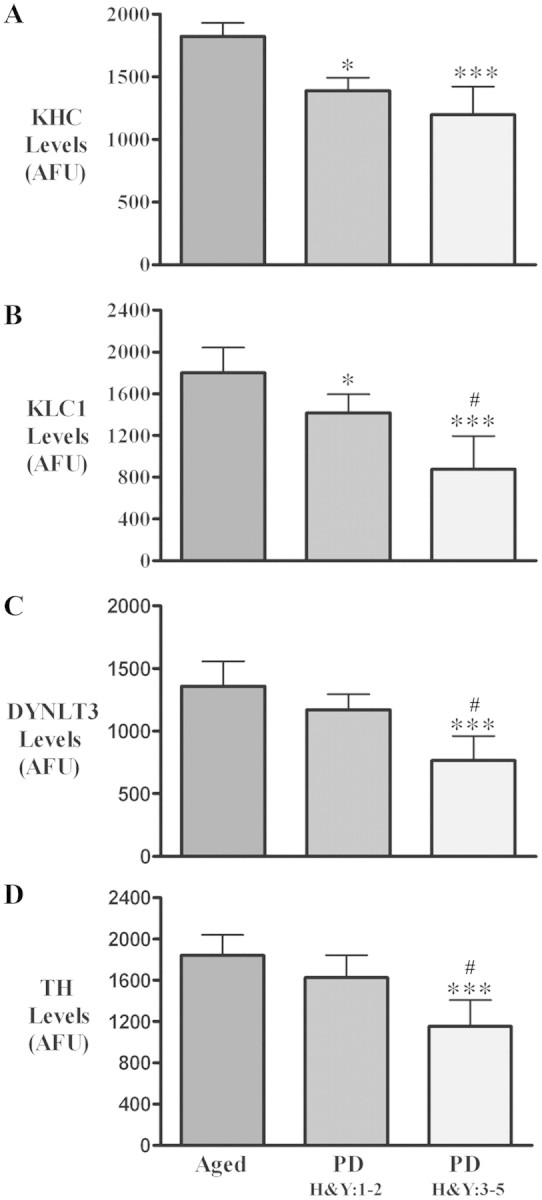
Histograms showing optical density values for KHC (A), KLC1 (B), DYNLT3 (C) and tyrosine hydroxylase (TH; D) fluorescent intensity within nigral neurons of the age-matched controls (aged; n = 9), Hoehn and Yahr stages 1–2 Parkinson’s disease (PD; n = 6) and Hoehn and Yahr stages 3–5 Parkinson’s disease (n = 10). Optical density values for KHC and KLC1, but not DYNLT3 and tyrosine hydroxylase, were significantly reduced in Hoehn and Yahr stages 1–2 Parkinson’s disease compared with age-matched controls. In Hoehn and Yahr stages 3–5 Parkinson’s disease, optical densities for KHC, KLC1, DYNLT3 and tyrosine hydroxylase were all significantly decreased relative to age-matched controls. [***P < 0.001, *P < 0.05 compared with age-matched controls; #P < 0.05 compared with Parkinson’s disease (Hoehn and Yahr 1–2)]. Data are mean ± SD. AFU = arbitrary fluorescence units.
Taken together, results from quantitative immunofluorescence analysis demonstrated significant reductions in conventional kinesin levels that precede tyrosine hydroxylase downregulation in remaining nigral neurons of cases with sporadic Parkinson’s disease. Downregulation of dynein DYNLT3 was observed in the late stage of Parkinson’s disease but not in the early stage of Parkinson’s disease.
Co-localization and quantitative analysis of axonal transport markers and α-synuclein in the nigrostriatal system in Parkinson’s disease
The results above indicated that a marked reduction in levels of critical axonal transport proteins in tyrosine hydroxylase-immunopositive neurons in sporadic Parkinson’s disease, whereas other neurons still displayed detectable-normal staining patterns. On the basis of results from our previous work (Chu et al., 2006, 2009), we hypothesized that the selective reductions in levels of axonal transport motor subunits were related to α-synuclein aggregation. To evaluate this possibility, we performed double labelling with anti-α-synuclein and either anti-KLC1 or anti-DYNLT3 antibodies and obtained the optical density measurements for KLC1 and DYNLT3 for neurons with or without α-synuclein-immunoreactive inclusions in sporadic Parkinson’s disease. Co-localization studies revealed that neurons both with or without α-synuclein-immunoreactive inclusions had significantly lower density of KLC1 immunoreactivity in Parkinson’s disease compared with age-matched controls (Fig. 5). Although cytoplasmic non-aggregated α-synuclein (Fig. 5B) was present within most nigral neurons in age-matched controls, levels of perikaryal KLC1-immunoreactive were similar among α-synuclein-positive and -negative neurons (Fig. 5C). In contrast, a marked reduction in levels of DYNLT3 immunoreactivity (Supplementary Fig. 3F) was only observed in Parkinson’s disease nigral neurons featuring α-synuclein-immunoreactive inclusions. On the basis of these findings, we hypothesized that Lewy neurites may be associated with reduced levels of axonal transport motor proteins. To assess this possibility, an antibody detecting the aggregation prone, pS129-α-synuclein was used (Gorbatyuk et al., 2008). Double-immunolabelling studies revealed that phosphorylated α-synuclein was mainly detected in aberrant tyrosine hydroxylase-immunoreactive fibres displaying swollen varicosities but not in morphologically normal tyrosine hydroxylase-immunoreactive fibres (Fig. 6A–F). Significantly, KHC1 immunoreactivity was absent in aberrant fibres displaying pS129-α-synuclein (Fig. 6G–I).
Figure 5.
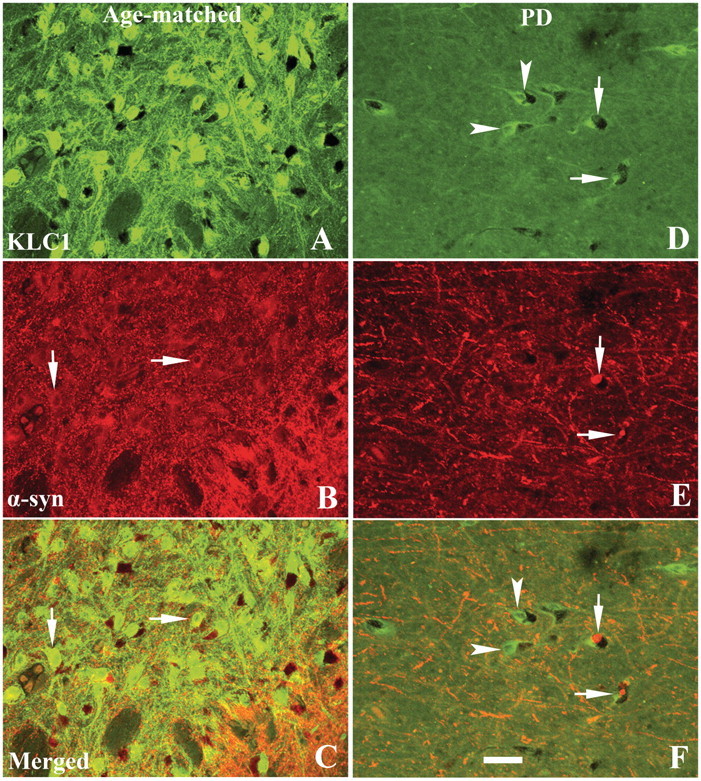
Confocal microscopic images of substantia nigra from age-matched control (A–C) and Hoehn and Yahr stage 3 Parkinson’s disease (PD) (D–F) illustrating immunostaining for KLC1 (green; A and D), α-synuclein (α-syn; red; B and E) and co-localization of KLC1 and α-synuclein (merged; C and F). Note that KLC1 immunofluorescence intensity was extensively reduced in both nigral neurons with (arrows, D–F) or without (arrowheads, D–F) α-synuclein inclusions, relative to age-matched controls (B and C). Nigral neurons labelled with cytoplasmic α-synuclein (no inclusion; arrows, B) in age-matched controls exhibited intensive KLC1 labelling that was similar in intensity to that of neurons without α-synuclein-immunoreactive cytoplasm. Scale bar = 80 µm (applies to all).
Figure 6.
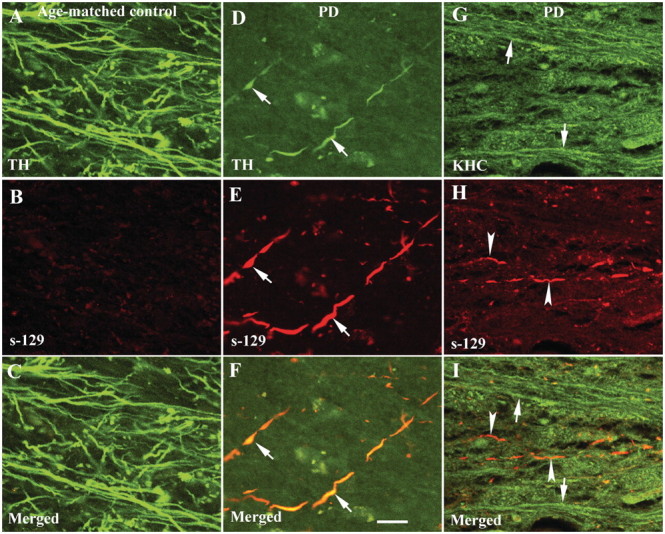
Confocal microscopic images of putamen from age-matched control (A–C) and Hoehn and Yahr stage 3 Parkinson’s disease (PD) (D–I) illustrating fibres labelled with tyrosine hydroxylase (TH; green; A and D), KHC (green; G), serine129-phosphorylated α-synuclein (s-129; red; B, E and H) and co-localization of tyrosine hydroxylase with s-129 (merged; C and F) and KHC with s-129 (merged; I). Note that processes featuring s-129 immunoreactivity (E and F) displayed light tyrosine hydroxylase labelling and swollen varicosities (arrows; D–F) compared with age-matched controls (A and C). Interestingly, there was no detectable KHC (arrowheads in I) in fibres filled with phosphorylated Ser-129 (arrowheads; H and I). Conversely, fibres stained with KHC (arrows; G and I) did not display detectable s-129 immunoreactivity. Scale bar = 20 µm (applies to all).
To unequivocally determine whether decreases in levels of axonal transport motors were associated α-synuclein inclusions in Parkinson’s disease, we analysed the relative intensities of KLC1 and DYNLT3 in nigral neurons that did or did not contain α-synuclein-positive inclusions. Kruskal–Wallis test revealed a statistically significant difference in optical density of KLC1-immunoreactive intensity across these groups (Fig. 7A; P < 0.001). Post hoc analyses revealed a significant decrease of KLC1-immunoreactive optical density in nigral neurons with (P < 0.001) and without (P < 0.001) α-synuclein-immunoreactive inclusions compared with controls. Furthermore, nigral neurons with α-synuclein inclusions in Parkinson’s disease displayed higher reductions in KLC1 immunoreactivity relative to Parkinson’s disease nigral neurons without α-synuclein inclusions (P < 0.05). Similarly, a significant difference in the optical density of DYNLT3-immunoreactive intensity was revealed between the three groups (Fig. 7B; P < 0.001). Interestingly, post hoc analyses revealed that the optical density of DYNLT3-immunoreactive intensity was only significantly decreased in nigral neurons with α-synuclein-immunoreactive inclusions (P < 0.001) but not in neurons without α-synuclein inclusions (P > 0.05) relative to age-matched controls.
Figure 7.

Histograms showing optical density values of KLC1 (A) and DYNLT3 (B) immunofluorescence intensity in nigral neurons with or without α-synuclein inclusions in both sporadic Parkinson’s disease (PD; n = 10) and age-matched control (n = 9) groups. Optical densities of KLC1 immunofluorescence intensity were significantly reduced in Parkinson’s disease neurons relative to age-matched controls, regardless of α-synuclein immunoreactivity levels, but neurons with α-synuclein inclusions displayed greater decreases of optical density of KLC1 immunofluorescence. The optical density of DYNLT3 immunofluorescence intensity was significantly reduced only in neurons with α-synuclein inclusions (B). (***P < 0.001; compared with age-matched controls; ###P < 0.001; ##P < 0.05 related to neurons without α-synuclein inclusion in Parkinson’s disease). Data are means ± SD. AFU = arbitrary fluorescence units.
These data indicate that in nigral neurons of cases with sporadic Parkinson’s disease, the decrease in levels of conventional kinesin subunits are independent of, but exacerbated by, α-synuclein inclusions, whereas reductions in DYNLT3 levels are selectively associated with accumulated α-synuclein inclusions.
Viral over-expression of α-synuclein in rat brain-induced alterations in tyrosine hydroxylase and axonal transport motor proteins
Results from immunohistochemical studies in sporadic Parkinson’s disease tissue led us to hypothesize that increased α-synuclein expression results in reduced levels of critical axonal transport proteins. To evaluate this possibility, we analysed a well-studied rat Parkinson’s disease model based on viral (rAAV) over-expression of mutant (A30P) α-synuclein within the substantia nigra (Kirik et al., 2003; Chu et al., 2011). Control rats received intranigral injections of rAAV encoding GFP. In control rats, robust GFP expression was seen in the substantia nigra and striatum (data not shown; Chu et al., 2011), but GFP expression did not affect tyrosine hydroxylase levels in the nigrostriatal system (Supplementary Fig. 4). The LB509 antibody, which selectively recognizes human α-synuclein protein (Chu et al., 2011), was used to examine the expression of viral-encoded human α-synuclein in rAAV-h-A30P-injected rats. Intense LB509 immunoreactivity was observed in the substantia nigra (Supplementary Fig. 4A and B) and striatum (Supplementary Fig. 4G and H) of rats injected with rAAV-h-A30P. As observed in cases with sporadic Parkinson’s disease, loss of tyrosine hydroxylase-immunoreactive terminals in striatum (Supplementary Fig. 4I and J) and neurons in substantia nigra (Supplementary Fig. 4C and D) was demonstrated in this Parkinson’s disease model (Chu et al., 2009, 2011; Ulusoy et al., 2010). Interestingly, tyrosine hydroxylase-immunoreactive fine fibres were dramatically decreased (Supplementary Fig. 4I and J) when α-synuclein-immunoreactive fibres were radically increased (Supplementary Fig. 4G) in the majority of the striatum of rAAV-h-A30P-injected rats. Some tyrosine hydroxylase-immunoreactive thick fibres still remained, but these displayed a distinct abnormal morphology characterized by swollen varicosities and intervaricose segments (Supplementary Fig. 4J). Thus, pathological alterations in rats that received rAAV-h-A30P mimicked the aberrant morphological features seen in cases with sporadic Parkinson’s disease.
Next, we examined the levels of axonal transport proteins in rAAV-h-A30P-injected rats. In substantia nigra of both uninjected and AAV-GFP-injected rats, robust KHC (Fig. 8E and Supplementary Fig. 5A) and DYNLT3 (Supplementary Figs. 5D and 6E) immunoreactivities were documented. KHC and DYNLT3 immunostaining profiles comprised both soma and processes of tyrosine hydroxylase-positive neurons. Double immunostaining revealed that virtually all tyrosine hydroxylase-immunoreactive nigral neurons were also KHC- (Supplementary Fig. 5C) and DYNLT3 positive (Supplementary Fig. 5F).
Figure 8.
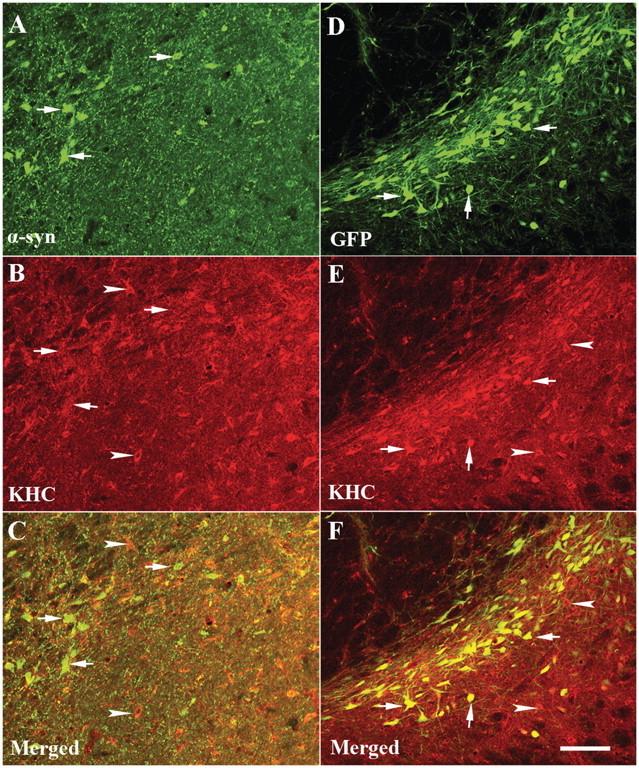
Laser confocal microscopy images of substantia nigra illustrating immunoreactivities for human α-synuclein (α-syn; green; A), KHC (red; B) and merged (C) from rats with targeted expression of human mutant (A30P) α-synuclein and green fluorescence protein (GFP; green; D), KHC (red; E) and merged (F) from rat with target expression GFP. Note that the KHC immunofluorescent intensity was diminished by targeting expression of α-synuclein. KHC immunoreactivity was observed in the cells without α-synuclein immunoreactivity (arrowheads; B and C) but not in cells with α-synuclein immunoreactivity (arrows; A–C). In contrast, GFP-positive neurons displayed intensive KHC immunostaining (arrows; E and F) similar to the GFP-negative cells (arrowheads). Scale bar = 110 µm (applies to all).
In the substantia nigra of AAV-h-A30P-injected rats, some neurons displayed a marked loss of KHC (Fig. 8B) and DYNLT3 (Supplementary Fig. 6B) staining, whereas others retained an intense immunoreactive profile. Double labelling of KHC and DYNLT3 with anti-α-synuclein antibody revealed two groups of neurons in AAV6-h-A30P-injected substantia nigra. One group did not display α-synuclein immunoreactivity, and these cells showed robust expression of both KHC and DYNLT3 markers. The second group expressed α-synuclein and displayed a marked reduction in KHC (Fig. 8C) and DYNLT3 (Supplementary Fig. 6C) staining levels. Aggregated pS129-α-synuclein was observed in swollen tyrosine hydroxylase-immunoreactive fibres (Fig. 9D–F) in the striatum of rats that received AAV6-h-A30P. KHC levels were undetectable in fibres labelled with phosphorylated α-synuclein (Fig. 9G–I), similar to what was observed in Parkinson’s disease striatum (Fig. 6).
Figure 9.
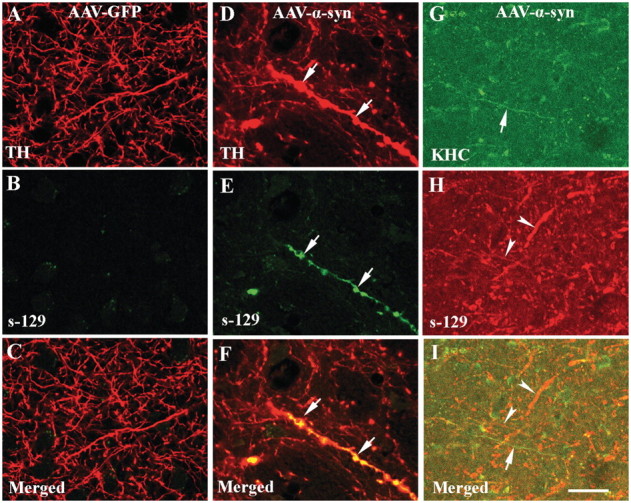
Confocal microscopic images of putamen from rats with targeted expression of either green fluorescent protein (AAV-GFP; A–C) or human mutant (A30P) α-synuclein (AAV-α-syn; D–I) illustrating fibres labelled with tyrosine hydroxylase (TH; red; A and D), KHC (green; G), phosphorylated Ser-129 α-synuclein (s-129; green; B and E, red; H) and co-localization of s-129 with tyrosine hydroxylase (merge; C and F) or s-129 with KHC (merged; I). Note that axonal fibres filled with phosphorylated Ser-129 (arrows; E) displayed swollen varicosities (D and F; arrows). Interestingly, KHC was undetectable in axonal fibres filled with phosphorylated Ser-129 (G–I; arrowheads) but abundant in fibres where phosphorylated Ser-129 labelling was absent (G and I; arrows). There was no significant immunoreactivity for phosphorylated Ser-129 in rats with targeted expression of GFP (B). Scale bar = 20 µm (applies to all).
Qualitative observations were confirmed by quantified fluorescence intensity measurements for KHC and DYNLT3. The optical density measurements of KHC and DYNLT3 were performed on the cell body on a per neuron basis in nigral neurons with and without α-synuclein or GFP accumulation as described earlier. Kruskal–Wallis test revealed a statistically significant difference in optical density of KHC-immunoreactive intensity (P < 0.001; Fig. 10A). Post hoc analyses revealed that the optical density of KHC was significantly decreased in nigral neurons with (P < 0.001) and without α-synuclein-immunoreactive (P < 0.001) accumulation in rats that received AAV6-h-A30P but not in GFP-positive (P > 0.05) and GFP-negative (P > 0.05) neurons compared with uninjected controls. Similarly, optical density of DYNLT3-immunoreactive intensity was significantly decreased (P < 0.001; Fig. 10B) in rats that received AAV6-h-A30P. Post hoc analyses revealed that optical density of DYNLT3-immunoreactive intensity was significantly reduced in both nigral neurons with and without α-synuclein-immunoreactive accumulation (P > 0.001) compared with controls. The neurons with α-synuclein-immunoreactive accumulation exhibited more reduction of DYNLT3-immunoreactive intensity (P < 0.05) relative to the neuron without α-synuclein-immunoreactive accumulation. There was no difference on optical density of DYNLT3-immunoreactive intensity in the GFP-positive (P > 0.05) and -negative (P > 0.05) neurons compared with controls.
Figure 10.

Histogram showing optical density values of KHC (A) and DYNLT3 (B) immunofluorescence intensity within nigral neurons from uninjected rats (control; n = 8), rats with targeted expression of GFP (n = 8) and rats with targeted expression of human mutant (A30P) α-synuclein (α-syn; n = 8). In rats with targeted expression of human A30P α-syn, optical density values of KHC (A) and DYNLT3 (B) immunofluorescence intensity were significantly reduced in neurons with α-synuclein (α-syn+) and without α-synuclein (α-syn−), relative to control rats. In rats with targeted expression of human GFP, optical densities of KHC and DYNLT3 were unchanged in both GFP-positive (GFP+) and -negative (GFP−) neurons, compared with control rats. (***P < 0.001; compared with controls; #P < 0.05 related to neurons without α-synuclein). Data are means ± SD. AFU = arbitrary fluorescence units.
Discussion
In this study, we examined the expression of critical axonal transport motor proteins in remaining nigral dopaminergic neurons throughout different stages of sporadic Parkinson’s disease. The major findings of this study are as follows: (i) conventional kinesin levels are significantly reduced in remaining nigral neurons in sporadic Parkinson’s disease. Significantly, this reduction occurs at very early Parkinson’s disease stages, suggesting that abnormalities in axonal transport might represent a critical pathological change in sporadic Parkinson’s disease. Interestingly, the reductions in conventional kinesin levels precede the alterations in dopaminergic phenotypic markers and the aberrant morphology of tyrosine hydroxylase-immunoreactive fibres characteristic of early (stages 1–2) Parkinson’s disease; (ii) the reductions in conventional kinesin levels appear in the presence or absence of perikaryal α-synuclein aggregations, the presence of α-synuclein inclusions exhibit much more decline; (iii) unlike conventional kinesin, reductions in the levels of the DYNLT3 subunit of cytoplasmic dynein were only observed at late Parkinson’s disease stages; and (iv) reductions in levels of conventional kinesin and cytoplasmic dynein subunits were recapitulated in a rat model of familial Parkinson’s disease based on over-expression of human mutant α-synuclein (A30P).
What remains to be established is whether the alterations in axonal transport result in the accumulation of α-synuclein or whether the accumulation of α-synuclein disturbs axonal transport. α-Synuclein normally functions as a synaptic protein (Iwai et al., 1995). It is synthesized in nigral perikarya and transported to the synapse so quickly that it is undetectable in nigral cell bodies in young non-human primates and young humans (Chu and Kordower, 2007). In aged monkeys and aged humans, α-synuclein can be detected, and this is a time in these primates’ life where axonal transport defects have been noted (Li et al., 2004; Cross et al., 2008). Supported by the present data, it is therefore tempting to speculate that defects in axonal transport precede other changes leading to an accumulation of α-synuclein within axon and perikarya. So why is this accumulation of α-synuclein not cleared like other unwanted proteins? We have previously shown that, in Parkinson’s disease, increases in α-synuclein are associated with decreases in lysosomal and proteosomal proteins that normally clear excess α-synuclein (Chu et al., 2009). These defects in autophagy-related proteins seen in Parkinson’s disease and associated specifically with α-synuclein accumulation can also be recapitulated with viral over-expression of α-synuclein. Taken together, these data support the conclusion that defects in axonal transport mechanisms are a primary event in nigral cell pathogenesis leading to an abnormal accumulation in perikaryal α-synuclein. This increase in α-synuclein leads to a defective autophagic clearance of this unwanted protein, starting a cycle of continually increasing α-synuclein, which is now poorly transported to its normal synaptic location and poorly cleared by perikaryal proteins such as LAMP1, the lysosomal protease cathepsin D and the 20S proteasome (Chu et al., 2009). We believe that this continued accumulation ultimately overwhelms these clearance mechanisms resulting in the misfolding, fibrillization and aggregation of α-synuclein, a process that exacerbates synaptic loss, loss of dopaminergic phenotype, striatal denervation and ultimately cell death. The fact that defects in kinesins can be seen in α-synuclein-positive and -negative nigral neurons supports this hypothesis. However, one can still not rule out the possibility that α-synuclein is the primary culprit, and it is α-synuclein that impairs axonal transport, setting this vicious cycle in motion. Indeed, this study demonstrates that viral over-expression of α-synuclein alone reduces the expression of kinesins and dynein. This scenario deserves further scrutiny as well.
Post-mortem studies must be interpreted with caution as factors such as disease heterogeneity and post-mortem interval can influence the results. To support our findings, we injected rats with viral vectors inducing the over-expression of human α-synuclein. rAAV-h-A30P caused reductions in kinesin and dynein accompanied by decrease of tyrosine hydroxylase immunoreactivity in striatum. Axons exhibit aberrant morphological profiles with axonal swelling and enlarged intervaricose segments, both hallmarks of axonal injury (Chung et al., 2009). The data from AAV-h-A30P-injected animals exhibited reductions in kinesin and dynein levels and abnormal axonal blockage were similar to those observed in early stage human Parkinson’s disease brains. These pre-clinical data support the accuracy of the human post-mortem findings.
A recent report indicated that viral over-expression of human mutant (A53T) α-synuclein results in a decrease in anterograde transport motor proteins in striatum but an increase in the substantia nigra 8 weeks after the injection (Chung et al., 2009). Our results from 6 weeks following viral α-synuclein over-expression differ from Chung et al. (2009) in that we observed decreases in both the striatum and nigra. There are a number of explanations for these divergent results including those that may be related to the use of different mutant human α-synuclein, different promoters and different methods of protein evaluation (western blots versus individual neuron evaluations taking into account the presence or absence of α-synuclein). Our data from rat genetic Parkinson's disease mode are similar to what we observed in the Parkinson's disease brain therefore supporting our preclinical data.
Natural α-synuclein can be co-localized with axonal transport proteins kinesin-1 and dynein and transported along axons (Jensen et al., 1999; Utton et al., 2005). Over-expression of human wild-type α-synuclein enhances axonal degeneration after peripheral nerve lesion in transgenic mice (Siebert et al., 2010). α-Synuclein accumulation was found at sites of axonal damage in the PNS (Moran et al., 2001; Siebert et al., 2010). These studies suggest that α-synuclein accumulation is related to axonal damage. Our results further demonstrate that α-synuclein accumulation in axons is associated with a decline in kinesin along with decline of striatal tyrosine hydroxylase immunoreactivity. Alternatively, on encountering terminal damage, vesicles containing α-synuclein might undergo retrograde transport to the somatodendritic compartment, where aberrant α-synuclein generation and deposition could occur. Facial nerve axotomy results in higher amounts of immunopositive α-synuclein in perikarya of neurons (Moran et al., 2001), verifying the hypotheses that α-synuclein might undergo retrograde transport to the somatodendritic compartments after axonal damage. This process could be enhanced by the reduction of kinesin and the increase of dynein. Our results revealed that there was a severe decrease of kinesin but not dynein in the early stage of Parkinson’s disease. If accumulation of α-synuclein occurs at sites of abnormal deposition of axonally transported cargos, reduced axonal transport may lead to local stimulation of phosphorylated α-synuclein processing and further inhibition of axonal transport. This proposed sequence of events would generate an autocatalytic spiral, in which processes leading to inhibition of axonal transport and phosphorylated α-synuclein production become mutually stimulatory, providing a rational explanation for early striatal synaptic loss in Parkinson’s disease.
In summary, the present data indicate that reductions in axonal transport mechanisms are present during early stages in nigrostriatal pathogenesis in Parkinson’s disease. Both anterograde and retrograde motors are affected with anterograde motors (kinesins) being defective first. These data were recapitulated in rats with targeted viral over-expression of α-synuclein. Taken together, these data suggest that therapies aimed at preventing or restoring defective axonal transport function should be investigated.
Funding
This work was supported by the Rush Neurological Sciences [internal grant to Y.C. and J.H.K].
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- DYNLT3
dynein light chain Tctex type 3
- KHC
kinesin heavy chain
- KLC1
kinesin light chain 1
- rAAV
recombinant adeno-associated virus
References
- Chu Y, Kordower JH. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson’s disease? Neurobiol Dis. 2007;25:134–49. doi: 10.1016/j.nbd.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis. 2009;35:385–98. doi: 10.1016/j.nbd.2009.05.023. [DOI] [PubMed] [Google Scholar]
- Chu Y, Le W, Kompoliti K, Jankovic J, Mufson EJ, Kordower JH. Nurr1 in Parkinson’s disease and related disorders. J Comp Neurol. 2006;494:495–514. doi: 10.1002/cne.20828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Mickiewicz AL, Kordower JH. α-Synuclein aggregation reduces nigral myocyte enhancer factor-2D in idiopathic and experimental Parkinson’s disease. Neurobiol Dis. 2011;41:71–82. doi: 10.1016/j.nbd.2010.08.022. [DOI] [PubMed] [Google Scholar]
- Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci. 2009;29:3365–73. doi: 10.1523/JNEUROSCI.5427-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DJ, Flexman JA, Anzai Y, Maravilla KR, Minoshima S. Age-related decrease in axonal transport measured by MR imaging in vivo. Neuroimage. 2008;39:915–26. doi: 10.1016/j.neuroimage.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. [Review] Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, et al. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–12. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, et al. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–38. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, et al. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci USA. 2008;105:763–8. doi: 10.1073/pnas.0711053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoeva FK, Bybikova EM, Minin AA. An isoform of kinesin light chain specific for the Golgi complex. J Cell Sci. 2000;113:2047–54. doi: 10.1242/jcs.113.11.2047. [DOI] [PubMed] [Google Scholar]
- Iseki E, Kato M, Marui W, Uéda K, Kosaka K. A neuropathological study of the disturbance of the nigro-amygdaloid connections in brains from patients with dementia with Lewy bodies. J Neurol Sci. 2001;185:129–34. doi: 10.1016/s0022-510x(01)00481-6. [DOI] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, et al. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–75. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- Jakes R, Crowther RA, Lee VM, Trojanowski JQ, Iwatsubo T, Goedert M. Epitope mapping of LB509, a monoclonal antibody directed against human alpha-synuclein. Neurosci Lett. 1999;269:13–6. doi: 10.1016/s0304-3940(99)00411-5. [DOI] [PubMed] [Google Scholar]
- Jensen PH, Li JY, Dahlström A, Dotti CG. Axonal transport of synucleins is mediated by all rate components. Eur J Neurosci. 1999;11:3369–76. doi: 10.1046/j.1460-9568.1999.00754.x. [DOI] [PubMed] [Google Scholar]
- Kanaan NM, Kordower JH, Collier TJ. Age-related accumulation of Marinesco bodies and lipofuscin in rhesus monkey midbrain dopamine neurons: relevance to selective neuronal vulnerability. J Comp Neurol. 2007;502:683–700. doi: 10.1002/cne.21333. [DOI] [PubMed] [Google Scholar]
- Kirik D, Annett LE, Burger C, Muzyczka N, Mandel RJ, Björklund A. Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson’s disease. Proc Natl Acad Sci USA. 2003;100:2884–9. doi: 10.1073/pnas.0536383100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Hoffman PN, Stirling W, Price DL, Lee MK. Axonal transport of human alpha-synuclein slows with aging but is not affected by familial Parkinson’s disease-linked mutations. J Neurochem. 2004;88:401–10. doi: 10.1046/j.1471-4159.2003.02166.x. [DOI] [PubMed] [Google Scholar]
- Lo KW, Kogoy JM, Pfister KK. The DYNLT3 light chain directly links cytoplasmic dynein to a spindle checkpoint protein, Bub3. J Biol Chem. 2007;282:11205–12. doi: 10.1074/jbc.M611279200. [DOI] [PubMed] [Google Scholar]
- Lykkebo S, Jensen PH. Alpha-synuclein and presynaptic function: implications for Parkinson’s disease.[Review] Neuromol Med. 2002;2:115–29. doi: 10.1385/NMM:2:2:115. [DOI] [PubMed] [Google Scholar]
- Madine J, Doig AJ, Middleton DA. A study of the regional effects of alpha-synuclein on the organization and stability of phospholipid bilayers. Biochemistry. 2006;45:5783–92. doi: 10.1021/bi052151q. [DOI] [PubMed] [Google Scholar]
- Mbefo MK, Paleologou KE, Boucharaba A, Oueslati A, Schell H, Fournier M, et al. Phosphorylation of synucleins by members of the Polo-like kinase family. J Biol Chem. 2010;285:2807–22. doi: 10.1074/jbc.M109.081950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran LB, Kösel S, Spitzer C, Schwaiger FW, Riess O, Kreutzberg GW, et al. Expression of alpha-synuclein in non-apoptotic, slowly degenerating facial motoneurones. J Neurocytol. 2001;30:515–21. doi: 10.1023/a:1015697318437. [DOI] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, et al. Axonal transport defects in neurodegenerative diseases. [Review]. J Neurosci. 2009;29:12776–86. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Opalach K, Serulle Y, Moreira JE, Sugimori M, et al. 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc Natl Acad Sci USA. 2007;104:2442–47. doi: 10.1073/pnas.0611231104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Lavine N, Jaffar S, Kordower JH, Quirion R, Saragovi HU. Reduction in p140-TrkA receptor protein within the nucleus basalis and cortex in Alzheimer’s disease. Exp Neurol. 1997;146:91–103. doi: 10.1006/exnr.1997.6504. [DOI] [PubMed] [Google Scholar]
- Perez SE, Lazarov O, Koprich JB, Chen EY, Rodriguez-Menendez V, Lipton JW, et al. Nigrostriatal dysfunction in familial Alzheimer’s disease-linked APPswe/PS1DeltaE9 transgenic mice. J Neurosci. 2005;25:10220–9. doi: 10.1523/JNEUROSCI.2773-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister KK, Benashski SE, Dillman JF, III, Patel-King RS, King SM. Identification and molecular characterization of the p24 dynactin light chain. Cell Motil Cytoskeleton. 1988;41:154–67. doi: 10.1002/(SICI)1097-0169(1998)41:2<154::AID-CM6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. [Review] Science. 2002;296:868–71. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Roy S, Zhang B, Lee VM, Trojanowski JQ. Axonal transport defects: a common theme in neurodegenerative diseases. [Review] Acta Neuropathol. 2005;109:5–13. doi: 10.1007/s00401-004-0952-x. [DOI] [PubMed] [Google Scholar]
- Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–95. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serulle Y, Morfini G, Pigino G, Moreira JE, Sugimori M, Brady ST, et al. 1-Methyl-4-phenylpyridinium induces synaptic dysfunction through a pathway involving caspase and PKCdelta enzymatic activities. Proc Natl Acad Sci USA. 2007;104:2437–41. doi: 10.1073/pnas.0611227104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert H, Kahle PJ, Kramer ML, Isik T, Schlüter OM, Schulz-Schaeffer WJ, et al. 2010) Over-expression of alpha-synuclein in the nervous system enhances axonal degeneration after peripheral nerve lesion in a transgenic mouse strain. J Neurochem. 2010;114:1007–18. doi: 10.1111/j.1471-4159.2010.06832.x. [DOI] [PubMed] [Google Scholar]
- Tomishige M, Stuurman N, Vale RD. Single-molecule observations of neck linker conformational changes in the kinesin motor protein. Nat Struct Mol Biol. 2006;13:887–94. doi: 10.1038/nsmb1151. [DOI] [PubMed] [Google Scholar]
- Towne C, Raoul C, Schneider BL, Aebischer P. Systemic AAV6 delivery mediating RNA interference against SOD1: neuromuscular transduction does not alter disease progression in fALS mice. Mol Ther. 2008;16:1018–25. doi: 10.1038/mt.2008.73. [DOI] [PubMed] [Google Scholar]
- Ulusoy A, Decressac M, Kirik D, Björklund A. Viral vector-mediated overexpression of α-synuclein as a progressive model of Parkinson’s disease. [Review] Prog Brain Res. 2010;184:89–111. doi: 10.1016/S0079-6123(10)84005-1. [DOI] [PubMed] [Google Scholar]
- Utton MA, Noble WJ, Hill JE, Anderton BH, Hanger DP. Molecular motors implicated in the axonal transport of tau and alpha-synuclein. J Cell Sci. 2005;118:4645–54. doi: 10.1242/jcs.02558. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.