

Abstract
Colorectal cancer can be prevented by the removal of adenomatous polyps during screening colonoscopy, but adequate bowel preparation is required. Oral sodium phosphate (OSP), an effective bowel purgative, is available over the counter and requires a substantially lower volume than polyethylene glycol-based preparative agents. Accumulating reports implicate OSP in electrolyte disturbances as well as acute kidney injury (AKI) in a syndrome termed phosphate nephropathy (a form of nephrocalcinosis). Despite published case reports and case series, the actual incidence, risk factors, and natural history of phosphate nephropathy remain largely undefined. Several recent observational studies have provided new information on these important issues while supporting a link between OSP and acute phosphate nephropathy as well as the development of chronic kidney disease in elderly patients, many of whom had a normal serum creatinine at the time of OSP ingestion. This review summarizes current knowledge about the renal complications of OSP, risk factors for its development, and the pathophysiology of acute and chronic kidney damage in nephrocalcinosis.
Approximately 14 million colonoscopies are performed in the United States yearly for colon cancer screening, and efforts to increase screening rates have included endorsements by celebrities (1,2). The diagnostic accuracy and cost-effectiveness of colonoscopy are closely related to the quality of the colon preparation, and yet methods to safely, effectively, quickly, comfortably, and affordably prepare the colon remain elusive (3). In clinical trials, nearly 75% of subjects undergoing bowel preparation report adverse events, most commonly abdominal distention, nausea, vomiting, abdominal pain, and dizziness (4).
Methods of bowel preparation have evolved from the traditional approach of dietary restriction and enemas, which although effective, are time-consuming and uncomfortable (5). An early preparative agent was mannitol, but it results in the production of methane, hydrogen, and other flammable gases and has been associated with fatal colonic explosions during polypectomy and electrocautery (6,7). Large volumes of saline or balanced electrolyte lavage solutions are also effective, but their use results in significant net fluid and electrolyte absorption. A significant advance occurred when Davis et al. at Baylor University substituted nonabsorbable sodium sulfate in place of sodium chloride and added polyethylene glycol for osmotic effect (8). They called this iso-osmolar, nonabsorbable solution “GoLytely,” although it is more generally known as polyethylene-glycol electrolyte lavage solution (PEG-ELS) (8). A version known as Nu-Lytely, formulated without the unpalatable ingredient sodium sulfate, became available in 1990 (9). Recent formulations have combined half-dose Nu-Lytely (2 L rather than 4 L) with Bisacodyl tablets (called Half-Lytely).
Despite the effectiveness and relative safety of PEG-ELS, many patients complain about the taste and the large volume they must drink. The search for alternatives to standard PEG-ELS preps led back to oral sodium phosphate (OSP) preparations, which have existed for many years. In early studies comparing the two approaches, patients and colonoscopists preferred OSP (10–12). A recent meta-analysis concluded that OSP preps result in better bowel cleansing, perhaps because patients are more likely to complete them (13). An industry-sponsored study that compared OSP tablets with PEG-ELS + bisacodyl concluded that OSP is more efficacious and better tolerated (4).
Like lactulose and milk of magnesia (magnesium hydroxide), OSP preparations are osmotic purgatives that obligate water excretion into the intestinal lumen to maintain its isotonicity with plasma (in contrast to the kidney, the proximal small intestine does not create or maintain an osmotic gradient between the luminal and blood side) (14). The retention of water in the bowel lumen results in peristalsis and colonic evacuation (15). It is the high sodium and phosphate content of OSP preps, which render them hyperosmolar. Each 45-ml dose of Fleet's Phospho-soda contains 5 g of sodium and 17.8 g of phosphate, yielding a solution with an osmolarity of 16,622 mOsm/L (748 mOsm/45 ml) (14). The amount of elemental phosphorus in two 45-ml doses is approximately 11.5 g, a substantial load compared with the typical daily dietary intake of 1 to 1.5 g (16). Two OSP tablet preparations are also approved for use: Visicol, which is similar in content to Fleet's Phospho-soda, and Osmo-Prep, which contains approximately 20% less phosphate than Visicol (15). All OSP preparations lead to both sodium and phosphate absorption. If phosphate absorption did not occur, a 100-ml dose would obligate 4.1 L of stool (17).
Metabolic Disturbances Associated with Bowel Preparation: a “Forgotten Menace?”
Metabolic disturbances, including hyperphosphatemia, hypocalcemia, hypernatremia, hyponatremia, hypokalemia, and anion-gap metabolic acidosis, have been reported in association with OSP preps, sometimes accompanied by volume depletion and AKI typically attributed to tubular injury (16,18–20). Because 1 to 1.8 L of hypotonic fluid is lost after the use of a 45-ml dose of OSP solution, dehydration, volume depletion, and hypernatremia are not uncommon (17,21,22). Osmotic diuresis resulting from the high concentration of intratubular phosphate may contribute to the volume depletion (23). Central pontine myelinolysis has been reported in a patient whose sodium rose to 180 mMol/L after an OSP prep (24).
In contrast, when excess water is retained during bowel preparation, severe hyponatremia and its associated complications can occur (25,26). Two deaths from hyponatremia have been reported in patients with end-stage renal disease who received PEG-ELS, which has also resulted in hyponatremic seizures in patients with normal renal function (26,27). In these cases, the affected patients consumed significant quantities of hypotonic fluids in addition to the PEG-ELS prep. The nonosmotic release of antidiuretic hormone, possibly as a result of nausea and stress, appears common during colonoscopy and likely impairs the ability to excrete free water (28). Patients with significant chronic kidney disease (CKD) may on rare occasion experience a significant increase in plasma volume after PEG-ELS and decompensated heart failure may result (29,30).
Hyperphosphatemia after OSP ingestion routinely occurs even in individuals with normal renal function, with one study reporting a rise in the mean serum phosphate from 3.7 to 7.3 mg/dl (10,31,32). In another study, calcium-phosphate products exceeding 65 were observed in more than one third of normal volunteers (21). Severe hyperphosphatemia complicated by hypocalcemia and tetany have been reported in those with both abnormal and normal renal function (Figure 1) (33–37). Older patients, those with abnormal gut motility (which enhances phosphate absorption) and those who have received repeated doses of OSP, have experienced particularly severe electrolyte disturbances and deaths (38–41). Enemas that contain phosphate can also result in severe hyperphosphatemia and AKI as well as other complications (42,43).
Figure 1.

Clinical course observed in a 74-yr-old who received 90 ml of oral sodium phosphate solution before colonoscopy. (36) Reproduced by permission, © Georg Thieme Verlag KG.
Some authorities recognized the risk of electrolyte disturbances from OSP early on and counseled against their use, particularly in patients with comorbid conditions such as kidney, liver, or heart disease (44,45). The first FDA review of the safety of OSP occurred in 2001 and resulted in a report urging physician awareness and recommending monitoring of electrolytes in high-risk patients (46). In the face of ongoing reports of metabolic complications, one group termed OSP “a forgotten menace” (36).
Kidney Injury after OSP Bowel Preps: Biopsy Studies
More recent attention has been directed to the possibility that in some patients the deposition of calcium-phosphate crystals after OSP administration may result in AKI. In a 2003 letter to the New England Journal of Medicine, Desmeules et al. reported a 71-yr-old whose creatinine rose from 1.0 to 4.5 mg/dl in a 10-wk period after the use of a OSP solution administered before colonoscopy (47). Analysis of the kidney biopsy specimen by light microscopy, scanning electron microscopy, and x-ray microanalysis confirmed the presence of intratubular calcium-phosphate deposits in the form of hydroxyapatite. Kidney function remained abnormal one year later. The authors called the patient's condition “phosphate nephropathy.”
In 2004, Markowitz et al. at Columbia University extended this report by describing 5 patients who developed AKI after OSP preps, all of whom had distal tubular injury and calcium-phosphate deposits demonstrated on kidney biopsy (48). Subsequently, the same group reviewed 7349 kidney biopsies and identified an additional 16 patients who appeared to have phosphate nephropathy associated with OSP (49). The mean age of the 21 total patients identified by Markowitz et al. was 64, and at baseline 17 of them had good renal function (mean creatinine <1.2 mg/dl). Two thirds (14 of 21) were receiving angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) at the time of OSP administration, and several were on nonsteroidal anti-inflammatory drugs or diuretics. Many of them were left with CKD: at follow-up roughly 17 mo after OSP exposure, the mean serum creatinine was 2.4, and 4 of 21 were dialysis dependent. Additional biopsy-proven reports of phosphate nephropathy have appeared, including one in which a patient had two kidney biopsies, the first of which showed membranous nephropathy and the second performed two months after the first, after an OSP prep, which showed membranous nephropathy plus de novo calcium-phosphate deposits not present on the first biopsy (50–53). Another patient with biopsy-proven phosphate nephropathy after OSP presented with acute visual loss from uremic optic neuropathy (54).
Kidney Injury after OSP: Observational Studies
Although dramatic, the biopsy-based case series of Markowitz et al. (48,49) and others leave unanswered important questions about the mechanisms by which OSP might result in phosphate nephropathy and do not address the risk of this complication in a population of patients who receive OSP. Recently, several observational studies of patients with preserved renal function who underwent bowel preparation have appeared and have reached conflicting conclusions about OSP and the risk of kidney injury (55–58).
Using a U.S. Department of Defense data system, Hurst et al. studied 9799 patients who underwent colonoscopy and identified 114 who had a >50% increase in baseline creatinine after the procedure (56). After adjustment for confounders in a multivariate analysis, the use of OSP was associated with increased risk of renal failure (odds ratio = 2.35). This association became stronger when the authors limited their analysis to patients with more severe kidney injury (defined as a doubling of serum creatinine). Older age was an independent risk factor for kidney injury from OSP. At follow-up an average of 8 months after the colonoscopy, the mean creatinine among the group with kidney injury (1.38) was below the peak value (1.78) but remained substantially above the preprocedure value (0.98), supporting the hypothesis that OSP causes CKD in addition to AKI. In the same issue, Brunelli et al. reported a case-control study using data from a cohort of 2237 patients who underwent colonoscopy at hospitals associated with the University of Pennsylvania (55). Using a definition of kidney injury that was somewhat less restrictive than that of Hurst et al. (56), the authors identified 116 patients with kidney injury after colonoscopy but were unable to find any association with OSP use. Patients who received OSP preps and were taking ACE inhibitors or angiotensin receptor blockers did appear to be at increased risk for kidney injury compared with those who were not.
Investigators from the Degge Group, a drug safety consulting firm based in Arlington, VA, working with collaborators and data from the Henry Ford Health System and grant support from the C.B. Fleet Company, studied 2352 patients with baseline normal renal function who underwent colonoscopy with OSP or PEG-ELS preparation and had creatinine determinations within 6 months of the procedure; 3.8% of OSP recipients and 3.3% of PEG-ELS recipients developed kidney injury, defined by a reduction in estimated glomerular filtration rate (GFR) to <60 ml/min. The adjusted odds ratio for GFR decline was 1.14 in the OSP group relative to PEG-ELS, but this difference was not statistically significant (confidence interval, 0.55 to 2.39). The authors concluded that the risk of renal impairment is similar with both preparations.
Khurana et al., who had previously reported a series of 12 cases of clinically diagnosed cases of phosphate nephropathy, subsequently reported a retrospective case-control study and found a statistically significant decline in GFR 6 months after OSP exposure (58,59). ACE and ARB use and diabetes mellitus were again identified as risk factors. Like the Hurst et al. study discussed earlier (56), this study also appears to identify a subset of patients who do not develop clinically manifest AKI but nonetheless end up with CKD as a result of the exposure. Of note, the control group included patients who had not received colonoscopy as well as patients who had not developed renal failure after colonoscopy. Selecting a control group by excluding patients with the outcome of interest will inevitably bias a study toward a positive result.
In conclusion, two of these four observational studies support an association between OSP and kidney injury and two do not. The reasons for these different results may lie in study methodologies, including the different definitions of kidney injury as well as the interval after colonoscopy at which the renal function was assessed. Selection of patients from different eras may have also influenced the results: whereas Hurst et al. (56) studied colonoscopy procedures conducted from 2002 through 2006, Brunelli et al. (55) assessed procedures from 2004 and 2005, many of which were performed coincident with or after the Markowitz et al. report (48), which might have biased providers against OSP. In each of these studies, patients who receive PEG-ELS appear at baseline to be at higher risk for kidney injury than patients who receive OSP, reflecting the widespread (although not universal) awareness among providers concerning the potential risk of OSP; thus, residual confounding or bias if present would skew the results in favor of OSP safety. Clearly, further studies are required to precisely determine the incidence of both AKI and CKD after OSP preparation. Randomized trials could eliminate the problem of residual confounding but would be limited to low-risk patients, which might not reflect the patients exposed to OSP in actual clinical practice. Studies that report 6- to 12-month follow-up of patients who develop metabolic complications after PEG-ELS are also absent from the literature. Further work is also required to define how and when CKD can complicate OSP in the absence of AKI, which will require stricter definition of the two entities. As discussed below, different pathophysiologic processes might explain these different clinical presentations.
Mechanism of Kidney Injury from OSP: Nephrocalcinosis
As is typical in clinical nephrology, the AKI associated with OSP is likely multifactorial. Advanced age and ACE/ARB use have been identified in both case series and population studies as risk factors. Additional putative risk factors, which have not been rigorously defined, are shown in Table 1 and include acute or CKD or the presence of a kidney transplant, excessive or repeated dosing, retention of OSP resulting from poor bowel motility or colitis, female gender, true or effective volume depletion from congestive heart failure or cirrhosis, a history of hypertension or diabetes, and diuretic, lithium, or nonsteroidal anti-inflammatory drug use (31,33,49,60). Although these factors are familiar to nephrologists as risks for prerenal azotemia and acute tubular injury, a unique aspect of kidney injury associated with OSP is the deposition of calcium-phosphate crystals, also known as nephrocalcinosis. It seems likely that mineral deposition is less reversible than the typical lesion of acute tubular necrosis and thus might predispose patients to the CKD observed by Markowitz et al. (48) and others.
Table 1.
Putative risk factors for phosphate nephropathy
| Preexisting acute or chronic kidney disease or a kidney transplant |
| Advanced age |
| Female gender |
| Hypertension or diabetes mellitus |
| True or effective volume depletion |
| Abnormal bowel motility |
| Angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, diuretic, lithium or nonsteroidal anti-inflammatory drug use |
| Excessive/repeated dosing of oral sodium phosphate |
Nephrocalcinosis is a tubulointerstitial nephropathy that either reflects a primary pathophysiologic process or results from severe tissue injury of any cause, so-called “dystrophic” calcification. In mild cases, plain x-rays may reveal nephrocalcinosis as small deposits of calcium salts in the calyces, resembling “pictures of the night sky” (61). Although CT scanning is the most sensitive imaging technique to detect nephrocalcinosis, severe cortical calcifications detected by kidneys, ureters, and bladder (KUB) plain film weeks or months after an ischemic kidney injury indicate renal cortical necrosis and is associated with nonrecovery of renal function (62). In cases of hyperparathyroidism, calcium salts are typically found along medullary tubular basement membranes, as concretions within tubules, and in the interstitium (62). The calcium-phosphate deposits seen after OSP are found primarily in the tubular lumens and the cytoplasm of tubular epithelial cells, with rare interstitial deposits as well (48). Paradoxically, many patients with nephrocalcinosis detected radiographically may have minimally impaired renal function, whereas nephrocalcinosis seen in association with OSP use, which can only be detected by kidney biopsy, may be associated with acute and CKD.
Nephrocalcinosis may be diagnosed incidentally or may present like any tubulointerstitial nephropathy, with low-grade proteinuria (<1 g/d), a bland urine sediment, or an unexplained rise in the serum creatinine. The patient may have a history of nephrolithiasis or renal colic. Hypercalcemia and hypercalciuria associated with hyperparathyroidism or malignancy are the most common causes, whereas granulomatous disease, immobilization, or vitamin D intoxication may on occasion be implicated. Through a variety of mechanisms, distal renal tubular acidosis may cause nephrocalcinosis, although nephrocalcinosis itself may cause distal acidification defects, thus confusing the association. Particularly in children, medullary sponge kidney and nephrocalcinosis may coexist.
Nephrocalcinosis occurs when calcium precipitates in conjunction with either oxalate or phosphate. The phosphates in calcium-phosphate deposits are detected in paraffin sections by the von Kossa stain (Figure 2). This stain does not detect pure calcium oxalates. Calcium phosphate crystals are not birefringent, whereas calcium oxalate crystals are birefringent upon examination under polarized light. Hypercalciuria is a well-established risk factor for calcium crystal deposition, but nephrocalcinosis can occur in the setting of normal calcium excretion, particularly in the presence of primary or secondary hyperoxaluria (63). Patients with primary hyperoxaluria overproduce oxalate due to inherited enzyme defects and develop severe nephrocalcinosis and nephrolithiasis (64). Secondary hyperoxaluria in patients with inflammatory bowel disease or malabsorption is also a common cause of nephrocalcinosis and may on occasion be associated with atherosclerosis due to calcium-oxalate deposition in the vasculature. Recently, calcium-oxalate nephropathy has been reported in a patient taking orlistat, an over-the-counter medication that inhibits intestinal lipid metabolism and results in secondary hyperoxaluria (65). Ethylene glycol is metabolized to oxalate and can also result in kidney injury due to calcium-oxalate deposition.
Figure 2.
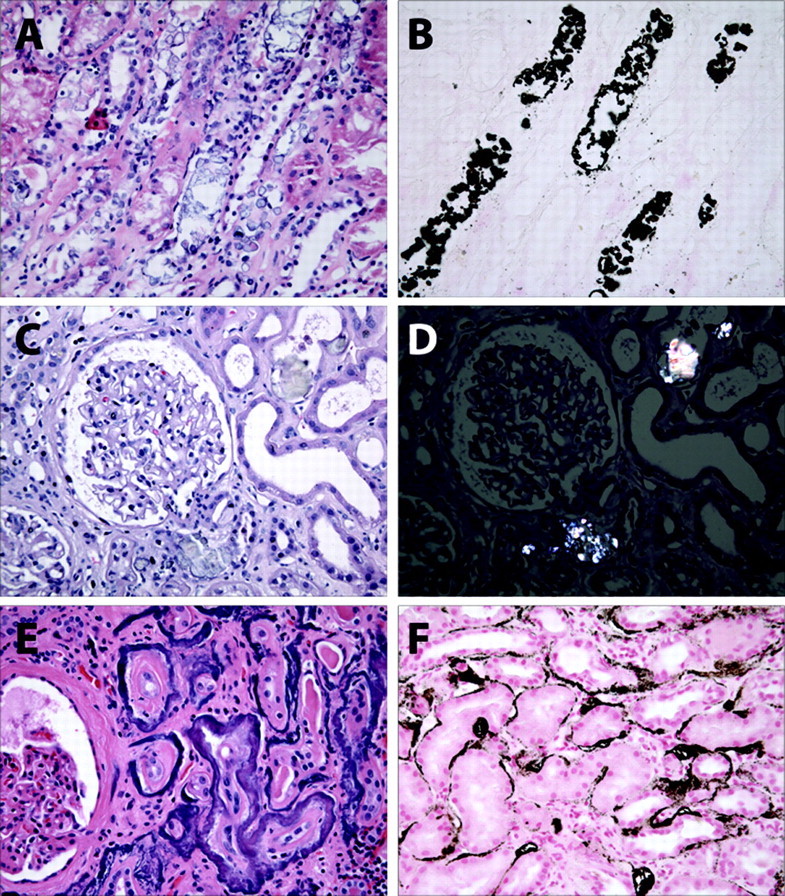
Renal pathology in nephrocalcinosis. (A) Hematoxylin and eosin-stained biopsy from a patient with phosphate nephropathy. Note the evidence of acute tubular injury with simplified epithelial cell brush borders and necrotic luminal cell debris. (B) von Kossa stain of the same biopsy specimen reveals abundant intraluminal calcium crystals. (C) Hematoxylin and eosin-stained glomerulus from a patient with acute kidney injury and inflammatory bowel disease. (D) On polarized light, the calcium oxalate crystals from the same section are positively birefringent. (E) Hematoxylin and eosin-stained section from a patient with advanced malignancy with bony metastases and hypercalcemia. Note the thickening of tubular basement membranes. (F) von Kossa stain from the same biopsy reveals typical findings in metastatic calcification with punctate and linear tubular basement calcium phosphate crystals.
In many cases, the initial event in the development of both nephrocalcinosis and nephrolithiasis may be the formation of calcium-phosphate deposits in the thin limb of Henle, called “Randall's plaques,” which form largely due to supersaturation of calcium and phosphate in that portion of the tubule (66,67). When these plaques rupture into the urinary space, they serve as a nidus for calcium-oxalate crystallization, and nephrocalcinosis results when the urothelium overgrows and encapsulates the crystals (63,68). The observation that most calcium-oxalate stones contain some calcium-phosphate supports this mechanism (61,66). Throughout much of the kidney tubule, calcium and phosphate exist in an “unstable compromise” of ionic supersaturation that is maintained by inhibitors, such as citrate, Tamm-Horsfall protein, and pyrophosphate (69). Once calcium-phosphate crystals form, urinary crystals of any type can promote crystal growth, which explains why treatment of hyperuricemia with allopurinol can prevent calcium stone formation (70). Despite these mechanistic similarities, clinical experience shows that some patients with nephrolithiasis have no nephrocalcinosis whereas other patients display the opposite pattern.
The deposition of calcium oxalate and calcium phosphate can be viewed within the broader context of renal crystal deposition. Under even normal conditions, the progressive concentration of urine through the removal of water and the extremes of urinary pH can create an environment conducive to crystal formation (66). Uric acid and cystine crystals form most readily in an acid urine, whereas alkaline conditions favor calcium-phosphate crystals. In contrast, calcium oxalate crystal formation is pH independent. Medications that are concentrated in the urine can precipitate and contribute to kidney injury. Well-recognized offenders include acyclovir, foscarnet, methotrexate, the sulfonamides, and indinavir (71). Recently, the deposition in kidney and other tissues of the cation gadolinium in association with phosphate and other anions has been implicated in the pathogenesis of nephrogenic systemic fibrosis (72). Gadolinium deposits are basophilic on hematoxylin and eosin staining and may be confused with nephrocalcinosis (73).
Hyperphosphaturia and Kidney Injury
Just as nephrocalcinosis can result from hyperoxaluria and the subsequent deposition of calcium-oxalate crystals, hyperphosphatemia and hyperphosphaturia can cause nephrocalcinosis and AKI through the deposition of calcium-phosphate crystals. This phenomenon, which has been described in diverse clinical settings as well as in animal models of hyperphosphaturia, lends support to the hypothesis that phosphaturia after OSP administration underlies the development of phosphate nephropathy.
In the 1930s and again in the 1960s, the use of phosphate to treat symptomatic hypercalcemia of varying causes was complicated by metastatic calcifications and AKI (74–78). Extraskeletal calcifications were occasionally demonstrated on plain x-rays and slit-lamp examination (79). Clinical investigations confirmed that phosphate administration lowered serum calcium primarily through Ca-Pi deposition (80,81).
In another example of exogenous phosphate therapy driving calcium-phosphate deposition in the kidney, patients with X-linked hypophosphatemic rickets (XLH) treated with phosphate supplementation (and calcitriol) commonly develop nephrocalcinosis. Verge et al. described 19 of 24 XLH patients with nephrocalcinosis detected by ultrasound and successfully correlated the grade of nephrocalcinosis with the mean dose of exogenous phosphate (82). Three children with XLH reported by Alon et al. had kidney biopsies, which demonstrated calcium phosphate deposition by von Kossa staining (83). Stickler and Morgenstern described two XLH patients who developed end-stage renal disease as a result of nephrocalcinosis (84). Hypercalciuria induced by calcitriol may also have contributed to the development of nephrocalcinosis in these patients.
Recently, Patel et al. at Baylor reported that urinary calcium excretion declined dramatically in five normal subjects who received OSP (23). The authors conclude that their findings are consistent with calcium-phosphate precipitation within the kidney, an effect similar to that seen in the other settings of exogenous phosphate administration described above.
The tumor lysis syndrome is a well-described cause of AKI, which usually results from the endogenous release of uric acid but can also result from phosphate release with subsequent crystal deposition in the renal tubules (85). Boles et al. reported a series of 34 patients with calcium-phosphate deposition associated with the tumor lysis syndrome, including the case of a 15-yr-old boy with acute leukemia who developed dialysis dependent AKI (86). Although alkalinization of the urine is often recommended to prevent uric acid deposition in the setting of tumor lysis, this therapy may increase the risk of calcium-phosphate deposition, which occurs more readily at an alkaline pH. The maintenance of a high flow of urine at a neutral pH may be the best approach to avoid either form of crystal deposition during tumor cell lysis.
Animal studies support the notion that hyperphosphaturia, independent of hypercalciuria, can result in nephrocalcinosis and kidney injury. In a mouse model of XLH, animals treated with phosphate and vitamin D develop nephrocalcinosis. Ritskes-Hoitinga et al. also described nephrocalcinosis in rats fed a high phosphate diet, an effect that was attenuated by parathyroidectomy and hypermagnesiuria, which may inhibit calcium-phosphate crystal deposition (87,88). Alternative mechanisms of kidney injury have also been suggested by animal studies. Using a rat model, Zager has shown that hyperphosphatemia potentiates kidney injury by inducing proximal tubular vacuolization and capillary collapse, without evident nephrocalcinosis (89).
Mechanism of Kidney Injury from OSP: Possible Role of the Immune System
By contrast, the pathophysiology of CKD after OSP administration is unknown and speculative. Despite the absence of experimental evidence, insight may be gained from other crystal-induced inflammatory and fibrotic diseases such as calcium crystal arthropathy, gout and nephrogenic systemic fibrosis (NSF). Increasing experimental data implicates the innate immune system in mediating the pro-inflammatory actions of tissue crystal deposition. Pattern recognition receptors, such as the Toll-like receptor (TLR), mediate monosodium urate monohydrate-dependent nitric oxide and IL-1β release from chondrocytes and macrophages, respectively. Given the ability of specific TLRs to recognize calcium crystals and the expression of a variety of TLRs in the adult kidney, we speculate that intraluminal calcium-phosphate crystal formation after OSP leads to recognition by specific epithelial TLRs with activation of the innate immune response (90,91). Persistence of these crystals could result in chronic inflammation with extracellular matrix deposition and interstitial fibrosis (92). Clearly, the cellular and molecular mechanisms of OSPS-mediated renal fibrosis require investigation.
An intriguing correlation between hyperphosphatemia, crystal deposition, and tissue fibrosis has been suggested in the pathogenesis of nephrogenic systemic fibrosis (72). Gadolinium-based contrast exposure is strongly implicated in the development of NSF, and patients developing this debilitating condition have Gd3+ tissue deposition (93). Excess serum phosphate may bind free Gd3+ with subsequent tissue deposition. Gd3+-phosphate complexes can be demonstrated in tissues of patients with NSF (72). The subsequent process of Gd3+-crystal-induced fibrosis may be similar to that evoked by calcium phosphate crystals, with phagocytosis of crystals into resident phagocytes (or perhaps epithelial cells) mediated by pattern recognition receptors, ultimately activating the innate immune response and causing a chronic inflammatory and pro-fibrotic process.
A Lower Dose Product and Remaining Questions
In response to the reports of phosphate nephropathy as well as warnings from government agencies concerning the use of OSP, the largest manufacturer of OSP products, the C.B. Fleet Company, discontinued the sale of the 90-ml OSP preparation (45 ml + 45 ml) and substituted a 75-ml dose product (45 ml + 30 ml), which reportedly has equal efficacy (94–96). Recommendations from the company continue to emphasize the importance of separating the two doses by an interval approaching 12 h and of maintaining adequate hydration before, during, and after OSP bowel preparation (97).
Although lower-dose OSP preparations appear to cause more modest elevations in serum phosphate, it is unknown whether they are safer than standard dose preparations. Similarly, it unknown whether optimal oral hydration can replace OSP-associated losses while maintaining normal electrolyte levels and preventing phosphate nephropathy. Recommendations for volume repletion with OSP preps vary from 0.7 to 2.2 L (Fleet currently recommends a “minimum” of 72 oz), but the optimal amount may exceed 3.7 L (98,99). Even carefully selected patients who were given explicit instructions for oral hydration and were studied in the setting of a clinical trial lost an average of 2.3 lbs of body weight after sodium phosphate bowel prep, although weight loss has also been observed with the combination of PEG-ELS + bisacodyl (4,94). In addition, the benefits of increased water intake need to be balanced against the risk of hyponatremia (23,25,26). It is also unknown whether water is adequate for oral hydration or whether electrolyte-containing oral rehydration solutions would be safer (97).
Other significant gaps exist in our knowledge of OSP-associated kidney injury. Although some estimate that phosphate nephropathy may affect as many as 1400 to 7000 Americans each year, prospectively collected data defining the true incidence of phosphate nephropathy are needed (100). Such data would also provide better information concerning putative risk factors, such as hypertension, diabetes, female gender, and renin-angiotensin blockade. The role of OSP-induced kidney injury in the development CKD, in the absence of diagnosed AKI, needs investigation, as does the role of parathyroid hormone and vitamin D status as potential risk factors (101).
Conclusion
Adequate bowel preparation is mandatory for colorectal cancer screening, and clinicians selecting bowel-cleansing agents must consider the potential metabolic consequences of OSP and PEG-ELS use. Although the ethical imperative to “do no harm” and the large and growing number of colonoscopies performed each year compel physicians to favor bowel purgatives that pass a very high standard of safety, the renal risks posed by OSP raised in this article need not change clinical practice in low-risk patients. Table 2 summarizes our suggested absolute and relative contraindications for OSP prescription. Multiple causative factors may contribute to kidney injury after bowel preparation, especially in patients with impaired intrarenal hemodynamics at baseline or volume depletion. Phosphate nephropathy is an important, although probably rare, cause of acute and chronic kidney injury after OSP use, particularly when prescribed to well-selected subjects. The risk of phosphate nephropathy from OSP may be mitigated by adequate volume repletion and the use of the minimal effective dose. Caution is warranted when any bowel preparative is administered to medically fragile patients, particularly those on maintenance hemodialysis. The ongoing use of OSP preps in high-risk patients, despite literally hundreds of published adverse reactions over a decade or more, underscores the limitations of manufacturers’ recommendations and the extant medical literature in improving prescribing safety (57).
Table 2.
Suggested contraindications to oral sodium phosphate administration
| Absolute contraindications |
| Estimated glomerular filtration rate <60 ml/min |
| Significant kidney disease with preserved GFR (e.g., nephrotic syndrome) |
| Preexisting hyperphosphatemia of any cause |
| Clinically significant congestive heart failure or cardiomyopathy |
| Clinically significant cirrhosis and/or ascites |
| Gastrointestinal disease, as detailed on package label including obstruction, megacolon, perforation, ileus, inflammatory bowel disease |
| Hypersensitivity to the ingredients |
| Relative contraindications |
| Age <18 or >60 to 70 or clinically significant debilitation |
| Use of diuretics, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, or nonsteroidal anti-inflammatory drugs, especially if in combination |
| Dehydration or risk for dehydration (e.g., nausea, vomiting, diarrhea, low salt diet, inability to take adequate oral fluids). |
| Uncorrected electrolyte abnormalities |
| Cardiac disease as detailed on package labeling, including recent MI, prolonged QT interval, or drugs that prolong interval, arrhythmia, unstable angina. |
| Pregnancy or nursing a child, as per package labeling. |
Disclosures
B.D.H. serves on the Scientific Advisory Board for C.B. Fleet and Co. S.O.T. serves on the boards of directors of Merck & Co., Inc. and Charles River Laboratories, Inc.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Seeff LC, Richards TB, Shapiro JA, Nadel MR, Manninen DL, Given LS, Dong FB, Winges LD, McKenna MT: How many endoscopies are performed for colorectal cancer screening? Results from CDC's survey of endoscopic capacity. Gastroenterology 127 :1670– 1677,2004 [DOI] [PubMed] [Google Scholar]
- 2.Cram P, Fendrick AM, Inadomi J, Cowen ME, Carpenter D, Vijan S: The impact of a celebrity promotional campaign on the use of colon cancer screening: the Katie Couric effect. Arch Intern Med 163 :1601– 1605,2003 [DOI] [PubMed] [Google Scholar]
- 3.Rex DK, Imperiale TF, Latinovich DR, Bratcher LL: Impact of bowel preparation on efficiency and cost of colonoscopy. Am J Gastroenterol 97 :1696– 1700,2002 [DOI] [PubMed] [Google Scholar]
- 4.Johanson JF, Popp JW Jr, Cohen LB, Lottes SR, Forbes WP, Walker K, Carter E, Zhang B, Rose M: A randomized, multicenter study comparing the safety and efficacy of sodium phosphate tablets with 2L polyethylene glycol solution plus bisacodyl tablets for colon cleansing. Am J Gastroenterol 102 :2238– 2246,2007 [DOI] [PubMed] [Google Scholar]
- 5.American Society of Colon, Rectal Surgeons (ASCRS), American Society for Gastrointestinal Endoscopy (ASGE), Society of American Gastrointestinal, Endoscopic Surgeons (SAGES), Wexner SD, Beck DE, Baron TH, Fanelli RD, Hyman N, Shen B, Wasco KE: A consensus document on bowel preparation before colonoscopy: prepared by a task force from the American Society of Colon and Rectal Surgeons (ASCRS), the American Society for Gastrointestinal Endoscopy (ASGE), and the Society of American Gastrointestinal and Endoscopic Surgeons (SAGES). Surg Endosc 20 :1147– 1160,2006 [DOI] [PubMed] [Google Scholar]
- 6.Bigard MA, Gaucher P, Lassalle C: Fatal colonic explosion during colonoscopic polypectomy. Gastroenterology 77 :1307– 1310,1979 [PubMed] [Google Scholar]
- 7.Zanoni CE, Bergamini C, Bertoncini M, Bertoncini L, Garbini A: Whole-gut lavage for surgery: a case of intraoperative colonic explosion after administration of mannitol. Dis Colon Rectum 25 :580– 581,1982 [DOI] [PubMed] [Google Scholar]
- 8.Davis GR, Santa Ana CA, Morawski SG, Fordtran JS: Development of a lavage solution associated with minimal water and electrolyte absorption or secretion. Gastroenterology 78 :991– 995,1980 [PubMed] [Google Scholar]
- 9.Fordtran JS, Santa Ana CA, Cleveland M: A low-sodium solution for gastrointestinal lavage. Gastroenterology 98 :11– 16,1990 [DOI] [PubMed] [Google Scholar]
- 10.Vanner SJ, MacDonald PH, Paterson WG, Prentice RS, Da Costa LR, Beck IT: A randomized prospective trial comparing oral sodium phosphate with standard polyethylene glycol-based lavage solution (Golytely) in the preparation of patients for colonoscopy. Am J Gastroenterol 85 :422– 427,1990 [PubMed] [Google Scholar]
- 11.Kolts BE, Lyles WE, Achem SR, Burton L, Geller AJ, MacMath T: A comparison of the effectiveness and patient tolerance of oral sodium phosphate, castor oil, and standard electrolyte lavage for colonoscopy or sigmoidoscopy preparation. Am J Gastroenterol 88 :1218– 1223,1993 [PubMed] [Google Scholar]
- 12.Hsu CW, Imperiale TF: Meta-analysis and cost comparison of polyethylene glycol lavage versus sodium phosphate for colonoscopy preparation. Gastrointest Endosc 48 :276– 282,1998 [DOI] [PubMed] [Google Scholar]
- 13.Tan JJ, Tjandra JJ: Which is the optimal bowel preparation for colonoscopy: a meta-analysis. Colorectal Dis 8 :247– 258,2006 [DOI] [PubMed] [Google Scholar]
- 14.http://www.phospho-soda.com/professionals/pdf/KeyFacts.pdf. Accessed January 10,2008
- 15.Rex DK: Dosing considerations in the use of sodium phosphate bowel preparations for colonoscopy. Ann Pharmacother 41 :1466– 1475,2007 [DOI] [PubMed] [Google Scholar]
- 16.Ahmed M, Raval P, Buganza G: Oral sodium phosphate catharsis and acute renal failure. Am J Gastroenterol 91 :1261– 1262,1996 [PubMed] [Google Scholar]
- 17.Schiller LR: Clinical pharmacology and use of laxatives and lavage solutions. J Clin Gastroenterol 28 :11– 18,1999 [DOI] [PubMed] [Google Scholar]
- 18.Fass R, Do S, Hixson LJ: Fatal hyperphosphatemia following fleet phospo-soda in a patient with colonic ileus. Am J Gastroenterol 88 :929– 932,1993 [PubMed] [Google Scholar]
- 19.Fine A, Patterson J: Severe hyperphosphatemia following phosphate administration for bowel preparation in patients with renal failure: two cases and a review of the literature. Am J Kidney Dis 29 :103– 105,1997 [DOI] [PubMed] [Google Scholar]
- 20.Kirschbaum B: The acidosis of exogenous phosphate intoxication. Arch Intern Med 158 :405– 408,1998 [DOI] [PubMed] [Google Scholar]
- 21.Lieberman DA, Ghormley J, Flora K: Effect of oral sodium phosphate colon preparation on serum electrolytes in patients with normal serum creatinine. Gastrointest Endosc 43 :467– 469,1996 [DOI] [PubMed] [Google Scholar]
- 22.Aradhye S, Brensilver JM: Sodium phosphate-induced hypernatremia in an elderly patient: a complex pathophysiologic state. Am J Kidney Dis 18 :609– 611,1991 [DOI] [PubMed] [Google Scholar]
- 23.Patel V, Emmett M, Santa Ana CA, Fordtran JS: Pathogenesis of nephrocalcinosis after sodium phosphate catharsis to prepare for colonoscopy: intestinal phosphate absorption and its effect on urine mineral and electrolyte excretion. Hum Pathol 38 :193– 194; author reply 194–195,2007 [DOI] [PubMed] [Google Scholar]
- 24.Tan HL, Liew QY, Loo S, Hawkins R: Severe hyperphosphataemia and associated electrolyte and metabolic derangement following the administration of sodium phosphate for bowel preparation. Anaesthesia 57 :478– 483,2002 [DOI] [PubMed] [Google Scholar]
- 25.Frizelle FA, Colls BM: Hyponatremia and seizures after bowel preparation: report of three cases. Dis Colon Rectum 48 :393– 396,2005 [DOI] [PubMed] [Google Scholar]
- 26.Ayus JC, Levine R, Arieff AI: Fatal dysnatraemia caused by elective colonoscopy. BMJ 326 :382– 384,2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schroppel B, Segerer S, Keuneke C, Cohen CD, Schlondorff D: Hyponatremic encephalopathy after preparation for colonoscopy. Gastrointest Endosc 53 :527– 529,2001 [DOI] [PubMed] [Google Scholar]
- 28.Cohen CD, Keuneke C, Schiemann U, Schroppel B, Siegert S, Rascher W, Gross M, Schlondorff D: Hyponatraemia as a complication of colonoscopy. Lancet 357 :282– 283,2001 [DOI] [PubMed] [Google Scholar]
- 29.Turnage RH, Guice KS, Gannon P, Gross M: The effect of polyethylene glycol gavage on plasma volume. J Surg Res 57 :284– 288,1994 [DOI] [PubMed] [Google Scholar]
- 30.Granberry MC, White LM, Gardner SF: Exacerbation of congestive heart failure after administration of polyethylene glycol-electrolyte lavage solution. Ann Pharmacother 29 :1232– 1235,1995 [DOI] [PubMed] [Google Scholar]
- 31.Gumurdulu Y, Serin E, Ozer B, Gokcel A, Boyacioglu S: Age as a predictor of hyperphosphatemia after oral phosphosoda administration for colon preparation. J Gastroenterol Hepatol 19 :68– 72,2004 [DOI] [PubMed] [Google Scholar]
- 32.Clarkston WK, Tsen TN, Dies DF, Schratz CL, Vaswani SK, Bjerregaard P: Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 43 :42– 48,1996 [DOI] [PubMed] [Google Scholar]
- 33.Vukasin P, Weston LA, Beart RW: Oral fleet phospho-soda laxative-induced hyperphosphatemia and hypocalcemic tetany in an adult: report of a case. Dis Colon Rectum 40 :497– 499,1997 [DOI] [PubMed] [Google Scholar]
- 34.Biberstein M, Parker BA: Enema-induced hyperphosphatemia. Am J Med 79 :645– 646,1985 [DOI] [PubMed] [Google Scholar]
- 35.Korzets A, Dicker D, Chaimoff C, Zevin D: Life-threatening hyperphosphatemia and hypocalcemic tetany following the use of fleet enemas. J Am Geriatr Soc 40 :620– 621,1992 [DOI] [PubMed] [Google Scholar]
- 36.Ma KK, Ng CS, Mui LM, Chan KC, Ng EK, Chung SC: Severe hyperphosphatemia and hypocalcemia following sodium phosphate bowel preparation: a forgotten menace. Endoscopy 35 :717 ,2003 [DOI] [PubMed] [Google Scholar]
- 37.Mishra R, Kaufman D, Mattern J 3rd, Dutta SK: Severe hyperphosphatemia and hypocalcemia caused by bowel preparation for colonoscopy using oral sodium phosphate in end-stage renal disease. Endoscopy 37 :1259– 1260,2005 [DOI] [PubMed] [Google Scholar]
- 38.Rohack JJ, Mehta BR, Subramanyam K: Hyperphosphatemia and hypocalcemic coma associated with phosphate enema. South Med J 78 :1241– 1242,1985 [DOI] [PubMed] [Google Scholar]
- 39.Ullah N, Yeh R, Ehrinpreis M: Fatal hyperphosphatemia from a phosphosoda bowel preparation. J Clin Gastroenterol 34 :457– 458,2002 [DOI] [PubMed] [Google Scholar]
- 40.Wechsler A, Schneider R, Sapojnikov M, Zamir D, Polyshuk I, Yagil Y: Bowel cleansing in patients with chronic renal failure: an often overlooked hazard. Nephrol Dial Transplant 21 :1133– 1134,2006 [DOI] [PubMed] [Google Scholar]
- 41.Aydogan T, Kanbay M, Uz B, Kaya A, Isik A, Bozalan R, Erkman M, Akcay A: Fatal hyperphosphatemia secondary to a phosphosoda bowel preparation in a geriatric patient with normal renal function. J Clin Gastroenterol 40 :177 ,2006 [DOI] [PubMed] [Google Scholar]
- 42.Orias M, Mahnensmith RL, Perazella MA: Extreme hyperphosphatemia and acute renal failure after a phosphorus-containing bowel regimen. Am J Nephrol 19 :60– 63,1999 [DOI] [PubMed] [Google Scholar]
- 43.Eckstein J, Savic S, Eugster T, Pargger H, Gurke L, Pechula M, Steiger J, Mayr M: Extensive calcifications induced by hyperphosphataemia caused by phosphate-based enema in a patient after kidney transplantation. Nephrol Dial Transplant 21 :2013– 2016,2006 [DOI] [PubMed] [Google Scholar]
- 44.Keeffe EB: Colonoscopy preps: what's best? Gastrointest Endosc 43 :524– 528,1996 [DOI] [PubMed] [Google Scholar]
- 45.Di Palma JA, Brady CE 3rd: Colon cleansing for diagnostic and surgical procedures: polyethylene glycol-electrolyte lavage solution. Am J Gastroenterol 84 :1008– 1016,1989 [PubMed] [Google Scholar]
- 46.Schwetz BA: From the Food and Drug Administration. JAMA 286 :2660 ,2001 [PubMed] [Google Scholar]
- 47.Desmeules S, Bergeron MJ, Isenring P: Acute phosphate nephropathy and renal failure. N Engl J Med 349 :1006– 1007,2003 [DOI] [PubMed] [Google Scholar]
- 48.Markowitz GS, Nasr SH, Klein P, Anderson H, Stack JI, Alterman L, Price B, Radhakrishnan J, D'Agati VD: Renal failure due to acute nephrocalcinosis following oral sodium phosphate bowel cleansing. Hum Pathol 35 :675– 684,2004 [DOI] [PubMed] [Google Scholar]
- 49.Markowitz GS, Stokes MB, Radhakrishnan J, D'Agati VD: Acute phosphate nephropathy following oral sodium phosphate bowel purgative: an underrecognized cause of chronic renal failure. J Am Soc Nephrol 16 :3389– 3396,2005 [DOI] [PubMed] [Google Scholar]
- 50.Aasebo W, Scott H, Ganss R: Kidney biopsies taken before and after oral sodium phosphate bowel cleansing. Nephrol Dial Transplant 22 :920– 922,2007 [DOI] [PubMed] [Google Scholar]
- 51.Gonlusen G, Akgun H, Ertan A, Olivero J, Truong LD: Renal failure and nephrocalcinosis associated with oral sodium phosphate bowel cleansing: clinical patterns and renal biopsy findings. Arch Pathol Lab Med 130 :101– 106,2006 [DOI] [PubMed] [Google Scholar]
- 52.Beyea A, Block C, Schned A: Acute phosphate nephropathy following oral sodium phosphate solution to cleanse the bowel for colonoscopy. Am J Kidney Dis 50 :151– 154,2007 [DOI] [PubMed] [Google Scholar]
- 53.Schattner A, Kopolovic J, Melzer E, Rapoport J: A 71-year-old woman with abdominal pain and acute renal failure. CMAJ 177 :454– 455,2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manley P, Somerfield J, Simpson I, Barber A, Zwi J: Bilateral uraemic optic neuritis complicating acute nephrocalcinosis. Nephrol Dial Transplant 21 :2957– 2958,2006 [DOI] [PubMed] [Google Scholar]
- 55.Brunelli SM, Lewis JD, Gupta M, Latif SM, Weiner MG, Feldman HI: Risk of kidney injury following oral phosphosoda bowel preparations. J Am Soc Nephrol 18 :3199– 3205,2007 [DOI] [PubMed] [Google Scholar]
- 56.Hurst FP, Bohen EM, Osgard EM, Oliver DK, Das NP, Gao SW, Abbott KC: Association of oral sodium phosphate purgative use with acute kidney injury. J Am Soc Nephrol 18 :3192– 3198,2007 [DOI] [PubMed] [Google Scholar]
- 57.Russmann S, Lamerato L, Marfatia A, Motsko SP, Pezzullo JC, Olds G, Jones JK: Risk of impaired renal function after colonoscopy: a cohort study in patients receiving either oral sodium phosphate or polyethylene glycol. Am J Gastroenterol 102 :2655– 2663,2007 [DOI] [PubMed] [Google Scholar]
- 58.Khurana A, McLean L, Atkinson S, Foulks CJ: The effect of oral sodium phosphate drug products on renal function in adults undergoing bowel endoscopy. Arch Intern Med 168 :593– 597,2008 [DOI] [PubMed] [Google Scholar]
- 59.Khurana A, McLean LB, Concepcion LA, Nickel AE, Foulks CJ: Acute phosphate nephropathy [poster 163A]. Presented at Renal Week: San Diego, CA, November 14–19,2006
- 60.Ma RC, Chow CC, Yeung VT, So WY, Kong AP, Tong PC, Cockram CS, Chan JC: Acute renal failure following oral sodium phosphate bowel preparation in diabetes. Diabetes Care 30 :182– 183,2007 [DOI] [PubMed] [Google Scholar]
- 61.Coe FL, Parks JH, Asplin JR: The pathogenesis and treatment of kidney stones. N Engl J Med 327 :1141– 1152,1992 [DOI] [PubMed] [Google Scholar]
- 62.Jennette JC, Olson JL, Schwartz MM, Silva FG: Heptinstall's Pathology of the Kidney, Philadelphia, Lippincott Williams & Wilkins,1998. , pp893– 895
- 63.Sayer JA, Carr G, Simmons NL: Nephrocalcinosis: molecular insights into calcium precipitation within the kidney. Clin Sci (Lond) 106 :549– 561,2004 [DOI] [PubMed] [Google Scholar]
- 64.Broyer M, Jouvet P, Niaudet P, Daudon M, Revillon Y: Management of oxalosis. Kidney Int Suppl 53[ Suppl]:93– 98,1996 [PubMed] [Google Scholar]
- 65.Singh A, Sarkar SR, Gaber LW, Perazella MA: Acute oxalate nephropathy associated with orlistat, a gastrointestinal lipase inhibitor. Am J Kidney Dis 49 :153– 157,2007 [DOI] [PubMed] [Google Scholar]
- 66.Asplin JR, Mandel NS, Coe FL: Evidence of calcium phosphate supersaturation in the loop of Henle. Am J Physiol 270 :F604– F613,1996 [DOI] [PubMed] [Google Scholar]
- 67.Evan AP, Lingeman JE, Coe FL, Parks JH, Bledsoe SB, Shao Y, Sommer AJ, Paterson RF, Kuo RL, Grynpas M: Randall's plaque of patients with nephrolithiasis begins in basement membranes of thin loops of Henle. J Clin Invest 111 :607– 616,2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prien EL: The riddle of Randall's plaques. J Urol 114 :500– 507,1975 [DOI] [PubMed] [Google Scholar]
- 69.Coe F, Parks JH: Defenses of an unstable compromise: crystallization inhibitors and the kidney's role in mineral regulation. Kidney Int 38 :625– 631,1990 [DOI] [PubMed] [Google Scholar]
- 70.Ettinger B, Tang A, Citron JT, Livermore B, Williams T: Randomized trial of allopurinol in the prevention of calcium oxalate calculi. N Engl J Med 315 :1386– 1389,1986 [DOI] [PubMed] [Google Scholar]
- 71.Perazella MA: Crystal-induced acute renal failure. Am J Med 106 :459– 465,1999 [DOI] [PubMed] [Google Scholar]
- 72.Perazella MA: Tissue deposition of gadolinium and development of NSF: a convergence of factors. Semin Dial 21 :150– 154,2008 [DOI] [PubMed] [Google Scholar]
- 73.Kay J, Bazari H, Avery LL, Koreishi AF: Case records of the Massachusetts General Hospital (case 6–2008): a 46-year-old woman with renal failure and stiffness of the joints and skin. N Engl J Med 358 :827– 838,2008 [DOI] [PubMed] [Google Scholar]
- 74.Goldsmith RS, Ingbar SH: Inorganic phosphate treatment of hypercalcemia of diverse etiologies. N Engl J Med 274 :1– 7,1966 [DOI] [PubMed] [Google Scholar]
- 75.Ayala G, Chertow BS, Shah JH, Williams GA, Kukreja SC: Acute hyperphosphatemia and acute persistent renal insufficiency induced by oral phosphate therapy [Letter]. Ann Intern Med 83 :520– 521,1975 [DOI] [PubMed] [Google Scholar]
- 76.Breuer RI, LeBauer J: Caution in the use of phosphates in the treatment of severe hypercalcemia. J Clin Endocrinol Metab 27 :695– 698,1967 [DOI] [PubMed] [Google Scholar]
- 77.Shackney S, Hasson J: Precipitous fall in serum calcium, hypotension, and acute renal failure after intravenous phosphate therapy for hypercalcemia: report of two cases. Ann Intern Med 66 :906– 916,1967 [DOI] [PubMed] [Google Scholar]
- 78.Carey RW, Schmitt GW, Kopald HH, Kantrowitz PA: Massive extraskeletal calcification during phosphate treatment of hypercalcemia. Arch Intern Med 122 :150– 155,1968 [PubMed] [Google Scholar]
- 79.Dudley FJ, Blackburn CR: Extraskeletal calcification complicating oral neutral-phosphate therapy. Lancet 2 :628– 630,1970 [DOI] [PubMed] [Google Scholar]
- 80.Hebert LA, Lemann J Jr, Petersen JR, Lennon EJ: Studies of the mechanism by which phosphate infusion lowers serum calcium concentration. J Clin Invest 45 :1886– 1894,1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sutters M, Gaboury CL, Bennett WM: Severe hyperphosphatemia and hypocalcemia: a dilemma in patient management. J Am Soc Nephrol 7 :2056– 2061,1996 [PubMed] [Google Scholar]
- 82.Verge CF, Lam A, Simpson JM, Cowell CT, Howard NJ, Silink M: Effects of therapy in X-linked hypophosphatemic rickets. N Engl J Med 325 :1843– 1848,1991 [DOI] [PubMed] [Google Scholar]
- 83.Alon U, Donaldson DL, Hellerstein S, Warady BA, Harris DJ: Metabolic and histologic investigation of the nature of nephrocalcinosis in children with hypophosphatemic rickets and in the hyp mouse. J Pediatr 120 :899– 905,1992 [DOI] [PubMed] [Google Scholar]
- 84.Stickler GB, Morgenstern BZ: Hypophosphataemic rickets: final height and clinical symptoms in adults. Lancet 2 :902– 905,1989 [DOI] [PubMed] [Google Scholar]
- 85.Ettinger DS, Harker WG, Gerry HW, Sanders RC, Saral R: Hyperphosphatemia, hypocalcemia, and transient renal failure: results of cytotoxic treatment of acute lymphoblastic leukemia. JAMA 239 :2472– 2474,1978 [DOI] [PubMed] [Google Scholar]
- 86.Boles JM, Dutel JL, Briere J, Mialon P, Robasckiewicz M, Garre M, Briere J: Acute renal failure caused by extreme hyperphosphatemia after chemotherapy of an acute lymphoblastic leukemia. Cancer 53 :2425– 2429,1984 [DOI] [PubMed] [Google Scholar]
- 87.Ritskes-Hoitinga J, Lemmens AG, Danse LH, Beynen AC: Phosphorus-induced nephrocalcinosis and kidney function in female rats. J Nutr 119 :1423– 1431,1989 [DOI] [PubMed] [Google Scholar]
- 88.Ritskes-Hoitinga J, Mathot JN, Van Zutphen LF, Beynen AC: Inbred strains of rats have differential sensitivity to dietary phosphorus-induced nephrocalcinosis. J Nutr 122 :1682– 1692,1992 [DOI] [PubMed] [Google Scholar]
- 89.Zager RA: Hyperphosphatemia: a factor that provokes severe experimental acute renal failure. J Lab Clin Med 100 :230– 239,1982 [PubMed] [Google Scholar]
- 90.Liu-Bryan R, Pritzker K, Firestein GS, Terkeltaub R: TLR2 signaling in chondrocytes drives calcium pyrophosphate dihydrate and monosodium urate crystal-induced nitric oxide generation. J Immunol 174 :5016– 5023,2005 [DOI] [PubMed] [Google Scholar]
- 91.Anders HJ, Schlondorff D: Toll-like receptors: emerging concepts in kidney disease. Curr Opin Nephrol Hypertens 16 :177– 183,2007 [DOI] [PubMed] [Google Scholar]
- 92.Wynn TA: Cellular and molecular mechanisms of fibrosis. J Pathol 214 :199– 210,2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boyd AS, Zic JA, Abraham JL: Gadolinium deposition in nephrogenic fibrosing dermopathy. J Am Acad Dermatol 56 :27– 30,2007 [DOI] [PubMed] [Google Scholar]
- 94.Rex D, Malik P, Chasen R, Thompson WO, Galt DJB: A randomized, single blind evaluation of oral sodium phosphate solution (OSPS) as bowel preparation with different hydration and dosing regimens [Abstract]. Am J Gastroenterol 101[ Suppl]:504 ,2006 [Google Scholar]
- 95.www.fda.gov/cder/drug/infopage/osp_solution. Accessed May 30,2008
- 96.http://www.hc-sc.gc.ca/dhp-mps/medeff/advisories-avis/prof/2005/phosphate_solutions_2_hpc-cps_e.html. Accessed May 30,2008
- 97.http://www.phospho-soda.com/professionals/pdf/UpdatedRecommendationsOSPS.pdf. Accessed May 30,2008
- 98.http://www.fda.gov/cder/drug/infopage/osp_solution/backgrounder.htm. Accessed May 30,2008
- 99.Holte K, Nielsen KG, Madsen JL, Kehlet H: Physiologic effects of bowel preparation. Dis Colon Rectum 47 :1397– 1402,2004 [DOI] [PubMed] [Google Scholar]
- 100.Markowitz GS, Radhakrishnan J, D'Agati VD: Towards the incidence of acute phosphate nephropathy. J Am Soc Nephrol 18 :3020– 3022,2007 [DOI] [PubMed] [Google Scholar]
- 101.Stratta P, Barbieri S, Lazzarich E, Fenoglio R, Quaglia M: Nephrocalcinosis in phosphate nephropathy following oral phosphate purgative: a role for underlying subclinical primary hyperparathyroidism? Am J Kidney Dis 50 :1053 ,2007. ; author reply 1053–1054, 2007 [DOI] [PubMed] [Google Scholar]