

Abstract
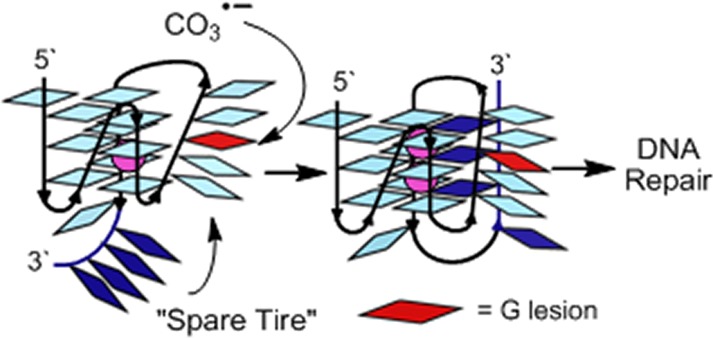
Uncontrolled inflammation or oxidative stress generates electron-deficient species that oxidize the genome increasing its instability in cancer. The G-quadruplex (G4) sequences regulating the c-MYC, KRAS, VEGF, BCL-2, HIF-1α, and RET oncogenes, as examples, are targets for oxidation at loop and 5′-core guanines (G) as showcased in this study by CO3•– oxidation of the VEGF G4. Products observed include 8-oxo-7,8-dihydroguanine (OG), spiroiminodihydantoin (Sp), and 5-guanidinohydantoin (Gh). Our previous studies found that OG and Gh, when present in the four G-tracks of the solved structure for VEGF and c-MYC, were not substrates for the base excision repair (BER) DNA glycosylases in biologically relevant KCl solutions. We now hypothesize that a fifth G-track found a few nucleotides distant from the G4 tracks involved in folding can act as a “spare tire,” facilitating extrusion of a damaged G-run into a large loop that then becomes a substrate for BER. Thermodynamic, spectroscopic, and DMS footprinting studies verified the fifth domain replacing a damaged G-track with OG or Gh at a loop or core position in the VEGF G4. These new “spare tire”-containing strands with Gh in loops are now found to be substrates for initiation of BER with the NEIL1, NEIL2, and NEIL3 DNA glycosylases. The results support a hypothesis in which regulatory G4s carry a “spare-tire” fifth G-track for aiding in the repair process when these sequences are damaged by radical oxygen species, a feature observed in a large number of these sequences. Furthermore, formation and repair of oxidized bases in promoter regions may constitute an additional example of epigenetic modification, in this case of guanine bases, to regulate gene expression in which the G4 sequences act as sensors of oxidative stress.
Short abstract
Oxidation of guanine in promoter G-quadruplexes can disrupt a structure comprised of four G tracks. A fifth G track or “spare tire” domain can reestablish folding leading to recruitment of NEIL1 and NEIL3 for initiation of DNA repair.
Introduction
Instability of the genome is a characteristic of nearly all human cancers.1 The detailed molecular basis and stage of cancer development at which this instability arises is actively being pursued. A key hallmark of genome instability is DNA damage resulting from enhanced oxidative stress. Damage imposed by free radicals resulting from oxidative stress and hyperinflammation observed in cancer cells preferentially oxidizes guanine (G) in DNA. This claim has been confirmed in mouse livers with inflammation-induced colitis leading to hepatocarcinoma.2 The products resulting from G oxidation in these liver cells were 8-oxo-7,8-dihydroguanine (OG) and its hyperoxidation products 5-guanidinohydantoin (Gh) and spiroiminodihydantoin (Sp, Figure 1A). Evolution has selected an intricate repair system to evade these damaged nucleotides in the genome,3,4 and failure of this system leads to greater genome instability.1 Inspection of the genome for likely sites of increased G oxidation identifies G-rich sequences that form G-quadruplex (G4) folds as likely candidates for modification (Figure 1B,C). This hypothesis is supported by experimental and bioinformatics studies that have revealed common instability sites in the genome near G4 sequences.5,6 A recent demonstration identified G4 sequences adopting G4 folds predominantly during the S phase of cellular division.7 Replication occurring during S phase renders DNA subject to the greatest levels of oxidative damage due to its relaxed state.8 Lastly, of the ∼375 000 potential G4 sequences in the human genome, a strong preference for their location in gene promoters, and more specifically oncogene promoter sequences, is observed.9 The G4s in the VEGF, c-MYC, KRAS, BCL-2, and HIF-1α oncogene promoters, to name a few, are also involved in transcriptional regulation of these genes.9
Figure 1.

Guanine oxidation to OG, Sp, or Gh in the four repeat VEGF G4 are not substrates for repair, while the addition of a “spare tire,” or fifth G-run, allows base excision repair. (A) Pathway for oxidation of G. (B) G-Tetrad. (C) The VEGF-4 sequence and model; damage at sites in red cannot be repaired as a G4 fold, and (D) the VEGF-5 sequence and model illustrating the “spare tire” hypothesis.
Evolutionary selection of easily damaged sequences that regulate oncogenes (i.e., G4s) appears at first glance to be a misstep of Darwinian evolution, particularly based on recent findings. For instance, the VEGF G4 is a target for hypoxia-induced oxidation of G to OG in cells.10 Double and triple point mutations of the VEGF G4 that cause it to misfold lead to unregulated gene transcription;11 loss of G4 structure was also observed upon site-specific introduction of OG or Gh at a G4 core nucleotide (VEGF-4, Figure 1B).12 In our studies, the four G-track VEGF G4 sequence with OG or Gh site-specifically synthesized at two sites were not removed by the base excision repair (BER) DNA glycosylases, OGG1, NEIL1, NEIL2, NEIL3, or NTH1 (Figure 1C).12 On the basis of these observations, one would conclude that the VEGF promoter G4 is a site of G to OG oxidation in cells that may cause gene transcription to go awry, particularly when the damage is not repaired. This example paints a gloomy picture for genome stability at critical sites responsible for regulation of genes involved in cancer initiation and progression. Or, has the genome evolved to select sequences that offset this poor prognosis?
In this report, we hypothesize and take initial steps to support a sequence-based evolutionary model that counteracts genome instability resulting from oxidative stress. Examination of the VEGF promoter region flanking the four G-tracks involved in G4 formation identifies a fifth G-track 7 nucleotides distant to the sequence in the 3′ direction (VEGF-5, Figure 1D). If oxidative damage to the G4 sequence occurs leading to impaired folding, unregulated transcription, and no BER repair, we ask whether the fifth G-track can act in the role of a “spare tire” to maintain the G4 fold allowing repair of the damage. Additionally, do other critical oncogene promoters with G4 regulatory domains carry a “spare tire” to minimize oxidative threats leading to genomic instability? This model provides molecular details of the genome that may have evolved in tandem with unique secondary structures (e.g., G4s), providing a safety net for these structures that fine-tune cellular processes.
Results and Discussion
Inspection of oncogene promoter G4s and others described in the literature point to a significant number of them carrying the “spare tire” domain. The addition of a fifth G-track is observed in the VEGF, c-MYC, HIF-1α, KRAS, BCL-2, RET, HSP90, PDGF, and AR promoter G4s, as examples (Table 1).13,14 This partial list highlights G4s with a fifth domain; more examples can be found in Table S1. To build experimental support for our hypothesis that G oxidative damage in G4s recruits the fifth domain, we turned our attention to the VEGF G4. This G4 sequence provides a compelling example of a sequence prone to G oxidation that subsequently alters transcription supporting cancer metastasis;10 additionally, a number of the other G4-regulated genes have been demonstrated to respond as a function of oxidative stress.10
Table 1. Oncogene Promoter G4 Sequences with a Fifth G-Track or Potential “Spare Tires”a,13,14.
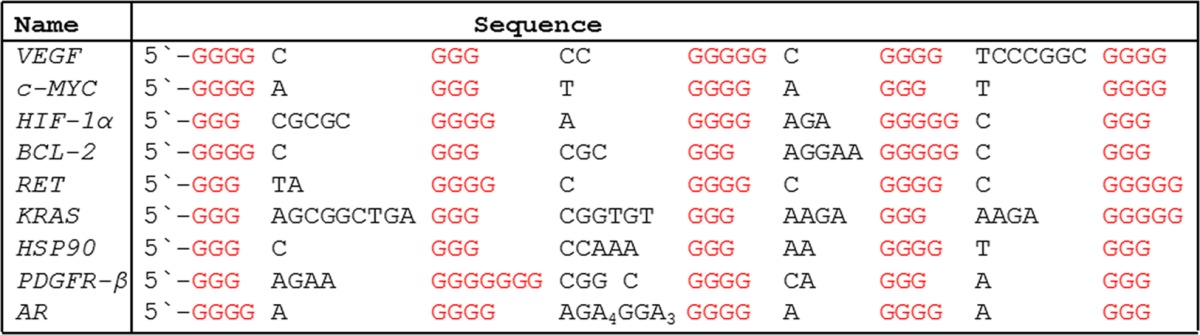
A more complete list of promoter G4 sequences along with all references to these sequences can be found in Table S1.
The VEGF G4 solution structure harnesses the first four G-tracks (VEGF-4) on the 5′ end of the sequence,15 a structure consistent with plasmid supercoiling studies.16 Experiments were conducted to determine the preferential sites of G oxidation and the products observed when the VEGF-4 sequence was oxidized in the G4 fold or alternatively as a duplex with its C-rich complement. The oxidant selected was CO3•– that is a major reactive oxygen species (ROS) found in inflammation and oxidative stress resulting from decomposition of ONOO– in cells.17 Oxidation of VEGF-4 in the G4 fold furnished hotspots for oxidation at G nucleotides in loops, based on the NMR solution structure,15 and those at the 5′ face of the G-tetrad core (Figures 2A and S1). The products identified without added N-acetylcysteine (NAC, a glutathione mimic) include Sp as the major product (∼72%), and the mass balance was completed by lower yields of Gh (∼19%), OG (2%), and oxazolone, Z (5%, Figure 2C). When NAC (3 mM) was added to the mixture, the sites of oxidation remained the same, and the product distribution included ∼12-fold more OG (15%) and 5-fold less Z (<1%), and similar amounts of Sp and Gh (Figures 2C and S2). NAC presumably quenches superoxide that is a reaction partner with oxidized G leading to Z, and therefore, minimizes the yield of Z while increasing the yield of OG, an observation we previously described.18 In contrast, oxidation of the duplex context yielded preferential damage at 5′-G nucleotides that did not change with NAC concentration (Figure 2B), and the product distribution now included Gh (67%) as the major product with and without NAC (Figure 2C). Further, the addition of NAC increased OG by ∼10-fold and decreased Z by ∼3-fold. These reaction sites and products can be chemically explained by previous reports highlighting solvent-exposed Gs in the G4 contexts, and Gs stacked 5′ to another G as favorable oxidation sites.18,19 In summary, the context- and reductant-dependent product yields and sites of lesion formation are consistent with previous studies.
Figure 2.

Oxidation (CO3•–) of the VEGF-4 sequence folded as a G4 or as a duplex identifies loop and core Gs as hotspots for damage leading to OG and hydantoin products. (A) Oxidation sites in the G4 fold; underlined Gs are involved in the major G4 structure characterized.15 (B) Oxidation sites in the duplex fold. (C) Products observed after oxidation of each context. Oxidations were conducted at physiological salt concentrations (140 mM KCl and 12 mM NaCl) buffered at pH 7.4 (20 mM cacodylate) at 37 °C with the oxidant CO3•–, with and without reductant (NAC, 3 mM, Figures S1 and S2).17
The oxidation reactions identified differences in sites and products between the two contexts. The G4 context favors Sp formation at loop Gs and 5′ Gs, while the duplex context yields damage at 5′ Gs and provides Gh in high yield. An intermediate in the formation of Sp and Gh is OG, whose yield significantly increased when oxidations were conducted under reducing conditions similar to those found in cells. These studies point to possible sites in the VEGF promoter G4 sequence in which G oxidation may predominantly be observed. Reactive sites include the G4 loop position 12 and core positions 7 and 14 that were selected for studying the “spare tire” hypothesis (Figure S3). Oligomers containing OG or Gh incorporated at these three positions were synthesized in the VEGF-4 and VEGF-5 sequences to compare the thermodynamic stability, spectroscopic signatures, dimethyl sulfate (DMS) footprinting behavior, and BER activity among these three sequences.
The first step to demonstrating that the fifth G-track could replace a damaged track was achieved using thermal melting (Tm) analysis by comparison of values for VEGF-4 and VEGF-5 with loop vs core damages. The melting midpoint monitored at 295 nm determined the Tm value in physiological salts and buffer (140 mM KCl and 12 mM NaCl buffered with 20 mM cacodylate at pH 7.4, Figures 3A and S4). For VEGF-4 when either OG or Gh damage was present in the loop position (G12), the Tm values were similar to the wild-type (WT) sequence; however, when either OG or Gh damage was placed at a core position (7 or 14) the Tm values were significantly reduced (−25 °C). When VEGF-5 was analyzed with damage at the loop or core, the Tm values were slightly higher (+2–3 °C) than the WT sequence (Figure 3A) in any of the locations. These observations support the idea that damage does not impede the ability of VEGF-5 to form a stable G4, as it does for the VEGF-4 sequence. The melting profiles were found to nearly superimpose upon heating and cooling, supporting the reversibility of the unfolding processes required for further model-dependent thermodynamic analysis (Figure S4). These Tm curves were then used to determine van’t Hoff-derived thermodynamic parameters.20 For VEGF-4, damage in the loop gave similar ΔGvH values as observed for the WT sequence, while damage in the core positions significantly impacted the ΔGvH value (+4–6 kcal/mol, Figure 3B). When the fifth domain was incorporated, the ΔGvH values for the damage-containing sequences had slightly lower values (−1–3 kcal/mol) than the WT sequence (Figure 3B). These results validate the utility of the fifth domain in maintaining the G4 stability when damage is present.
Figure 3.

Measured Tm values and CD spectra for VEGF-4 and -5 with either OG or Gh at a loop or a core position provide initial support for the “spare tire” hypothesis. (A) A plot of Tm values for each strand studied. (B) A table of van’t Hoff-derived ΔGvH and K+ ion coordination (ΔnK+) values for each strand. The CD spectra recorded for VEGF-4 (C) and VEGF-5 (D) WT and OG-containing sequences. Spectra for the Gh-containing strands can be found in Figure S4; they are nearly identical to the OG strands. The CD, Tm, and ΔGvH values were determined in buffered physiological salt conditions (20 mM cacodylate pH 7.4, 140 mM KCl, and 12 mM NaCl), while the ΔnK+ values were determined in buffered (20 mM KPi pH 7.4) KCl solution.
The use of Tm measurements while varying the KCl concentration was previously shown to allow estimation of the number of bound K+ ions in the G4 channel (ΔnK+).20 Analysis of the Tm values in 20–200 mM KCl solutions with no NaCl present was conducted with each sequence. The VEGF-4 WT sequence was found to bind 2.3 K+ ions, a number that is consistent with similar G4 sequences.20 A similar ΔnK+ value was observed for damage incorporated at the loop position; in contrast, when damage was incorporated at a core site, the number of bound K+ ions decreased by ∼1 (Figures 3B and S5). This observation supports a claim that damage is tolerated in the loop, while damage to a core G leads to an altered structure that binds one less K+ ion in the VEGF-4 sequence. It was previously proposed that G4s bearing nucleotides that cannot Hoogsteen base pair (Figure 1B) lead to highly unstable structures, possibly a triplex-like fold that only binds 1 K+ ion.12,21,22 When the same analysis was conducted with the VEGF-5 sequence, the WT, loop-, and core-containing damaged sequences all gave ΔnK+ values >2 (Figure 3B). The similar ΔnK+ values measured for the VEGF-5 sequences provide additional support for the “spare tire” replacing the damaged G-run. Compilation of these thermal studies demonstrates that introduction of G oxidation products (OG or Gh) that cannot form G-tetrads in the VEGF-4 sequence significantly perturbs the thermodynamics of the fold, while recruitment of the fifth domain restores these properties.
In the second part of our study, the structures of VEGF-4 and VEGF-5 were probed. Circular dichrosim (CD) spectra provide a glimpse of the structures in solution. Spectra for OG or Gh at the same position gave nearly identical spectra, and therefore, only spectra for the OG-containing strands vs the WT sequence are displayed in the text for brevity (see Figure S6 for Gh-containing G4 CD spectra). The CD spectra for VEGF-4 with loop damage (position 12) and the WT sequence gave identical shapes but slightly different intensities in buffered physiological salt concentrations (λmax = 260 nm and λmin = 242 nm, Figure 3C). Both spectra are consistent with a parallel-folded G4, as previously reported for the WT sequence.15 The VEGF-4 sequences with core damage (positions 7 and 14) lead to spectra with a λmax red-shifted by 5 nm (Figure 3C). This subtle shift in the CD spectrum is consistent with a triplex-like fold23 that was characterized in the hTelo sequence both computationally and experimentally.24,25 Additionally, a triplex structure is consistent with the KCl concentration-dependent Tm studies that identified loss of one coordinated K+ ion (Figure 3B). In contrast to the differences observed for the VEGF-4 sequences, the CD spectra observed for the VEGF-5 sequences gave λmax = 262 nm with a broad shoulder at 290 nm with positive signal, and a λmin = 240 nm (Figure 3D). These spectra have a different profile than observed for VEGF-4. The Phan laboratory found the human telomerase reverse transcriptase G4 sequence to have a similar CD profile as the VEGF-5 sequences, in which their NMR studies identified a mixture of parallel and hybrid folds in solution;26 therefore, on the basis of the similarity in CD spectra, we propose a mixture of folds in solution for the VEGF-5 sequences with and without damage. More importantly, the presence of OG or Gh in the VEGF-5 sequence at all three positions studied did not induce significant distortions in the shape of the spectra and only minor changes in the peak intensities (Figures 3D and S6). This observation is in line with the Tm and thermodynamic properties of these sequences described above (Figure 3A,B).
The final set of evidence for G lesions distorting the four G-track quadruplex and allowing recruitment of the fifth G-track to reestablish a folded state was achieved by DMS footprinting. Alkylation of G by DMS occurs at N7, which is blocked by Hoogsteen base pairing, and it therefore only reacts with Gs in loops of G4s. Because DMS sites of alkylation are revealed by hot piperidine workup, Gh-containing strands were not studied due to the piperidine lability of this lesion;27 however, OG is not sensitive to alkaline cleavage. For the VEGF-4 WT and loop damaged sequences, DMS footprinting showed predominantly alkylation at the loop Gs, as identified by the NMR studies (Figures 4 and S7).15 When OG was placed at the disruptive core positions, DMS cleavage bands were observed at all G nucleotides, further supporting a highly disrupted or dynamic structure. When the WT VEGF-5 sequence was studied with DMS, no significant protection of Gs from alkylation was observed, consistent with a heterogeneous mixture of folds (Figure 4). Introduction of OG at the loop position of VEGF-5 did not significantly change the DMS profile compared to the WT sequence. In contrast, when OG was placed at the core positions, the fifth domain showed nearly complete protection from DMS alkylation, and hyperreactivity was observed at the Gs located in the G-track with the damage (Figure 4, only a structure for OG at position 14 is shown). These DMS studies provide additional support that a damaged G-track can be looped out and replaced with the “spare tire” fifth domain. Extrusion of an OG into a loop was previously shown in the c-MYC sequence,28 consistent with these VEGF results.
Figure 4.

Structural model to describe DMS footprinting and thermodynamic results for the VEGF-5 sequences with and without OG placed at loop or core positions, supporting the “spare tire” hypothesis. (A) The structure switching model. For simplicity, only the VEGF-5 model is provided. (B) The pixel intensities for the bands on the PAGE after footprinting analysis.
The last experiment conducted was to demonstrate that recruitment of the “spare tire” domain for replacement of a damaged G-track in the VEGF G4 could lead to a new biochemical outcome. In our previous studies with the VEGF-4 sequence in KCl solution, OG or Gh at positions 12 or 14 showed that the lesions were not substrates for the DNA glycosylases OGG1, NEIL1, NEIL2, NEIL3, or NTH1.12 In the present experiments, VEGF-5 with OG or Gh at a loop position (12) and two different core positions (7 and 14) were evaluated for initiation of BER with the same suite of DNA glycosylases (Figure 5). These studies led to many striking observations. First, Gh was removed by NEIL1, NEIL2, and NEIL3 when placed at a loop or at either core position (Figure 5A). As anticipated from the structural studies, NEIL glycosylase removal was more efficient when Gh was at a core site than at a loop site, likely because damage to a core site more efficiently recruits the fifth domain. This biochemical experiment provides critical verification for the role that the “spare tire” domain plays in looping out the damage-containing G-track producing a single-stranded-like region allowing the glycosylases to find a substrate. This ability to initiate repair is not possible without the fifth domain when the G4 folds on a K+ ion,12 the relevant intracellular cation. Second, NEIL1, NEIL2, and NEIL3 showed nearly equal preference for initiating repair of Gh, an observation in contrast to our previous studies with the human telomere G4, in which NEIL1 and NEIL3 showed greater activity than NEIL2.12 Time-course experiments for Gh removal by NEIL1 identified a plateau in the removal efficiency (Figures 5C and S8). After a 40 min reaction, 30% of the core damage to VEGF-5 (7 and 14) was removed, while 10% of the loop damage was removed. The plateau in the time-course studies indicates that not all structures formed by the fifth domain provide kinetically competent folds for DNA repair. Lastly, as expected, NTH1 was not a good DNA glycosylase for initiation of repair of these purine lesions. When the damaged G4s were folded with the complementary strand to form duplexes, NEIL1 was able to remove nearly all Gh lesions (Figure 5A), an expected result.12
Figure 5.

Efficiencies for removal of Gh from a loop (12) or core positions (7 and 14) in the VEGF-5 sequence. The NEIL glycosylases were only active on Gh lesions when the “spare tire” G-track was present. (A) A representative gel to demonstrate the removal of OG or Gh lesions in the VEGF-5 context. (B) Quantification of Gh removal by a suite of DNA glycosylases. (C) Time-course analysis of Gh removal from the loop (12) or core (7 and 14) sites of VEGF-5 by NEIL1.
Next, OGG1 activity on the VEGF-5 structures with OG at positions 7, 12, or 14 was studied. OGG1 showed some activity on folded VEGF-5 sequences, especially on VEGF-5-OG7 (Figure S9). Because OGG1 has a strong preference for duplex DNA containing an OG·C base pair and very little activity on single-stranded DNA,3,4 the VEGF-5 sequences with OG may have adopted a transient duplex structure after the damage-containing G-track was replaced by the fifth G-track. Indeed, the VEGF-5 sequence (5′-CGGGGCGGGCCGGGGGCGGGGTCCCGGCGGGGC-3′) houses a run of Cs (underlined) that might be capable of forming a transient hairpin structure with one of the G tracks. This hairpin could be the actual structure operated on by OGG1, a glycosylase that normally acts on OG in duplex DNA. In all of the structural studies conducted, we proposed a mixture of folds, and these studies identify that damage in some of these folds can be processed by DNA glycosylases. In summary, when oxidatively damaged lesions to G are formed in 4-track G4s, such as VEGF-4, the resulting structures prevent the BER process;12 conversely, rolling the “spare tire” domain into the damaged G4 (i.e., VEGF-5) provides a new structure allowing initiation of BER. In our previous studies, the human telomere repeat sequence was studied with respect to repair of oxidized G nucleotides and a fifth G-track.12 These studies found the human telomere repeat sequence could roll the G-tracks to put the damage in a loop that allowed BER to occur; however, this extrusion process was not required for initiation of BER as it was for the promoter G4 VEGF. These observations may provide molecular details about mechanisms in the genome for combating oxidative insults at critical sites.
Oxidation of duplex DNA at G yielding Gh significantly distorts duplex DNA based on Tm analysis (−17 °C).29 In contrast, if Gh is located in the loop region of G4s or if it occurs at a G4 core site, a “spare tire” domain is recruited to maintain a stable fold, as demonstrated with the VEGF sequence in the present study (Figure 4). The thermal stability of these G4s compared to duplexes suggests a structural switch that extrudes Gh, allowing binding of NEIL DNA glycosylases, particularly NEIL2 and NEIL3 that do not efficiently operate on duplex DNA.12 Our previous results with the 4-track VEGF sequence led to a puzzling result in KCl solution with damage at loop and core sites yielding no repair activity. By taking a broader view of the sequence context and including the fifth track, we find it plays a key role in structural stability by refolding the G4 and allowing the NEIL enzymes to be recruited to initiate repair. The plasticity of DNA to adopt new structures allowing repair of damage will enhance the overall stability of the genome.
A hallmark of cancer is instability of the genome resulting from a number of sources, of which DNA damage from oxidative stress is a major contributor.1 Regions of the genome with G4 sequences are anticipated to be hotspots for G oxidation on the basis of in vitro studies (Figure 2)18 and are sites of double-strand breaks in vivo.6 Interestingly, a number of G4 folds regulate transcription of genes involved in critical pathways for cancer initiation and progression.9,13 Mutations that prevent these G4s from folding cause unregulated transcription.30 The 5′ core Gs and loop Gs of G4s are hotspots for oxidative damage (Figure 2) leading to structures that mask the damage from the BER repair process, and if unrepaired would cause mutations at these G4s. We previously established in the 4 G-track VEGF and c-MYC G4s that damaged nucleotides (OG or Gh) cannot be removed by the DNA glycosylases OGG1, NEIL1, NEIL2, or NEIL3, a poor prognosis for minimizing oxidatively damage-induced mutations.12 In the present study, we hypothesized and demonstrated that the damaged nucleotide could be revealed by the fifth G-track (i.e., “spare tire”) via a structural transition. This transition extrudes the lesion into a large loop allowing faithful BER to be initiated. Removal of the damaged nucleotide to yield a strand break (monofunctional glycosylases require APE-1 to yield the strand break)12 should relax the G4 allowing it to reform the duplex state and finish the repair process. Thus, these studies suggest that critical sites in the genome are wise to travel with a “spare tire”, and based on the large number of G4s with this feature (Tables 1 and S1), it appears the genome evolved to play it safe with regulatory G4s, bringing a backup G-track in case one becomes damaged. Furthermore, this feature minimizes instability at genomic sites that otherwise could be very unstable.
Further support for this hypothesis is found in the literature. Studies using massive human genome sequencing and mining of the data found promoter G4s to be less polymorphic and more conserved than other regions of the genome.30,31 Therefore, one hypothesis is that the fifth domain G-tracks allow damaged G4 structures to refold to maintain a quadruplex motif while enhancing BER after free radical attack. Consequently, the fifth domain acts to maximize genomic stability from oxidative threats by aiding the BER process. Alternatively, evolution of the fifth G-run could have been driven by the need to incorporate a specific transcription factor binding site. Or, both may be true. That is, the oxidized guanine base may itself behave as an epigenetic marker that alters DNA structure, changing gene expression levels by enhancing the formation of a specific secondary structure that recruits a BER enzyme as a transcription factor. The idea that BER enzymes and oxidatively damaged DNA can also function as transcription factors and epigenetic markers was proposed for OGG1 and OG;32 our studies may have identified a mechanism through which the NEIL glycosylases and Gh, and likely Sp,33 have the same capabilities (i.e., a DNA-glycosylase transcription factor and an oxidized guanine base epigenetic marker). An epigenetic function for nucleotides that were originally thought to be DNA lesions in eukaryotes was recently corrected. It was found that N6-methyl-dA under DNA methyltransferase regulation has regulatory capabilities in eukaryotes.34,35 Perhaps this same feature is operating for the hydantoins, Gh and Sp, in conjunction with the NEIL DNA glycosylases.
The “spare tire” feature in G4 motifs is also likely operational in RNA that is not restricted by a complementary strand. Oxidation of RNA occurs at a greater frequency than DNA oxidation,2 and the structural plasticity of these strands is critical in their cellular functions,36 which is a perfect place to look next for utilization of “spare tire” domains and oxidatively derived nucleobases that serve regulatory functions.
Conclusions
Cellular reactive oxygen species show a strong preference for oxidation of G yielding products with altered base-pairing properties. The G-rich sequences of oncogene promoter G4s should be susceptible to G oxidation. Our previous studies determined that c-MYC and VEGF G4s with four G-tracks, when oxidized to the G lesions OG or Gh, were not substrates for glycosylases to initiate BER. With respect to genome stability at critical sites, this at first glance appears to be problematic for the long-term integrity of the genome. We identified that a large number of oncogene G4s harbor a fifth G-track slightly removed from the other 4 (Tables 1 and S1), and we hypothesize that this fifth track is present to act in the role of a “spare tire” for aiding in repair of damaged G4s. Biophysical studies were conducted to elucidate that the fifth G-track was capable of swapping out a G-run containing a damaged base, placing the lesion in a large loop. Looping out the damage led to faithful BER and recovery from oxidative insults. This observation has led to a hypothesis about evolution of genomic sequences that favor regulatory G4s carrying a “spare tire” domain to facilitate repair of damaged nucleotides, and to a role for G-rich sequences as sensors of oxidative stress leading to epigenetic effects on gene expression. Observation of nucleobase oxidative damage inducing a shape shift in secondary structure leading to protein binding and function constitutes a possible transcription factor-like quality for the NEIL DNA glycosylases binding the epigenetic-like base lesion Gh (and likely Sp).
Acknowledgments
Funding for this research was provided by the National Institutes of Health (R01 CA090689 to C.J.B., and P01 CA098993 to S.S.W).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00202.
Experimental methods, Tm, CD, PAGE data, and extended table of G4 sequences (PDF)
Author Contributions
# A.F.M. and J.Z. contributed equally to the conception of this manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Negrini S.; Gorgoulis V. G.; Halazonetis T. D. Genomic instability--an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [DOI] [PubMed] [Google Scholar]
- Mangerich A.; Knutson C. G.; Parry N. M.; Muthupalani S.; Ye W.; Prestwich E.; Cui L.; McFaline J. L.; Mobley M.; Ge Z.; Taghizadeh K.; Wishnok J. S.; Wogan G. N.; Fox J. G.; Tannenbaum S. R.; Dedon P. C. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, E1820–E1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace S. S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S.; O’Shea V.; Kundu S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquette M. L.; Huber M. D.; Maizels N. G-rich proto-oncogenes are targeted for genomic instability in B-cell lymphomas. Cancer Res. 2007, 67, 2586–2594. [DOI] [PubMed] [Google Scholar]
- De S.; Michor F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nat. Struct. Mol. Biol. 2011, 18, 950–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G.; Tannahill D.; McCafferty J.; Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burhans W. C.; Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian S.; Hurley L. H.; Neidle S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy?. Nat. Rev. Drug Discovery 2011, 10, 261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark D. W.; Phang T.; Edwards M. G.; Geraci M. W.; Gillespie M. N. Promoter G-quadruplex sequences are targets for base oxidation and strand cleavage during hypoxia-induced transcription. Free Radical Biol. Med. 2012, 53, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer G.; Cramer T.; Suske G.; Kemmner W.; Wiedenmann B.; Hocker M. Oxidative stress regulates vascular endothelial growth factor-A gene transcription through Sp1- and Sp3-dependent activation of two proximal GC-rich promoter elements. J. Biol. Chem. 2003, 278, 8190–8198. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Fleming A. M.; Averill A. M.; Burrows C. J.; Wallace S. S. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 2015, 43, 4039–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks T. A.; Kendrick S.; Hurley L. Making sense of G-quadruplex and i-motif functions in oncogene promoters. FEBS J. 2010, 277, 3459–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D. J.; Phan A. T.; Kuryavyi V. Human telomere, oncogenic promoter and 5′-UTR G-quadruplexes: diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007, 35, 7429–7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal P.; Hatzakis E.; Guo K.; Carver M.; Yang D. Solution structure of the major G-quadruplex formed in the human VEGF promoter in K+: insights into loop interactions of the parallel G-quadruplexes. Nucleic Acids Res. 2013, 41, 10584–10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D.; Guo K.; Rusche J. J.; Hurley L. H. Facilitation of a structural transition in the polypurine/polypyrimidine tract within the proximal promoter region of the human VEGF gene by the presence of potassium and G-quadruplex-interactive agents. Nucleic Acids Res. 2005, 33, 6070–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin Y.; Cloutier J. F.; Shafirovich V.; Geacintov N. E.; Dedon P. C. Paradoxical hotspots for guanine oxidation by a chemical mediator of inflammation. Nat. Chem. Biol. 2006, 2, 365–366. [DOI] [PubMed] [Google Scholar]
- Fleming A. M.; Burrows C. J. G-Quadruplex folds of the human telomere sequence alter the site reactivity and reaction pathway of guanine oxidation compared to duplex DNA. Chem. Res. Toxicol. 2013, 26, 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genereux J. C.; Barton J. K. Mechanisms for DNA charge transport. Chem. Rev. 2010, 110, 1642–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen C. M.; Gmeiner W. H.; Marky L. A. Unfolding of G-quadruplexes: Energetic, and ion and water contributions of G-quartet stacking. J. Phys. Chem. B 2006, 110, 6962–6969. [DOI] [PubMed] [Google Scholar]
- Lech C. J.; Cheow Lim J. K.; Wen Lim J. M.; Amrane S.; Heddi B.; Phan A. T. Effects of site-specific guanine C8-modifications on an intramolecular DNA G-quadruplex. Biophys. J. 2011, 101, 1987–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala D.; Mashimo T.; Sannohe Y.; Yu Z.; Mao H.; Sugiyama H. Intramolecular folding in three tandem guanine repeats of human telomeric DNA. Chem. Commun. 2012, 48, 2006–2008. [DOI] [PubMed] [Google Scholar]
- Gray R. D.; Trent J. O.; Chaires J. B. Folding and unfolding pathways of the human telomeric G-quadruplex. J. Mol. Biol. 2014, 426, 1629–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T.; Yagi H.; Sannohe Y.; Rajendran A.; Sugiyama H. Folding Pathways of Human Telomeric Type-1 and Type-2 G-Quadruplex Structures. J. Am. Chem. Soc. 2010, 132, 14910–14918. [DOI] [PubMed] [Google Scholar]
- Gray R. D.; Buscaglia R.; Chaires J. B. Populated Intermediates in the Thermal Unfolding of the Human Telomeric Quadruplex. J. Am. Chem. Soc. 2012, 134, 16834–16844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K. W.; Lacroix L.; Yue D. J.; Lim J. K.; Lim J. M.; Phan A. T. Coexistence of two distinct G-quadruplex conformations in the hTERT promoter. J. Am. Chem. Soc. 2010, 132, 12331–12342. [DOI] [PubMed] [Google Scholar]
- Fleming A. M.; Alshykhly O.; Zhu J.; Muller J. G.; Burrows C. J. Rates of chemical cleavage of DNA and RNA oligomers containing guanine oxidation products. Chem. Res. Toxicol. 2015, 28, 1292–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett J.; Burns J.; Broxson C.; Tornaletti S. Spontaneous DNA lesions modulate DNA structural transitions occurring at nuclease hypersensitive element III(1) of the human c-myc proto-oncogene. Biochemistry 2012, 51, 5257–5268. [DOI] [PubMed] [Google Scholar]
- Yennie C. J.; Delaney S. Thermodynamic consequences of the hyperoxidized guanine lesion guanidinohydantoin in duplex DNA. Chem. Res. Toxicol. 2012, 25, 1732–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baral A.; Kumar P.; Halder R.; Mani P.; Yadav V. K.; Singh A.; Das S. K.; Chowdhury S. Quadruplex-single nucleotide polymorphisms (Quad-SNP) influence gene expression difference among individuals. Nucleic Acids Res. 2012, 40, 3800–3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakken S.; Rognes T.; Hovig E. The disruptive positions in human G-quadruplex motifs are less polymorphic and more conserved than their neutral counterparts. Nucleic Acids Res. 2009, 37, 5749–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba X.; Bacsi A.; Luo J.; Aguilera-Aguirre L.; Zeng X.; Radak Z.; Brasier A. R.; Boldogh I. 8-oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J. Immunol. 2014, 192, 2384–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Bandaru V.; Bond J. P.; Jaruga P.; Zhao X.; Christov P. P.; Burrows C. J.; Rizzo C. J.; Dizdaroglu M.; Wallace S. S. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4925–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Huang H.; Liu D.; Cheng Y.; Liu X.; Zhang W.; Yin R.; Zhang D.; Zhang P.; Liu J.; Li C.; Liu B.; Luo Y.; Zhu Y.; Zhang N.; He S.; He C.; Wang H.; Chen D. N(6)-methyladenine DNA modification in Drosophila. Cell 2015, 161, 893–906. [DOI] [PubMed] [Google Scholar]
- Greer E. L.; Blanco M. A.; Gu L.; Sendinc E.; Liu J.; Aristizabal-Corrales D.; Hsu C. H.; Aravind L.; He C.; Shi Y. DNA methylation on N(6)-adenine in C. elegans. Cell 2015, 161, 868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethoff E. A.; Chugh J.; Mustoe A. M.; Al-Hashimi H. M. Functional complexity and regulation through RNA dynamics. Nature 2012, 482, 322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.