

Abstract
Advanced breast cancer requires systemic treatment, therefore developing an efficient and safe strategy is urgently needed. To ensure the success of target therapy, we have developed a breast cancer-specific construct (T-VISA) composed of the human telomerase reverse transcriptase (hTERT; T) promoter and a versatile transgene amplification vector VISA (VP16-GAL4-WPRE integrated systemic amplifier) to target PEA-15 (Phosphoprotein enriched in astrocytes) in advanced breast tumors. PEA-15 contains a death effector domain that sequesters extracellular signal-regulated kinase (ERK) in cytoplasm, thereby inhibiting cell proliferation and inducing apoptosis. T-VISA-PEA-15 was found to be highly specific, selectively express PEA-15 in breast cancer cells, and induce cancer-cell killing in vitro and in vivo without affecting normal cells. Moreover, intravenously treatment with T-VISA-PEA-15 coupled with liposome nanoparticles attenuated tumor growth and prolonged survival in mice bearing advanced breast tumors. Importantly, there was virtually no severe toxicity when PEA-15 is expressed by our T-VISA system compared with cytomegalovirus (CMV) promoter. Thus, our findings demonstrate an effective cancer-targeted therapy that is worthy of development in clinical trials eradicating advanced breast cancer.
Keywords: Breast cancer, T-VISA system, PEA-15, Target therapy
1. Introduction
Breast cancer is the first most common cancer among women and one of the leading causes of death due to cancer all over the world[1]. Many advanced breast cancer patients develop local recurrence or distant metastasis during the course of the disease[2; 3]. Therefore, additional treatment strategies are needed. Targeted gene therapy is an attractive strategy due to the benefit of cancer-specific expression of therapeutic genes[4; 5]. We previously showed that T-VISA vector containing hTERT promoter and VISA (VP16-GAL4-WPRE integrated systemic amplifier) system could target gene expression specifically in human breast tumor, without severe toxicity in normal tissues[6].
Recently, our studies showed that PEA-15 (phosphoprotein enriched in astrocytes) is a multifunctional protein, which can regulate cell proliferation, autophagy, and apoptosis[7; 8; 9]. PEA-15 controls cell proliferation by interfering with the extracellular signal–regulate kinase (ERK) pathway[10]. Other authors also demonstrated that high expression level of PEA-15 in cancer cells correlated with longer survival of cancer patients, such as astrocytic tumors and colorectal cancer[11; 12]. However, the role of PEA-15 in breast cancer has not been completely illuminated in the use for cancer targeted therapy.
It has previously been shown that PEA-15 can suppress the growth of triple-negative breast cancer cells[7]. Therefore, to investigate thoroughly the role of PEA-15 for therapeutic efficacy, we selectively expressed PEA-15 by integrating it into the T-VISA vector to generate T-VISA-PEA-15, and discussed its cancer-killing efficacy and safety. Fortunately, our study demonstrates that T-VISA-PEA-15 gene therapy not only has significant anti-cancer effects, but also assured the tumor-specific targeting effects without systemic toxicity in normal cells. Thus, the current study provides a promising treatment strategy worthy of further development in clinical trials for treating advanced breast cancer by a cancer-targeted gene therapy.
2. Materials and Methods
2.1 Cell lines and culture
Both breast cancer cell lines (T47D, MCF-7, MDA-MB-231, MDA-MB-468, MDA-MB-361, MDA-MB-453, BT474, 4T1, SKBR-3) and immortalized normal mammary epithelial (184A1, and MCF-10A) were obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained according to the manufacturer’s instructions. The above cell lines were passaged in our lab for less than six months after resuscitation of frozen aliquots. All cell lines were authenticated by short tandem repeat DNA profiling before use and found to be free of Mycoplasma infection. Primary human breast cancer cell lines (P1, P2 and P3) were obtained from patients with breast cancer who underwent modified radical mastectomy. This study was approved by the Institutional Review Board of Sun-Yat Sen University Cancer Center of China. Tissues were collected under patients’ agreement. Stable cell lines MDA-MB-231-Luc expressing firefly luciferase were constructed and maintained as described before[6].
2.2 Western blot analysis and immunohistochemistry
Western blotting was performed as described previously[7; 13] using anti-PEA-15(Cell Signaling Technology, Cambridge, MA), anti-ERK, anti-phospho-ERK (Ser217/Ser221), Elk-1, AP-1, caspase-3 and caspase-7(Cell Signaling Technology, Danvers, MA). The membranes were stripped and re-blotted with a Rabbit monoclonal anti-GAPDH antibody (Sigma, St Louis, MO) as a loading control.
The formalin-fixed, paraffin-embedded tissues were sectioned at Five-micrometer and immunostaining for PEA-15 was performed as our previous protocol by using the rabbit anti-PEA-15 antibody (Abcam), horseradish peroxidase-conjugated avidin biotin complex (Vector Laboratories, Burlingame, CA, USA) and 0.03% diaminobenzidine (DAB) chromagen.
2.3 Preparation of plasmids and liposome
To engineer the treatment plasmids, we replaced the luciferase gene with PEA-15, which was digested with BglII/NheI, followed by ligation to the vector backbone. In order to avoid ampicillin resistance in the clinic, the pUK21-T-VISA-PEA-15 containing kanamycin resistance gene was created by inserting the functional part of pGL3-T-VISA-PEA-15 vector into Notl and SalI sites of the pUK21 vector. Therapeutic plasmids purified by Qiagen Endo-Free Mega Prep kit (Qiagen, Valencia, CA, USA) according to the manufacturer’ protocol. The endotoxin level was determined to be <10 endotoxin units/mg of DNA (QCL-1000 kit, BioWhittaker, Walkersville, MD, USA). Plasmid was incorporated into extruded DOTAP/Cholesterol liposome that was produced in our laboratory according to the previously published protocol[5; 14].
2.4 Luciferase reporter assay
Cells were transiently co-transfected with 2 μg indicated plasmid and 0.1 μg CMV-Luc (internal control) in 12-well plates using Lipofectamine (Invitrogen) for cell killing experiment. Forty-eight hours later, cells were subjected to lysis and assayed for luciferase activity using the dual-luciferase reporter assay system (Promega) according to the manufacturer’s protocol.
2.5 FACS analysis
To evaluate apoptosis, cells were incubated with Annexin V and PI solution (BD Biosciences) according to the manufacturer’s protocol. Flow cytometric analysis was performed immediately using a FACScalibur TM (Becton Dickinson, Heidelberg, Germany).
2.6 Breast tumor animal model and in vivo gene targeted therapy
Female BALB/c nu/nu mice at 4–6 weeks of age were used to establish human xenograft tumors model. All mice were maintained in a specific pathogen-free environment at the Animal Experiment Center of Sun Yat-Sen University, and all procedures were approved by the Animal Care and Use Committee of Sun Yat-Sen University and conformed to the legal mandates and national guidelines for the care and maintenance of laboratory animals. To test the antitumor effect of CMV-PEA-15 and T-VISA-PEA-15 in vivo, luciferase-expressing MDA-MB-231 cells (5×106) were injected into the left fourth inguinal mammary gland of mice. After inoculation, mice were noninvasively imaged by In vivo Imaging System (Xenogen) to assess tumor growth and then randomly assigned to one of three groups for every tumor model. Each group of mice received 100 μl of DNA:liposome complexes that contained 20 μg of pUK21-T-VISA (control), pUK21-CMV-PEA-15, or pUK21-T-VISA-PEA-15 administered through tail-vein injection, twice per week for 3 consecutive weeks. The growth of tumors was imaged as previously described. Before each imaging, mice were anesthetized and intraperitoneally injected with 100 ul of D-luciferin (Xenogen; 15 mg/ml in phosphate-buffered saline) and then images of mice were taken by the IVIS imaging system, and Living Imaging software (both from Xenogen) was used to quantify the signals.
2.7 Tunel staining analysis
To determine tissue undergoing apoptosis after PEA-15 expression, an in situ Apoptosis Detection Kit, peroxidase (POD) was conducted according to the manufacturer’s instructions. The percentage of apoptotic cells were calculated by random selecting 3 fields.
2.8 Acute toxicity analysis
The analysis of acute toxicity was performed as previously described. Briefly, high doses (50 or 100 μg) of DNA-liposome complex were injected into the intact Balb/c mice. Blood samples from mice were collected at indicated time and measured by automatic analyzer (Roche Cobas Mira Plus, Roche, Mannheim, Germany).
2.9 Statistical analysis
Data are given as mean ± SD. Student’s t-test was done to evaluate the differences between different groups. Survival curves were calculated by the Kaplan–Meier method. The difference in mouse survival time between two groups was assessed with the log-rank test. The significance level was set at a value of P < 0.05.
3. Results
3.1 Expression of PEA-15 in paired breast tumor tissues and adjacent normal breast tissues
In our study, we examined the expression of PEA-15 protein by IHC analysis in 8 pairs of fresh invasive breast tumor tissues (T) compared with adjacent normal breast tissues (N), respectively (Fig. 1). The results showed that protein expression had significantly higher levels in adjacent normal tissues than that in breast tumor tissues (P < 0.01).
Fig. 1.
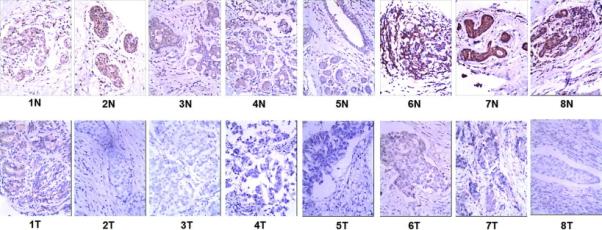
Expression of PEA-15 is downregulated in breast tumor tissues compared with adjacent morphologically normal breast epithelial tissues from the same patient.
IHC confirmed that PEA-15 protein was downregulated in the breast tumor tissue (T) compared with the paired adjacent normal breast epithelium (N) from the same patient.
3.2 T-VISA-PEA-15 selectively inhibits breast cancer cell proliferation
Up to now, PEA-15 has been reported to be correlated with low proliferation and our previous study have demonstrated that overexpression of PEA-15 can suppress triple-negative breast cancer cell growth[7]. In addition, our constructed T-VISA vector has been assured to be a robust cancer-specific promoter, and it hold the ability to drive target gene expression highly and specifically in breast cancer cells[6]. Therefore, to enhance the gene expression activity and specificity, we incorporated PEA-15 into our T-VISA system and then evaluated the potential anti-tumor effects of T-VISA-PEA-15 in numerous breast cancer cells (Fig. 2A). In order to balance the difference of gene transfection efficiency, we transiently co-transfected 9 human breast cancer cell lines with CMV-PEA-15, T-VISA-PEA-15 plus the indicator plasmid pGL3-CMV-Luc. The cell killing effects of T-VISA-PEA-15 were assessed by assaying luciferase activity 48 hours after transfection (Fig. 2B and 2C). The results showed that T-VISA-PEA15 significantly suppressed these cancer cells dose-dependently (Fig. 2B), but the killing effects has no obvious differences compared with CMV-PEA-15 treatment group (Fig. 2B and 2C). We further demonstrated that T-VISA-PEA-15 efficiently killed primary breast cancer cells obtained from three individual patients (P1, P2 and P3) compared with the control vector (Fig. 2D). Thus, T-VISA-PEA-15 has the ability to preferentially kill breast cancer cell lines and primary breast tumor cells. However, T-VISA-PEA-15 did not kill normal breast cell lines MCF10A and 184A1 (Fig. 2E). In contrast, CMV-PEA-15 induced death of not only breast cancer cells (Fig. 2B, 2C and 2D) but also primary breast tumor cells and normal cells tested (Figure 2E). In brief, T-VISA-driven PEA-15 selectively kills breast cancer cells in vitro.
Fig. 2.
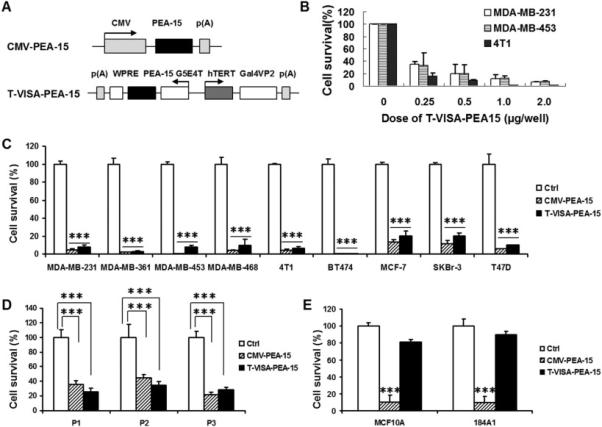
T-VISA-PEA-15 selectively exerts an inhibitory effect in multiple breast cancer cell lines.
(A) Schematic diagram of CMV-PEA-15 and T-VISA-PEA-15 constructs in the pUK21 backbone.
(B) Different advanced breast cancer cells were respectively cotransfected increasing amount (0, 0.25, 0.5, 1.0, or 2 μg/well) of T-VISA-PEA-15 and 100 ng of CMV-Luc.
(C) T-VISA-PEA-15 effectively inhibits breast tumor cells proliferation. The results are expressed as a percentage of T-VISA control transfected cells (100%) and represent the mean ± SD of three independent experiments. ***indicates P < 0.001.
(D) PEA-15 expression driven by T-VISA also kills primary human breast tumor cells effectively. ***indicates P < 0.001.
(E) The killing effects of T-VISA-PEA-15 on normal breast cells are slower than CMV-PEA-15. ***indicates P < 0.001.
3.3 T-VISA-PEA-15 sequesters extracellular signal-regulated kinase (ERK) in cytoplasm, and then induces cancer cell apoptosis
To investigate the mechanisms of cell killing effects of our T-VISA-PEA-15:liposome complex, we conducted western blotting to test the expression of Erk1/2 in cytoplasm. The results showed that both T-VISA-PEA-15 and CMV-PEA-15 sequesters pERK in cytoplasm, and the downstream nuclear transcription factor Elk-1 and AP-1 were all down-regulated (Fig.3A). Moreover, we found an increase in cleaved caspase 3 and cleaved caspase 7 protein expression (Fig. 3B), indicating that PEA-15 driven by CMV or T-VISA promoter does indeed increase apoptosis. Furthermore, we used Annexin V-FITC/PI staining and FACS to assess whether CMV-PEA-15 and T-VISA-PEA-15 induced cell apoptosis in MDA-MB-231 and MCF-7 cells. Consistent with our expectation, T-VISA-PEA-15-transfected cells had an obviously higher ratio of apoptotic cells compared with CMV-PEA-15 or Ctrl (T-VISA) (Fig.3C).
Fig. 3.
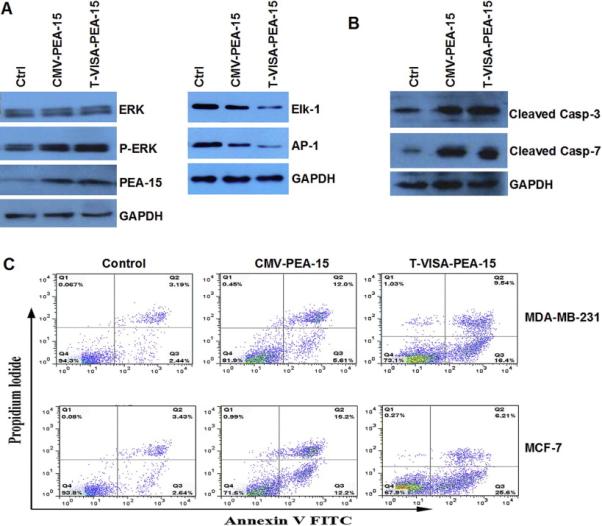
T-VISA-PEA-15 erk1/2 in cyto and induce the apoptosis.
(A) Western blot analysis of ERK, pERK and the downstream protein expression in MDA-MB-231 cell. GAPDH was used as an internal control.
(B) Western blotting verified cleaved caspase 3 and cleaved caspase 7 protein expression.
(C) Annexin V-FITC/propidium iodide staining and FACS quantification of the number of apoptotic cells in MDA-MB-231 and MCF-7 cells transfected with DNA(Ctrl, CMV-PEA-15 or T-VISA-PEA-15)-liposome complexes. FACS, fluorescence-activated cell sorting; FITC, fuorescein isothiocyanate.
3.4 T-VISA-PEA-15 nanoparticles suppress tumor growth and prolong mouse survival time more effectively than CMV-PEA-15 in breast tumor mouse model
To investigate the cell killing effects of T-VISA-PEA-15 in vivo, we established an orthotopic animal model with human breast cancer cell line, MDA-MB-231-Luc, and delivered the indicated plasmid DNA in liposome complexes when tumors were palpable. We monitored the tumor growth by noninvasive IVIS imaging device in real time and the survival information of mice were also recorded. In MDA-MB-231-Luc model, expression of PEA-15 driven by T-VISA or CMV treatment group demonstrated a strong reduction in tumor growth (both P < 0.05), but tumor inhibition by T-VISA-PEA-15 treatment was more substantial than CMV-PEA-15 treatment (Fig. 4A). What’s more, T-VISA-PEA-15 significantly prolonged the survival time of mice more effectively than CMV-PEA-15 (P <0.05), although the survival time of mice in treatment groups had all been extended compared to control group (P <0.05 in both treatment groups) (Fig. 4B). By terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL) assay, the results showed that T-VISA-PEA-15 induced apoptosis in tumor cells more remarkable than CMV-PEA-15 and Ctrl (Fig. 4C).
Fig. 4.
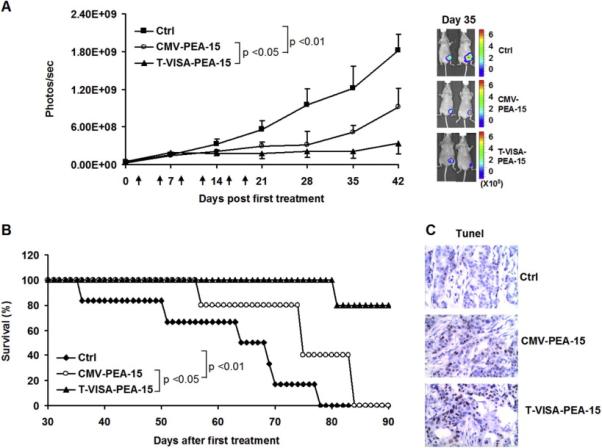
T-VISA-PEA-15:liposome complexes dramatically suppresses breast tumor growth and Significantly Improves Survival in the breast tumor model. BALB/c nude mice bearing MDA-MB-231-Luc tumors were treated with 20 μg of liposomal plasmid DNA (CMV-PEA-15, T-VISA-PEA-15 or Ctrl), twice weekly for three consecutive weeks.
(A) The tumor cell growth photon signals were quantified (left panel) with Xenogen’s software and representative images are presented (right panel). Error bars indicate SEM. Arrows indicate the treatment time points.
(B) Kaplan-Meier survival analysis from (A) was determined. Expression of PEA-15 driven by T-VISA vector prolonged mouse survival more effectively than PEA-15 driven by CMV promoter.
(C) Representative images of in vivo apoptosis of tumor by TUNEL assay.
3.5 T-VISA-PEA-15 nanoparticles has virtually no acute toxicity in normal mice compared with CMV-PEA-15
In order to compare the treatment safety profile between T-VISA-PEA-15 and CMV-PEA-15, a single high dose of 50 or 100 μg plasmid DNA was injected by tail veil of BALB/c mice (Fig. 5A and B). Blood samples were collected and serum levels of liver alanine aminotransferase (ALT), aspartate aminotransferase (AST), and blood creatinine (CRE) were measured for mice that received 50 μg plasmid DNA. In the CMV-PEA-15 treatment group, the serum level of ALT, AST and CRE were significantly higher than either T-VISA-PEA-15 or control group but decreased by Day 4 (Fig. 5C, 5D and 5E). Moreover, T-VISA-PEA-15-liposome complexes and control groups showed 100% event-free survival, while 85% or 70% event-free survival for CMV-PEA-15 treated mice at a dose of 50 or 100 μg DNA, respectively (Fig. 5A and B). These results indicate that T-VISA-PEA-15-liposome-mediated systemic gene therapy is safer than the commonly used CMV promoter-driven treatment strategy.
Fig. 5.
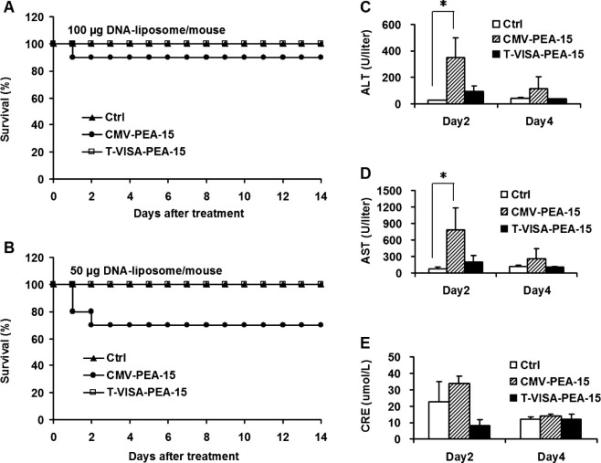
T-VISA-PEA-15 nanoparticles have no systemically acute toxicity compared with CMV-PEA-15 in immunocompetent mice.
(A and B) Kaplan-Meier survival analysis of female BALB/c mice who were treated with single doses of 100 μg or 50 μg plasmid in a liposomal complex via the tail vein. (C, D, and E) Kinetics serum level of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and blood creatinine (CRE) in mice after a single dose of 50 μg plasmid injection. Asterisk, P < 0.05.
4. Discussion
The delivery of therapeutic genes into tumor cells remains a key obstacle for cancer therapy[15; 16]. In this study, we examined the effectiveness of PEA-15 targeted therapy utilizing a T-VISA breast cancer specific promoter system. We first, examined the expression levels of protein levels of PEA-15 in the tissues of patients with breast cancer and the adjacent normal breast tissues of these patients. We observed that PEA-15 levels were significantly reduced in breast tumor tissues compared to their normal counterparts. The function of PEA-15 as tumor suppressor or as inhibitor of tumor cell growth and invasion in various cancers has been investigated[8; 9; 17; 18]. Our group and others reported that phosphorylation status of PEA-15 is associated with the anti-tumor functions of PEA-15. PEA-15 contains two major serine residues (Ser104 and Ser116) at C-terminus. When PEA-15 is unphosphorylated state, PEA-15 bind to ERK and reduces the transcriptional activity of ERK by inhibiting the transcription factor Elk-1[19; 20; 21; 22; 23]. However, phosphorylation at Ser104 blocks PEA-15 binding to ERK, or double phosphorylation at Ser104 and Ser116 switches the binding specificity of PEA-15 from ERK to FADD, thereby stabilizing its anti-apoptotic function[24; 25]. Based on these observations, we proposed that unphosporylated-PEA-15 at both Ser104 and Ser116 could result in more potent suppression of tumorigenicity than wild-type PEA-15 in breast cancer. We have shown that the double-unphosphorylated form (PEA-15-AA), strongly inhibited tumor growth and Ki-67 expression in an ovarian cancer xenograft model[8; 9]. Several other studies also have shown that unphosphorylated form of PEA-15 acts as a tumor suppressor in cervical cancer and lung cancer cells [26; 27]. Moreover, recent studies indicate that serous ovarian cancer has a genomic pattern very similar to that of triple-negative breast cancer [28] which may indicate that potent form of PEA-15, such as PEA-15-AA, can be selectively targeted for TNBC using breast cancer-specific promoter.
In general, gene delivery systems are comprised of viral as well as non-viral systems[29; 30; 31], Viral systems possess higher gene transfer efficiency, however, which have many unsolved problems, such as immunogenicity, cytopathic and recombinogenic effects[32], while non-viral systems are confronted with low packaging and gene transfection efficiency, and so on[14; 33]. Therefore, to enhance the transgene expression of non-viral vector, we previously constructed T-VISA promoter, which have been demonstrated to hold high activity and specificity compared to CMV promoter in breast cancer cell lines. Moreover, our T-VISA can targetedly drive the expression of miR-34a and pro-apoptosis gene BikDD in breast tumor in vitro and in vivo[13]. Looking for clinical application PEA-15, we incorporated the PEA-15 cDNA into our T-VISA promoter system and engineered T-VISA-PEA-15 plasmid. To our delighted, forced expression of PEA-15 by T-VISA vector in breast cancer cell lines lead to inhibition of proliferation compared to CMV promoter in vitro, while the cell killing effects on normal cell lines were significantly weaker than CMV. To intensive study the biological function of PEA-15 in breast cancer, we found that cytoplasmic pERK were down-regulated by CMV-PEA-15 and T-VISA-PEA-15, whereas the level of ERK was not changed. In short, PEA-15 sequestered extracellular signal-regulated kinase (ERK) in cytoplasm[7; 34], leading to apoptosis of breast cancer cells and suggesting that T-VISA-PEA-15 may exert its tumor suppressive effect by enhancing multiple pro-apoptosis gene expression.
Based on the above investigation, we then studied the effectiveness of non-viral delivery system in breast cancer mouse models. The T-VISA-PEA-15: liposome-nanoparticles treatment had better therapeutic effect than Ctrl and CMV-PEA-15 complexed with in the MDA-MB-231-Luc animal model. What’s more, the mouse survival in T-VISA-PEA-15 treatment group was prolonged more effectively than Ctrl and CMV-PEA-15 group. It could be explained by the differences of the duration of transgene expression and tumor cell apoptosis induced by CMV or T-VISA vector. In addition, the acute toxicity of T-VISA-PEA-15 treatment group in vivo was assessed and the results showed PEA-15 induced by T-VISA is more safer than CMV. The main mechanism of action of T-VISA-PEA-15 in breast cancer is summarized in Fig.6.
Fig.6.
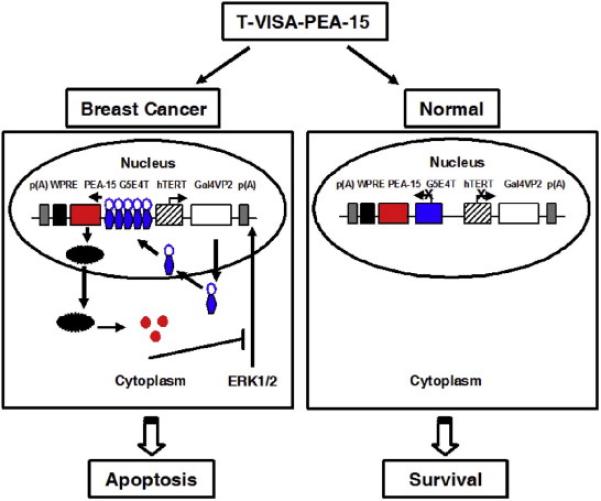
Diagrammatic sketch of the T-VISA-PEA-15 system targeting to breast cancer.
The T-VISA-PEA-15 system involves a series of steps in human breast cancer: Firstly, the hTERT promoter drives the GAL4-VP2 fusion protein specifically expression in breast tumor and then open target gene PEA-15 mRNA transcripts. Afterwards, the PEA-15 mRNA transcripts enhance and prolong expression duration of PEA-15 protein, which can sequesters extracellular signal-regulated kinase (ERK) in cytoplasm, thereby leading to apoptosis of breast cancer cells. However, there’s little non-specificity gene expression in normal cells for the activity of hTERT promoter is very lower, thereafter, normal cells survival.
Now that we can surmount the disadvantage of efficient personalized gene expression through modified the non-viral vector, T-VISA-PEA-15 nanoparticles would undoubtedly be an excellent candidate for future development into clinical trial and the approach described here could also be easily applied to other cancer cell types via modifying the suppressor gene and cancer-specific promoter.
Acknowledgements
This work was supported by funds from the National Natural Science Foundation of China (31100935, 81272514, 81302318, 81372133), the Key Program of the National Natural Science Foundation of China (31030061), the China Postdoctoral Science Foundation (2012M520075), the Science and Technology Planning Project of Guangzhou (2014J4100169), US NIH through Grant (CA127562), and through MD Anderson’s Cancer Center Support Grant (CA016672). We thank Dr Mark Ginsberg (University of California, San Diego, CA, USA) for providing plasmid constructs PEA-15.
Footnotes
5. Conflicts of interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- [2].Dou S, Yao YD, Yang XZ, Sun TM, Mao CQ, Song EW, Wang J. Anti-Her2 single-chain antibody mediated DNMTs-siRNA delivery for targeted breast cancer therapy. Journal of controlled release : official journal of the Controlled Release Society. 2012;161:875–883. doi: 10.1016/j.jconrel.2012.05.015. [DOI] [PubMed] [Google Scholar]
- [3].Yao YD, Sun TM, Huang SY, Dou S, Lin L, Chen JN, Ruan JB, Mao CQ, Yu FY, Zeng MS, Zang JY, Liu Q, Su FX, Zhang P, Lieberman J, Wang J, Song E. Targeted delivery of PLK1-siRNA by ScFv suppresses Her2+ breast cancer growth and metastasis. Science translational medicine. 2012;4:130–148. doi: 10.1126/scitranslmed.3003601. [DOI] [PubMed] [Google Scholar]
- [4].Xie X, Guo J, Kong Y, Xie GX, Li L, Lv N, Xiao X, Tang J, Wang X, Liu P, Yang M, Xie Z, Wei W, Spencer DM, Xie X. Targeted expression of Escherichia coli purine nucleoside phosphorylase and Fludara(R) for prostate cancer therapy. The journal of gene medicine. 2011;13:680–691. doi: 10.1002/jgm.1620. [DOI] [PubMed] [Google Scholar]
- [5].Lang JY, Hsu JL, Meric-Bernstam F, Chang CJ, Wang Q, Bao Y, Yamaguchi H, Xie X, Woodward WA, Yu D, Hortobagyi GN, Hung MC. BikDD eliminates breast cancer initiating cells and synergizes with lapatinib for breast cancer treatment. Cancer cell. 2011;20:341–356. doi: 10.1016/j.ccr.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Xie X, Li L, Xiao X, Guo J, Kong Y, Wu M, Liu W, Gao G, Hsu JL, Wei W, Hung MC, Xie X. Targeted expression of BikDD eliminates breast cancer with virtually no toxicity in noninvasive imaging models. Molecular cancer therapeutics. 2012;11:1915–1924. doi: 10.1158/1535-7163.MCT-12-0191. [DOI] [PubMed] [Google Scholar]
- [7].Bartholomeusz C, Gonzalez-Angulo AM, Kazansky A, Krishnamurthy S, Liu P, Yuan LX, Yamasaki F, Liu S, Hayashi N, Zhang D, Esteva FJ, Hortobagyi GN, Ueno NT. PEA-15 inhibits tumorigenesis in an MDA-MB-468 triple-negative breast cancer xenograft model through increased cytoplasmic localization of activated extracellular signal-regulated kinase. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:1802–1811. doi: 10.1158/1078-0432.CCR-09-1456. [DOI] [PubMed] [Google Scholar]
- [8].Xie X, Bartholomeusz C, Ahmed AA, Kazansky A, Diao L, Baggerly KA, Hortobagyi GN, Ueno NT. Bisphosphorylated PEA-15 sensitizes ovarian cancer cells to paclitaxel by impairing the microtubule-destabilizing effect of SCLIP. Molecular cancer therapeutics. 2013;12:1099–1111. doi: 10.1158/1535-7163.MCT-12-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lee J, Bartholomeusz C, Krishnamurthy S, Liu P, Saso H, Lafortune TA, Hortobagyi GN, Ueno NT. PEA-15 unphosphorylated at both serine 104 and serine 116 inhibits ovarian cancer cell tumorigenicity and progression through blocking beta-catenin. Oncogenesis. 2012;1:e22. doi: 10.1038/oncsis.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Callaway K, Abramczyk O, Martin L, Dalby KN. The anti-apoptotic protein PEA-15 is a tight binding inhibitor of ERK1 and ERK2, which blocks docking interactions at the D-recruitment site. Biochemistry. 2007;46:9187–9198. doi: 10.1021/bi700206u. [DOI] [PubMed] [Google Scholar]
- [11].Funke V, Lehmann-Koch J, Bickeboller M, Benner A, Tagscherer KE, Grund K, Pfeifer M, Herpel E, Schirmacher P, Chang-Claude J, Brenner H, Hoffmeister M, Roth W. The PEA-15/PED protein regulates cellular survival and invasiveness in colorectal carcinomas. Cancer letters. 2013;335:431–440. doi: 10.1016/j.canlet.2013.02.053. [DOI] [PubMed] [Google Scholar]
- [12].Watanabe Y, Yamasaki F, Kajiwara Y, Saito T, Nishimoto T, Bartholomeusz C, Ueno NT, Sugiyama K, Kurisu K. Expression of phosphoprotein enriched in astrocytes 15 kDa (PEA-15) in astrocytic tumors: a novel approach of correlating malignancy grade and prognosis. Journal of neuro-oncology. 2010;100:449–457. doi: 10.1007/s11060-010-0201-1. [DOI] [PubMed] [Google Scholar]
- [13].Li L, Xie X, Luo J, Liu M, Xi S, Guo J, Kong Y, Wu M, Gao J, Xie Z, Tang J, Wang X, Wei W, Yang M, Hung MC, Xie X. Targeted expression of miR-34a using the T-VISA system suppresses breast cancer cell growth and invasion. Molecular therapy : the journal of the American Society of Gene Therapy. 2012;20:2326–2334. doi: 10.1038/mt.2012.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xie X, Xia W, Li Z, Kuo HP, Liu Y, Li Z, Ding Q, Zhang S, Spohn B, Yang Y, Wei Y, Lang JY, Evans DB, Chiao PJ, Abbruzzese JL, Hung MC. Targeted expression of BikDD eradicates pancreatic tumors in noninvasive imaging models. Cancer cell. 2007;12:52–65. doi: 10.1016/j.ccr.2007.05.009. [DOI] [PubMed] [Google Scholar]
- [15].Hu YL, Huang B, Zhang TY, Miao PH, Tang GP, Tabata Y, Gao JQ. Mesenchymal stem cells as a novel carrier for targeted delivery of gene in cancer therapy based on nonviral transfection. Molecular pharmaceutics. 2012;9:2698–2709. doi: 10.1021/mp300254s. [DOI] [PubMed] [Google Scholar]
- [16].Klutz K, Willhauck MJ, Dohmen C, Wunderlich N, Knoop K, Zach C, Senekowitsch-Schmidtke R, Gildehaus FJ, Ziegler S, Furst S, Goke B, Wagner E, Ogris M, Spitzweg C. Image-guided tumor-selective radioiodine therapy of liver cancer after systemic nonviral delivery of the sodium iodide symporter gene. Human gene therapy. 2011;22:1563–1574. doi: 10.1089/hum.2011.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Botta G, Perruolo G, Libertini S, Cassese A, Abagnale A, Beguinot F, Formisano P, Portella G. PED/PEA-15 modulates coxsackievirus-adenovirus receptor expression and adenoviral infectivity via ERK-mediated signals in glioma cells. Human gene therapy. 2010;21:1067–1076. doi: 10.1089/hum.2009.181. [DOI] [PubMed] [Google Scholar]
- [18].Bartholomeusz C, Rosen D, Wei C, Kazansky A, Yamasaki F, Takahashi T, Itamochi H, Kondo S, Liu J, Ueno NT. PEA-15 induces autophagy in human ovarian cancer cells and is associated with prolonged overall survival. Cancer research. 2008;68:9302–9310. doi: 10.1158/0008-5472.CAN-08-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chou FL, Hill JM, Hsieh JC, Pouyssegur J, Brunet A, Glading A, Uberall F, Ramos JW, Werner MH, Ginsberg MH. PEA-15 binding to ERK1/2 MAPKs is required for its modulation of integrin activation. The Journal of biological chemistry. 2003;278:52587–52597. doi: 10.1074/jbc.M309322200. [DOI] [PubMed] [Google Scholar]
- [20].Formstecher E, Ramos JW, Fauquet M, Calderwood DA, Hsieh JC, Canton B, Nguyen XT, Barnier JV, Camonis J, Ginsberg MH, Chneiweiss H. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Developmental cell. 2001;1:239–250. doi: 10.1016/s1534-5807(01)00035-1. [DOI] [PubMed] [Google Scholar]
- [21].Bartholomeusz C, Itamochi H, Nitta M, Saya H, Ginsberg MH, Ueno NT. Antitumor effect of E1A in ovarian cancer by cytoplasmic sequestration of activated ERK by PEA15. Oncogene. 2006;25:79–90. doi: 10.1038/sj.onc.1209014. [DOI] [PubMed] [Google Scholar]
- [22].Hill JM, Vaidyanathan H, Ramos JW, Ginsberg MH, Werner MH. Recognition of ERK MAP kinase by PEA-15 reveals a common docking site within the death domain and death effector domain. The EMBO journal. 2002;21:6494–6504. doi: 10.1093/emboj/cdf641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Whitehurst AW, Robinson FL, Moore MS, Cobb MH. The death effector domain protein PEA-15 prevents nuclear entry of ERK2 by inhibiting required interactions. The Journal of biological chemistry. 2004;279:12840–12847. doi: 10.1074/jbc.M310031200. [DOI] [PubMed] [Google Scholar]
- [24].Renganathan H, Vaidyanathan H, Knapinska A, Ramos JW. Phosphorylation of PEA-15 switches its binding specificity from ERK/MAPK to FADD. The Biochemical journal. 2005;390:729–735. doi: 10.1042/BJ20050378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Trencia A, Perfetti A, Cassese A, Vigliotta G, Miele C, Oriente F, Santopietro S, Giacco F, Condorelli G, Formisano P, Beguinot F. Protein kinase B/Akt binds and phosphorylates PED/PEA-15, stabilizing its antiapoptotic action. Molecular and cellular biology. 2003;23:4511–4521. doi: 10.1128/MCB.23.13.4511-4521.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sulzmaier F, Opoku-Ansah J, Ramos JW. Phosphorylation is the switch that turns PEA-15 from tumor suppressor to tumor promoter. Small GTPases. 2012;3:173–177. doi: 10.4161/sgtp.20021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Quintavaller C, Costanzo S, Zanca C, Tasset I, Fraldi A, Incoronato M, Mirabelli P, Monti M, Ballabio A, Pucci P, Cuervo AM, Condorelli A. Phosphorylation-Regulated Degradation of the Tumor-Suppressor Form of PED by Chaperone-Mediated Autophagy in Lung Cancer Cells. J Cell Physiol. 2014 doi: 10.1002/jcp.24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].N. Cancer Genome Atlas Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sher YP, Chang CM, Juo CG, Chen CT, Hsu JL, Lin CY, Han Z, Shiah SG, Hung MC. Targeted endostatin-cytosine deaminase fusion gene therapy plus 5-fluorocytosine suppresses ovarian tumor growth. Oncogene. 2013;32:1082–1090. doi: 10.1038/onc.2012.134. [DOI] [PubMed] [Google Scholar]
- [30].Shen H, Mittal V, Ferrari M, Chang J. Delivery of gene silencing agents for breast cancer therapy. Breast cancer research : BCR. 2013;15:205. doi: 10.1186/bcr3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Steidl U, Steidl C, Ebralidze A, Chapuy B, Han HJ, Will B, Rosenbauer F, Becker A, Wagner K, Koschmieder S, Kobayashi S, Costa DB, Schulz T, O’Brien KB, Verhaak RG, Delwel R, Haase D, Trumper L, Krauter J, Kohwi-Shigematsu T, Griesinger F, Tenen DG. A distal single nucleotide polymorphism alters long-range regulation of the PU.1 gene in acute myeloid leukemia. The Journal of clinical investigation. 2007;117:2611–2620. doi: 10.1172/JCI30525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Giacca M, Zacchigna S. Virus-mediated gene delivery for human gene therapy. Journal of controlled release : official journal of the Controlled Release Society. 2012;161:377–388. doi: 10.1016/j.jconrel.2012.04.008. [DOI] [PubMed] [Google Scholar]
- [33].Rogers ML, Rush RA. Non-viral gene therapy for neurological diseases, with an emphasis on targeted gene delivery. Journal of controlled release : official journal of the Controlled Release Society. 2012;157:183–189. doi: 10.1016/j.jconrel.2011.08.026. [DOI] [PubMed] [Google Scholar]
- [34].Glading A, Koziol JA, Krueger J, Ginsberg MH. PEA-15 inhibits tumor cell invasion by binding to extracellular signal-regulated kinase 1/2. Cancer research. 2007;67:1536–1544. doi: 10.1158/0008-5472.CAN-06-1378. [DOI] [PubMed] [Google Scholar]