

Multiple Endocrine Neoplasia type 1 (MEN-1)
MEN-1 is inherited as an autosomal dominant disorder (1) (2). It has a prevalence of 2–3 per 100,000 (3) and is reported to be present in 0.22–0.25% of autopsies (4). The gene causing MEN-1 is located on the long arm of chromosome 11 (11q13) (5) (6) and is composed of 10 exons (9 coding) (2) (4). The MEN-1 gene is a tumor suppressor gene. It encodes a 610 amino acid nuclear protein called menin. Abnormalities of this gene result in mutations, deletions, and/or truncations of the menin protein (4) (7,8). The exact mechanism by which alterations of menin result in endocrine tumors is still unclear. Menin interacts with a large number of proteins many of which have important roles in transcriptional regulation, genomic stability, cell division and cell cycle control (2) (4) (7,8). The crystal structure of menin supports its role as a key scaffolding protein that regulates gene transcription (9) (10).
Patients with MEN-1 usually develop primary hyperparathyroidism (HPT) as the initial disorder of the syndrome (90–100%) (1) (11) (12) (13), followed by pancreatic neuroendocrine tumors (pNET) that can either be functional (20–70%), of which gastrinoma is the most common, or nonfunctional (80–100%), pituitary adenomas (20–65%), adrenal tumors (10–73%) and thyroid adenomas (0–10%) (2) (14) (12) (15). Patients with MEN-1 also have a high occurrence of other endocrine and non-endocrine tumors including carcinoid tumors (thymic 0–8%, gastric 7–35%, bronchial 0–8%, and rarely intestinal), skin and subcutaneous tumors (angiofibromas 88%, collagenomas 72%, lipomas 34% and melanoma), central nervous system tumors (meningiomas, ependymomas and schwannomas 0–8%) and smooth muscle tumors (leiomyomas and leiomyosarcomas 1–7%) (2) (15) (16) (17) (18) (19) (20) (21) (22) (23) (24) (25) (26). In early studies thyroid disease was also reported in MEN1 patients but in a recent cross-sectional study of 95 MEN1 patients (27), the rate of coourrence of a thyroid incidentoma was the same as matched non-MEN1 patients (45% vs 51%), respectively. In addition, other nonendocrine malignant tumors are also being reported to occur in MEN-1 (11). These include lymphomas, renal cell cancer, melanoma, leiomyosarcoma, thrombotic thrombocytopenia purpura, myeloma, ovarian tumors, gastrointestinal stromal tumor (28), seminoma, chondrosarcoma, mesothelioma and thymomas (11) (29) (30) (31) (32) (33) (34) (35) (36,37). However, whether the incidence of these nonendocrine tumors is truly increased or not is unclear.
Early Diagnosis of MEN-1
Before MEN-1 can be diagnosed it must be suspected. Suspicion should be raised in any patient with a family history of endocrine tumors of the pancreas, family members with pituitary or parathyroid disease or a family history of endocrinopathy; in patients with renal colic with NETs; in any patient with ZES (20–25% have it as part of the MEN-1 syndrome); with a young age onset of a functional pNET; with multiple pNETs; with hyperparathyroidism with multiple gland involvement or with hyperplasia or with a pNET associated with hypercalcemia or another endocrinopathy (1,2). In most patients (83%) MEN1 clinically presents after age 21 (38). In the 17% of patients with MEN1 presenting at <21 years, which should lead to suspicion of the diagnosis, the most frquent abnormalities were hyperparathyroidsm (75%), pituitary adenoma (34%), insulinoma (12%), nonfunctiona pNET (9%) and gastrinoma (2%) (38). Genetic screening for MEN-1 is recommended when an individual has 2 or more MEN-1 related tumors, multiple abnormal parathyroid glands before age 30 years, recurrent HPT at a young age, gastrinoma and hyperparathyroidism (HPT) or multiple pNETs at any age, plus a family history of kidney stones or endocrine tumors that are part of the syndrome (39,40). Genetic testing includes sequencing of the entire coding region of the MEN-1 gene (exons 2–10) and identifies mutations in about 80% of patients with familial MEN-1 (1,41,42) (Table 1).
Table 1.
Basic facts about the different endocrine neoplasia familial syndromes
| Fanilial Endocrine Syndrome | Chromosome | Gene | Pattern of inheritance | Glands Involved | Extraglandular manifestations | Cause of death |
|---|---|---|---|---|---|---|
| MEN-1 | 11 | Menin | Autosomal dominant | Parathyroid Pancreas Pituitary Thyroid Adrenal |
Bronchus Thymus Stomach Lipomas Skin tumors |
Pancreatic NET, Bronchial or thymic NET |
| MEN-2a | 10 | RET | Autosomal dominant | Thyroid Adrenal Parathyroid |
None | MTC |
| MEN-2b (MEN-3) | 10 | RET | Autosomal dominant | Thyroid Adrenal |
Marfanoid habitus, Mucosal neuromas, Corneal nerve hypertrophy, Intestinal ganglioneruomatosis. | MTC |
| FMTC | 10 | RET | Autosomal dominant | Thyroid | None | Natural causes |
| MEN-4 | 12 | CDKN1B/p27Kip1 | Autosomal dominant | Parathyroid GEP pituitary |
none | unclear |
Parathyroid disease in MEN-1
Primary hyperparathyroidism (HPT) is the most common endocrine abnormality in MEN-1. It reaches nearly 100% penetrance by the age of 50 years. HPT is usually the first manifestation of MEN-1 with a typical age of onset of 20–25 years. Decreased bone density and kidney stones are common. HPT often occurs at the same time as Zollinger-Ellison syndrome (ZES) and surgery to correct the HPT greatly ameliorates the clinical findings of ZES (43) (44). As in sporadic cases, biochemical testing for HPT is critical to the diagnosis (45) (46). Total or ionized serum level of calcium and intact serum parathyroid hormone levels are measured and both should be elevated. 24 hour urinary calcium should also be measured and will be elevated. Opinions differ as to the timing of parathyroid surgery in MEN-1 as well as to the type of operation that should be performed. Early surgery may delay the lifetime exposure to the biochemical manifestations of HPT, while waiting until the parathyroid glands enlarge may make the operation easier. Patients with MEN-1 and HPT typically have multiple abnormal glands. The tumors are asymmetric in size and should be considered as independent clonal adenomas (47). Imaging studies are not useful for initial operations because all four parathyroid glands must be identified, but are necessary and very useful in reoperations (Figure 1). Some have recommended in MEN1 patients a minimally invasive parathyroidectomy should be considered with selective removal of only the enlarged glands while others recommend subtoal parathyroidectomy (3.5 glands removed) or a total parathyriodectomy with inplant (13,40–48), The current operation that is recommended by most experts is a subtotal parathyroidectomy (3 and ½ gland resection) with removal of the cervical thymus. Intraoperative parathyroid hormone level monitoring is recommended to be certain that sufficient abnormal parathyroid tissue has been removed (45) (48). A viable 50 mg amount of normal parathyroid tissue should be left in the neck and marked with a hemoclip. Total parathyroid resection and transplantation of parathyroid tissue to the non-dominant forearm used to be recommended, but it has been shown to have too high an incidence of hypoparathyroidism due to graft failure (49). Because of the multiple abnormal parathyroid gland nature of this disease, there is a high probability of recurrent HPT years after surgery if less than 3 and ½ parathyroid gland resections are performed (50). Patients should be followed for this possibility. Calcium-sensing receptor agonists (calcimimetics) are a new class of drugs that can act directly on the parathyroid gland, decrease PTH release, and may even decrease parathyroid tissue growth. A small number of patients with MEN1 and hyperparathydoidism have been treated with the calcium sensing receptor agonist, cinacalcet and it has been effetive in control the hyperparathyroidism (51,52). These agents may play an important role in the management of these patients in the future (53). Parathyroid carcinoma has been reported in patients with MEN1 (54,55), however it is uncommon, occuring in only 0.28% of all MEN1 patients, and the clinical presentation is similar to that seen in patients without MEN1 with parathyroid carcinoma (55). Patients with MEN1 commonly develop bone disease with osteoporosis thought secondary to the hyperparathyroidism (56) (57). However, recent studies show that menin has important effects in bone particularly in regulating osteoblast activity which plays a key role in bone deveopment, remodeling and maintenance (58). Some MEN1 patients require multiple re-operations to control the recurrent or persistent hyperparathyroidism and reoperation can be difficult and associated with increased morbidity. Recently ethanol ablation of abnormal parathyroids (59) has been described to be able to successfully control the hyperparathyrroidsm in such patients with MEN1 with a low rate of hypocalcemia and no permanent complications.
Figure 1. Sestamibi scan (A), neck CT scan (B) and somatostatin receptor scintigraphy SRS (C) in an MEN-1 patient with recurrent primary hyperparathryoidism and metastatic pancreatic NET.

Patient is a 45 year old man with recurrent primary hyperparathyroidism status post an operation in which they removed two abnormal parathyroid glands and now he has recurrent disease. Sestamibi scan (A) shows an abnormal right inferior parathyroid gland (yellow arrow) and the same abnormal gland is demonstrated on a 4-D neck CT scan with intravenous contrast (yellow arrow) (B). Octreoscan shows an abnormal pancreatic NET in the tail of the pancreas with possible liver metastases (C) left at 4 hours and middle panel 24 hours after labelled octreotide), while 68Ga DOTATATE (C right panel) clearly shows more tumor and also a pituitary adenoma.
Pituitary Tumors in MEN-1
Anterior pituitary adenomas are the initial clinical manifestation of MEN-1 in 25% of cases (2,8). Its prevalence in MEN-1 is between 20 and 60% and in a recent study of all patients (N=144) with pituitary adenomas seen in one institution over a 6 year period, 7.7% had MEN-1 (60). Most of these anterior pituitary tumors are microadenomas (<1 cm in diameter). Every type of anterior pituitary tumor has been reported to occur in MEN-1 with the most common being a prolactinoma. Screening for anterior pituitary tumors requires measuring serum levels of prolactin, IGF-1 and magnetic resonance imaging of the pituitary. Patients should be questioned for loss of peripheral vision. Visual fields are assessed formally, if any suspicion of change or there is evidence of a pituitary tumor. Because these tumors occur in patients of child-bearing age, undiagnosed pregnancy may cause a confusingly elevated prolactin level. Treatment of pituitary tumors in MEN-1 is the same as sporadic pituitary tumors. Bromocriptine and cabergoline are used to treat prolactinomas. Octreotide and lanreotide are used to treat growth hormone secreting tumors. Transsphenoidal surgery is the treatment of choice for discrete pituitary microadenomas or macroadenomas and may be curative. The major surgical morbidity of transphenoidal hypophysectomy is permanent diabetes insipidus. Even in patients who are successfully treated, long-term follow-up is indicated because tumors may recur.
pNETs in MEN-1
The prevalence of pNETs in MEN-1 is between 30–75% clinically and 80–100% in post-mortem studies (2,11,61). The pathology of pNETs in MEN-1 is typically multicentric and multifocal with multiple endocrine tumors throughout the pancreas and the duodenum in patients with MEN-1/ZES (Figure 2). Unfortunately the only reliable method of establishing the presence of pNETs in MEN1 patients not associated with a clinical syndrome (NF-pNETs) is to perform detailed imaging studies. Although NF-pNETs can secrete a number of peptides includng chromogranin A, neuron specific enolase, pancreatic polypeptide, neurotensin or ghrelin, these don’t result in a distinct clincial syndrome and a recent study demonstrates their assesmment in plasma in MEN1 patients has low diagnostic accuracy for NF-pNETs (62). The frequency with which pNETs are detected by imaging depends to a large extent on the imaging modality. Cross-sectional imaging studies (CT, MRI, transabdominal ultrasound) frequently (35%) miss tumors <2 cm, octreoscan misses 25–30% <1 cm, 68Ga-DOTATOC/PET misses 20–30% <0.5 cm and endoscopic ultrasound (EUS) can detect pNETs down to 0.1–0.2 cm (63). 68Ga-DOTATE is less available than octreoscan but is more sensitive and frequently images more tumors than octreoscan (Figure 1). Studies in MEN-1 patients show that EUS is the most sensitive single modality to detect pNETs (64). In one recent comparative study (65) EUS was markedly superior to 11C-5-HTP scanning, somatostatin receptor scintigraphy and CT/MRI for the detection of pancreatic NET. However in another recent prospective study, the findings of MRI and EUS were complementary in MEN-1 patients for the detection of pNETs (66). Tumors vary from microadenomas, to adenomas, to carcinomas (Figure 2) with lymph node and liver metastases. The most characteristic MEN-1 pancreatic lesion on pathology studies is the presence of diffuse microadenomatosis (<0.5 mm) (67–69). Molecular studies in multiple pNETs or gastrinomas from MEN-1 patients assessing loss of heterozygosity at various loci and X-chromosome inactivation studies demonstrate that the multiple pNETs arise as independent events (70). Whereas microadenomatosis of the pancreas is almost invariably seen, in various studies only 0–13% of MEN-1 patients are reported to develop large (>2 cm) and/or symptomatic NF-pNETs (2), with many patients not developing larger pNETs even over long periods of time. At present the molecular events that lead to this variability in growth of pancreatic microadenomas in different MEN-1 patients are unknown and there are well-established predictive factors, requiring sequential imaging as the only means to evaluate the growth over time (2,71). Most studies in MEN1 patients show no genotype phenotype correlations (72) (73), however a few recent studies have reported various MEN1 gene muations more frequently in patients with aggressive tumors or with decreased survival, however they are not widely used or generally established for clinical use. These include inactivating mutations of MEN1 gene, presence of exon 2 or nonsense/frameshift muatation in exon 2,9,10; the presence of mututions affecting the June D interacting domain and presence of mutations affecting the checkpoint kinase 1 interacting domain (74–76). Islet cell hyperplasia is rare. Duodenal gastrinomas in MEN-1 are usually small (<1 cm), submucosal, and multifocal (77–80) and as seen in sporadic ZES, occur primarily in the proximal duodenum (81,82). Lymph node metas5tases occur in 45–95% of both duodenal and pancreatic gastrinomas demonstrating that they are equally malignant, however, liver metastases occur less frequently in patients with duodenal than pancreatic NETs, demonstrating that they are not equally aggressive (2,83). pNETs contain in decreasing frequency chromogranin A, pancreatic polypeptide, glucagon, insulin, proinsulin, somatostatin, gastrin, vasoactive intestinal polypeptide, serotonin, calcitonin, growth hormone releasing factor, and neurotensin (2,84). Just because tumors may stain positive for these hormones by immunohistochemistry does not mean that they are associated with a clinical syndrome (2,84,85). A functional pNET, by definition, demonstrates excessive secretion of hormones associated with symptoms of hormonal excess (86,87). Malignant pNETs are rare prior to the age of 30 years; however, 50% of middle age MEN-1 patients have evidence for a malignant pNET. A prospective study in patients with MEN-1/ZES demonstrated that 14% of all patients had pNETs/gastrinomas demonstrating aggressive growth which was associated with decreased survival (88), which is a lower rate than seen in patients with sporadic ZES, wherein 25% are reported to show an aggressive growth pattern (89).
Figure 2. Histologic appearance of the pancreas in two separate patients with MEN-1.

(A) Histologic sections of a pancreas in a patient with multiple neuroendocrine microadenomas in a background of islet cell hyperplasia. Panel 1 (40X): The H&E stain demonstrates clusters of enlarged microadenomas, the largest of which is 0.35 cm. Panel 2 (40X): Immunhistochemical staining for synaptophysin highlights the multiple (>4) neuroendocrine microadenomas. (B) Histologic sections of a pancreatic neuroendocrine tumor (WHO Grade 2) from another patient with MEN1. Panel 1 (400X): H&E stain shows tumor cells in a trabecular pattern with stippled chromatin and prominent nucleoli. Panel 2 (200X): The Ki67 prolifeation index is 13%. Panels 3 and 4 (200X): Immunohistochemical stainning for chromogranin A (GgA) (panel 3) and synaptophysin (panel 4) are postive. Coutesy of Drs Allison Zemek and Teri Longacre, Department of Pathology, Stanford University School of Medicine, Stanford, CA.
One of the most significant controversies concerning patients with MEN-1 is the role of surgery in the management of various aspects of pNETs (2) (1) (90) (91) (92) (83) (93) (80) (94). Almost all agree that surgical resection should be undertaken for MEN-1 patients who develop insulinomas (18%), or other rare functional pNETs such as VIPomas, glucagonomas, GRFomas (<5%), or very rarely functional somatostatinomas (<1%). However, there is no agreement on surgery’s role in patients with the most frequent functional pNET, gastrinomas causing the Zollinger-Ellison syndrome, that occur in 54% of patients or nonfunctional pNETs (NF-pNETs) that develop in 80–100% of patients (2) (1) (95) (96) (97) (98) (99) (100) (101). There are several reasons for this controversy. In contrast to patients with insulinoma or the other rarer functional pNETs, in which surgery is frequently curative, (2) (1) (100) (101)surgery for patients with MEN-1 with ZES or with NF-pNETs is seldom curative. Therefore, surgical interventions must be carefully considered in a true risk benefit analysis. This risk benefit ratio is difficult to calculate for a number of reasons. A primary factor is that the natural history of patients with ZES, pNET and MEN-1 is changing and largely unknown (1) (11) (Figure 3). In early series, before effective medical control of gastric acid hypersecretion due to ZES existed, most MEN-1 patients died from the complications of the uncontrolled acid hypersecretion (23) (102) (103) (Figure 4). With the development of effective medical therapies (Histamine H2-receptor antagonists-1970–80s, PPIs-1985-present) the gastric acid secretion became an uncommon cause of death (<20 %) (2) (11) (104) (11) (Figure 4). In contrast, death from the malignant behavior of a NET has increased in frequency to >60% in recent series (11) (105) (106) (107) (86) (Figure 4). Patients with MEN-1 are now living longer (mean age death- 55–60) (Figure 3) (11) (105) and the death rate from other malignant tumors is increasing (11). This is particularly true of thymic neuroendocrine tumors (carcinoids). Currently, thymic carcinoids are the cause of death in approximately 10–25% of MEN-1 patients (106) (11). These are the most lethal NET in MEN-1 (11) (18) (108) (23). Further, a wide range of other tumors are increasingly reported in MEN-1 patients. Even though malignant neuroendocrine tumors are reported as a cause of death of MEN-1 patients in various series, the exact source of the metastatic NET is usually not clearly established. This is illustrated by the fact that a given patient may have a pNET and a carcinoid tumor (gastric, thymic or pulmonary) so the source of the metastatic disease is unclear. This consideration is further complicated by the fact that small NF-pNETs have an excellent long term survival (2) (11) (90) (83) (92) and in fact, their survival is not different from patients without pNETs (90) (99). An additional factor affecting the role of surgery is the fact that patients with MEN-1 and pNETs may have life-threatening symptoms secondary to unregulated hormonal overproduction of the tumor. Examples include peptic ulcer disease and diarrhea secondary to gastrinoma, hypercortisolism secondary to ectopic ACTH secretion, and hypoglycemia secondary to an insulinoma. In the past surgery was frequently the only hope to ameliorate and control these symptoms. However, effective drugs can control these symptoms making surgery less necessary. Specifically, PPIs can control acid hypersecretion in all MEN-1/ZES patients (104), long acting somatostatin analogues can control severe diarrhea in VIPoma and necrolytic migratory pruritic rash in glucagonoma (109) and diazoxide can control hypoglycemia in insulinoma (2) (86) (87) (110). Lastly, surgery may be associated with both short-term and long-term complications that may result in morbidity and mortality. For example, some studies suggest that patients with MEN-1 have an increased incidence of diabetes mellitus (11) (111) (112) and that pancreatic resections exacerbate this condition (113). Furthermore, since microscopic pNETs are almost invariably left in the pancreas, curative surgery may require a total pancreatectomy, which can result in a greatly impaired quality of life and difficult management course. Each of these factors (unknown natural history, low cure rate without aggressive surgery, excellent prognosis for small pNETs-in MEN-1/ZES-NF-pNETs, good medical control of functional pNETs, short/long-term surgical side-effects) has made it difficult to calculate the true risk benefit ratio of surgery for pNETs in patients with MEN-1. There are currently no clear markers that identify the cases that are at greatest risk for progression and death due to a malignant pNET. In recent studies a number of clinical/laboratory/tumoral features are reported to have prognostic value in MEN-1 patients (11,32,88,90). These include: the presence of thymic carcinoids, the presence of more than one functional hormonal pNET syndrome, need for >3 parathyroid surgical procedures, presence of either liver metastases or distant metastases from the pNET, aggressive primary tumor growth (invasion into SMV, bile duct obstruction), large pNETs (>4 cm), pNETs with areas of poor vascular enhancement on CT (114), calcifications on CT (115) and serial imaging with evidence for progression by increasing tumor size or new lesions (11,23).
Figure 3. Survival of MEN-1 patients with pNETs.
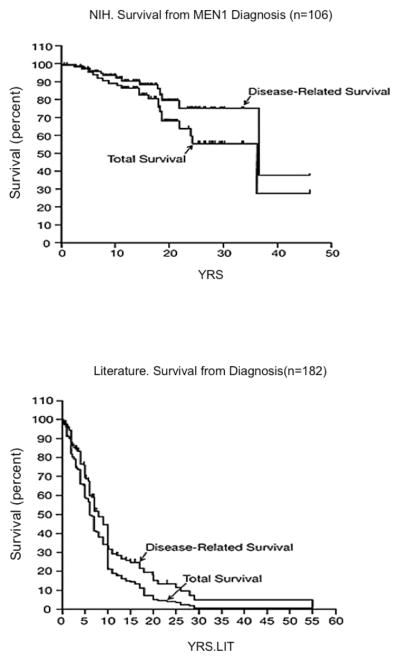
Shown are total survival and disease-related survival from two groups of patients with MEN-1 and pNETs. Data in the upper panel (A) are the survival from the time of diagnosis of MEN-1 for 106 patients with MEN-1/ZES followed at the NIH. The bottom panel (B) shows survival data from the time of MEN-1 diagnosis for 182 MEN-1 patients with pNETs from pooled literature data from case reports and small series. The survival curves are plotted as Kaplan/Meier plots. (Adapted from from Ito T, Igarashi H, Uehara H, Berna MJ, Jensen RT. Causes of death and prognostic factors in multiple endocrine neoplasia type 1: a prospective study: comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Medicine (Baltimore). 2013;92(3):135–181, with permission.)
Figure 4. Death rates and causes of death in patients with MEN-1 from different series over time.
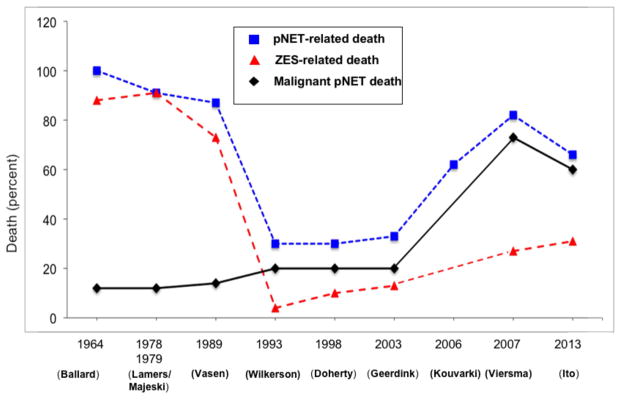
The percentage of patients in various studies of MEN-1 patients reported to have a pNET-related death (Death due to any pNET casue including hormonal syndrome or malinant tumor), death due to ZES (acid, tumor) or death due to a malignant pNET are shown. As a percentage of all deaths there was marked decrease with time in deaths due to ZES, primarily due to the increased ability to control gastric acid hypersecretion, which is also reflected in the decrease with time in the percnetage of all deaths that were pNET-related, and then with control of the hormone excess state there was an increase in the percentages of all deaths due to malignant pNETs or pNET-related. (Data from references 11, 23, 39, 102, 105, 237–240.)
Gastrinomas (Zollinger-Ellison Syndrome, ZES) are the most common functional pNET in MEN1 (1,95). Recent studies show the duodenal, but not pancreatic gastrinomas develop by hyperplasia of duodenal G cells (116). With increasing hyperplasia and subsequent LOH at the 11q13 MEN-1 locus, duodenal micro-gastrinomas arise (116). ZES is diagnosed by demonstrating an elevated fasting serum level of gastrin (off proton pump inhibitors) with a concomitant increased gastric acid output (>10 mEq/hr). Correlates for a poor prognosis with MEN-1/ZES are pancreatic tail primary tumors, hepatic metastases, tumors that make both gastrin and ACTH, distant metastases and severe hypergastrinemia (defined as a level > 3000) (11,88). Various surgical approaches have been performed in an attempt to cure MEN-1/ZES patients. However, MEN-1 patients typically have multiple, small duodenal gastrinomas associated with lymph node metastases in >50% of cases, (117,118) in addition to multiple pancreatic NETs, which are uncommonly the cause of the ZES (<15%) (79,81,119) (Figure 2). Almost all studies show that local removal of duodenal gastrinomas is associated with persistent ZES (1,5,119–123). However, Whipple pancreaticoduodenectomy has been reportedly associated with biochemical cure (2,118,124,125) (126,127). This latter approach is controversial because the symptoms of ZES are well controlled with PPIs, studies show MEN-1/ZES patients have an outstanding long term survival on PPIs (up to 100% at 15 years) and the immediate and long term morbidity/mortality of Whipple procedure may be too great (2,83,99) (87). An argument for the more aggressive surgical procedure is that with even longer follow-up a higher percentage of these MEN-1/ZES patients develop malignant gastric carcinoid tumors that may also affect survival (Figure 5b) (107). Therefore, the surgical approach to these patients will likely remain controversial until more data on the natural history becomes available. Our approach, because of their excellent quality of life and outlook, is to currently recommend only local resection (excision of duodenal tumors and/or enucleation of pancreatic head tumors with lymph node sampling and distal pancreatectomy for body/tail tumors that are greater than 2 cm). Lymph node sampling is recommended because it has prognostic value (128–130) and perhaps can increase the cure rate. Although uncommonly curative, it is associated with acceptable morbidity and mortality and long-term tumor control and survival with high quality of life. If the patient has larger (>2–2.5 cm) pancreatic head tumors with positive lymph nodes, then we agree that more radical resection (proximal pancreaticoduodenectomy) may be considered in selected cases, although our usual operation is to locally resect these tumors and not to perform a pancreaticoduodenectomy (131). Our recommendations are consistent with those of both large neuroendocrine tumor society guidelines recently published [i.e., NANETs (127,132) and ENETs (87)] as well as the clinical practice guidelines for MEN-1 published by the Endocrine Society (1).
Figure 5. (A) Thymic carcinoid on CT, (B) type 2 gastric carcinoids on endoscopy.
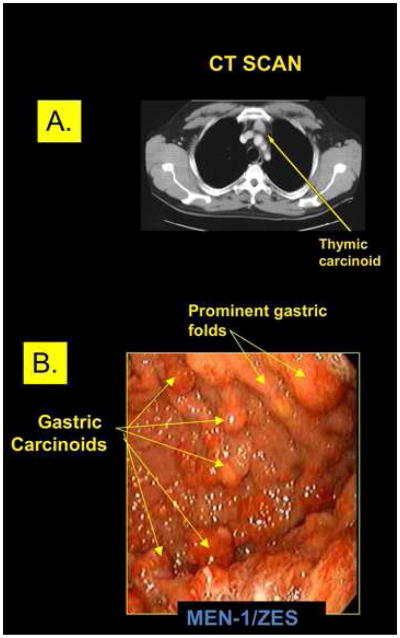
(A) CT scan of a small resectable thymic carcinoid in the anterior mediastinum of a patient with MEN-1. Unfortunately this is an uncommon finding in MEN-1 patients with these tumors that are usually first discovered when advanced disease is present. (B) Upper endoscopy of the stomach of a patient with Zollinger-Ellison syndrome (ZES) and MEN-1 who had been treated with proton pump inhibitors to reduce gastric acid secretion for many years. Image shows prominent gastric folds (arrows top right) consistent with ZES and multiple gastric carcinoids (lower right arrows) each of which is labeled. This patient illustrates the difficulty of removing all NETs endoscopically in many of these MEN-1 patients with Type 2 gastric carcinoids.
Insulinoma is the second most common functional pNET in MEN-1 (2,133,134) (131). Approximately 10–18% of MEN-1 patients will develop insulinoma and 5–10% of insulinomas occur in the setting of MEN-1. Insulinoma is the first manifestation of MEN-1 in up to 10% of patients (2,135). Patients have hypoglycemia and neuroglycopenic symptoms (altered mental status and seizures). This characteristically occurs at a young age (<35 years). Fasting hypoglycemia (glucose < 45 mg/dl) and concomitant hyperinsulinemia (levels > 5 uU/ml) are diagnostic (86). Insulinomas are generally small (<2 cm) and distributed uniformly throughout the pancreas (1,135). Since patients with MEN-1 have multiple pNETS, it may not be clear which tumor is secreting the excessive insulin. However, studies have reported that the insulinoma in MEN-1 is most commonly a dominant (>2cm) pNET that is frequently identified by conventional imaging studies like CT or MRI, usually in the body and tail of the pancreas (100,136,137) (138). Medical control of the hypoglycemia is not as effective as that with other functional tumors making surgery more important for symptom relief. Insulinomas in the pancreatic body and tail are removed by a distal pancreatectomy. Tumors in the head are enucleated. Preoperative endoscopic and intraoperative ultrasound can provide precise localization of the tumor and may facilitate excision by imaging the relationship to the pancreatic duct. MEN-1 patients with an insulinoma and no imaging evidence of a dominant tumor, should undergo calcium angiogram (139,140). This will localize the section of the pancreas containing the insulioma. Surgical resection can usually be done laparoscopically if the lesion is well localized preoperatively. Patients need to be followed for subsequent development of recurrent tumor and hypoglycemia (135). In a recent study (141)of 73 MEN1 patients with insulinoma after distal pancreatectomy (63%), total/cephalic pancreatectomy (12%) or eneucleation (25%), at a median followup of 9 years, 82% remained hypoglycemia free. The recurrence rate was higher with enucleation but the long term complication rate was much greater (43–55%) with resections than with enucleation (0%) (p=0.002) (141). This lead to the conclusion that while resection is associated with a lower recurence rate, enucleation alone may be an alternative because of its lower long term morbidity (141).
Nonfunctional pNETs (NF-pNETs) are a frequent problem in patients with MEN-1, occurring as microadenomas in 80–100% on pathology examination (Figure 2), in 25–60% by imaging studies and causing symptoms in up to 12% in different studies (2,90,99,142). For NF-pNETs in asymptomatic patients with MEN-1, there is controversy over the role of surgery, the timing of surgery and the type of surgery performed. Studies have demonstrated that patients with small NF-pNETs (<2 cm) have an excellent long term survival and in fact, it is the same as MEN-1 patients without any pNET (90) (99) (83). Several groups recommend to avoid surgery if the NF-pNET is < 2 cm or slowly growing (90) (99) (83,89). Others recommend surgery for tumors that are 1 cm (143) (144). Still others recommend routine surgery for all patients in whom a pNET is identified by any method, even if it is identified biochemically and not imaged (93). Another approach (145) (64) is to perform serial EUS on patients with small pNETs (<1–2 cm) and operate if growth occurs. At present there is no consensus as to what size or change in size should be used for intervention. This lack of consensus is due in large part to a lack of information on the natural history of NF-pNETs, especially those <1.5–2 cm in size that are asymptomatic. Recently a number of studies have attempted to address this question by following MEN1 patients with serial EUS’s which have been shown to be reliable for assessing changes in pNET size (146) and to be able to detect additional small (<0.5 cm) new NF-pNETs as well as changes in existing NF-pNETs (64,71,147,148). These studies show that the majority of NF-pNETs<1.5/2 cm remain stable or even decrease in size, with most of the rest only increase in size slowly. At present too few patients have been studied in this manner to allow specific criteria about what rate of change or other alteration in the NF-pNET should lead to surgical intervention. It has been suggested that if change occurs biopsy may help identify which patients should undergo surgery. The goal of surgery in MEN-1 with NF-pNETs is to control tumor growth and prevent progression. Recent studies (11) (134) identify the following factors as suggestive of poor prognosis: higher fasting serum levels of gastrin, presence of more than one functional hormonal syndrome, need for >3 parathyroid surgical procedures, presence of distant metastases from pNET, aggressive primary tumor growth (invasion into SMV, bile duct obstruction), large pNETs (> 4 cm), pNETs with areas of poor vascular enhancement on CT (114), calcifications (115) imaging evidence of progression (11). Endoscopic ultrasound and fine needle or core needle biopsy can be especially useful for determining malignant potential by measuring Ki67 rate (Figure 2) (145) (66) (149) (127). Further, gallium 68 DOTATOC PET/CT can be used to better stage the true extent of tumor in MEN-1 patients (41). It is very usefulr because it is a sensitive whole body study and it may identify other primary NETs like pituitary, bronchus, thymus, stomach and ileum (150) (151) (152). It is more sensitive than octreoscan which uses 111In-labeled pentreotide with SPECT imaging (Figure 1). Ga 68 DOTATOC PET/CT was originally available only in Europe and now it is becoming available in the United States. The standard operation for NF-pNETs is distal or subtotal pancreatic resection with intraoperative ultrasound and enucleation of tumors from the pancreatic head and duodenum. For bulky (>2 to 2.5 cm) tumors in the head of the pancreas proximal pancreaticoduodenectomy or Whipple procedure may be necessary. Dissection of lymph nodes along the celiac axis and hepato-duodenal ligament is also indicated. Extensive pancreaticoduodenal resection is associated with increased risk and is primarily indicated for clearly malignant tumors, as these tumors must be controlled. At present because these patients have excellent long-term survival (Figure 3), we recommend surgical exploration only in MEN-1 patients with NF-pNETs >2 cm or symptomatic. This recommendation is consistent with the guidelines by both large neuroendocrine tumor societies [i.e., NANETs (127,132) and ENETs (87) as well as the clinical practice guidelines for MEN-1 published by the Endocrine Society (1).
Management of rare functional pNETs in MEN-1
Glucagonomas occur in 3% (range 1–6%) of MEN-1 patients, VIPomas in 3% (range 1–12%), GRFomas and somatostatinomas in <1% (2) (153). All guidelines agree that if unresectable disease is not present and the patient has no medical contraindication to surgery (113), these patients should undergo surgical exploration for potential cure and control of the malignant nature of the pNET (1) (2) (127) (132) (153).
Stomach, thymic, and bronchial NETs and adrenal cortical tumors
Patients with MEN-1 develop type II gastric carcinoid tumors (7–35%) (Figure 5), bronchial carcinoid tumors (0–8%) (Figure 6), thymic carcinoid tumors (0–8%) and adrenal cortical tumors (27–36%, <2% symptomatic) (2) (11) (17) (26) (154) (155). In a recent study of MEN-1 patients (2) who died 45% had adrenal cortical tumors, 19% gastric carcinoid tumors, 10% lung carcinoid tumor, and 6% thymic carcinoid tumor. Of these less common tumors listed, the tumor that appears to cause death most frequently was the thymic carcinoid tumor (Figure 5). It was second only to pNETs as the cause of death in MEN-1 patients (11). Unfortunately, thymic NETs in MEN-1 patients are rarely discovered when completely resectable and most present with advanced disease encasing great vessels, invading surrounding tissues and frequently with bone and/or liver metastases (2) (11) (18) (156). In contrast to thymic carcinoids occuring in a non MEN1 setting, thymic carcinoids occuring in MEN1 patients are rarely associated with ectopic hormone production such as Cushing syndrome [2,10,17]. Early diagnosis by awareness and imaging with 68Ga-DOTATE, CT or MRI followed by complete surgical resection are the mainstays of treatment.
Figure 6. MRI axial view (top) and coronal view bottow of right lower lobe bronchial NET in patient with mEN-1.

Axial and coronal MRI images, respectively, of a small bronchial carcinoid tumor (Lung NET) in the right lower lobe of the lung in a MEN-1 patient who presented for routine follow-up. She underwent a right lower lobectomy and had positive lymph node metastases at surgery.
Type 2 gastric carcinoid tumors occur almost entirely in MEN-1 patients with ZES (2) (157) and are thought secondary to the trophic action of chronic hypergastrinemia on the gastric ECL cells combined with the presence of LOH at the MEN-1 locus on 11q13 (158) (Figure 5). Studies show that almost all patients with MEN1/ZES have gastric ECL cell proliferative changes, which are currently thought to be precursor changes leading to the development of gastric carcinoids in these patients (25). A large prospective study (25) found gastric ECL proliferative changes were present in all MEN-1/ZES patients studied, the changes were advanced in >50% of the patients and more severe that seen in patients with sporadic ZES without MEN-1. The natural history is unclear because prior to the mid 1980’s most ZES patients underwent a total gastrectomy and thus did not develop these tumors. More recent studies show these Type 2 gastric carcinoids (5–6% all gastric carcinoids) are more aggressive that the more common Type 1 found in patients without MEN-1 with atrophic gastritis/pernicious anemia (70–80% all gastric carcinoids). Type 2 metastasize to the liver at a higher rate (10–30%) (107,157) (26). Those with localized disease (>70–90%%) are usually excised endoscopically after assessing the extent of invasion by EUS, but in some patients the tumors are present in excessive numbers and larger size. In these latter patients the gastric NETs can be invasive so that additional treatments may be needed. In some cases aggressive surgical resection (subtotal or total gastrectomy and D-2 lymph node dissection) is recommended and additional treatment with long-acting somatostatin analogues or CCK-B receptor antagonists is administered (1) (157,159,160). Because most gastric carcinods develop asymptomatically and because they can be more aggressive than Type 1 gastric carcinods that occur in atrophic gastritis, it is recommended that all patients with ZES/MEN1 undergo regular endoscopic evaluation (annually).
Curative resection (lobectomy with lymph node dissection) is recommended for bronchopulmonary NETs (Figure 6) and radical median sternotomy with thymectomy for thymic carcinoid tumors (1) (11) (Figure 5). Whereas most bronchopulmonary NETs can be completely resected (155), unfortunately this is not the case for most thymic carcinoids, which are very aggressive tumors and metastasize early, especially to bone (2) (11,18) (156). Thymic carcinoids were not recognized as part of the MEN-1 syndrome until the 1980s so there is only limited information on their natural history (11) (161). Controversy exists as to the best method for their early detection, whether surgical resection should be performed even if distant metastatic disease is present, whether the recommendation of routine thymic resection during parathyroid surgery for hyperparathyroidism reduces the risk of the subsequent development of thymic carcinoids, and whether radiation or some other anti-tumor treatment should be given after surgical resection. (2) (11) (161) (18). Current guidelines recommend screening CT or MRI screening of the chest every 1–2 years (1), continued cervical thymectomy when parathyroid glands are removed and many recommend aggressive resection even with metastatic disease to prevent local complications (1) (18).
Adrenal tumors in MEN1 patients are frequent (27–36%), however they are usually small (<3–4 cm) (>80%), nonfunctional (85%), benign (>86%) and asymptomatic (>98%) (2) (11) (131). These tumors may cause primary hyperaldosteronism, primary hypercortisolism and may become malignant (131). At present adrenal tumors in MEN-1 patients are generally treated as that for non-MEN-1 adrenal tumors (1). Some studies suggest important differences in adrenal tumors in MEN-1 and non-MEN-1 patients (131). At present the exact screening time, tumor size that should be appropriated for surgical intervention and frequency of assessment of functionality have not been systematically studied and defined to detect early malignant change or functionality in MEN-1 patients.
Management of Metastatic Disease in MEN-1
Of the MEN-1 associated tumors, metastatic disease most commonly occurs with pancreatic, thymic and bronchial NETs, and occasionally in gastric carcinoid tumors. For example, approximately 60% of patients with duodenal gastrinomas or pNETs have metastatic disease at the time of diagnosis (11) (162). The presence of liver metastases initially or their development subsequently is associated with a poor prognosis and decreasing survival (2,11) (18) (83) (156). One of the unique problems confounding the treatment of MEN-1 patients, is often the fact that the exact primary source of the liver metastases is unclear. It may be a pNET, a duodenal gastrinoma, thymic carcinoid or another carcinoid like the stomach or bronchial. For example, there are a number of reports of MEN-1/ZES patients with liver metastases from a NET which is not the gastrinoma (163) (164). Liver metastases in a patient with MEN-1 and a pNET and without apparent carcinoid tumors like thymic, pulmonary, or stomach are almost always attributed to the pNET. This ambiguity may be resolved beause recently pathologists are using different immunohistochemical studies to try to determine the true primary source of the metastasis (i.e. PAX8, TTF-1, CDX-2, etc) (165) (166). This distinction of the true source of the metastases is becoming more important, because recent studies with a number of therapies used in patients with advanced metastatic NET disease [somatostatin analogues, everolimus, sunitinib, chemotherapeutic agents) demonstrate that pNETs and GI-NETS (carcinoids) respond differently (132) (164) (167) (168). Management of metastatic disease in patients with MEN-1 is generally comparable to the management of patients without MEN-1 (132) (164,167) (168). However, for patients with multiple primary endocrine tumors, vigilance must be taken when evaluating new findings on cross-sectional imaging to be certain which tumor is metastatic and/or progressing. The oncologic management of metastatic neuroendocrine tumors has improved with new drugs showing response rates primarily in pNETs. The FDA has recently approved two agents for the treatment of metastatic pNETs, the mTOR inhibitor everolimus (169) and the tyrosine kinase inhibitor sunitinib (170). Both have shown promising anti-tumor effects in recent studies. For advanced GI-NETs, two double-blind, randomized, placebo-controlled somatostatin analogues studies (PROMID (171), CLARINET (172)) as well as other studies (167) show both the somatostatin-long-acting analogue, octreotide-LAR and lanreotide autogel have antiproliferative effects, respectively. In addition, other novel therapies are being evaluated for the treatment of advanced pNETs and GI-carcinoids including the use of radiolabeled somostatatin analogues [90Yttrium, 177Lutetium-labeled-analogues] using the finding that almost all well-differentiated GI-NETs overexpress at least one subtype of somatostatin receptor which can be used to image as well as, target these cytotoxic agents, and this approach is currently undergoing Phase III trials (173).
In summary, the importance of our intial comments regarding timely diagnosis of MEN-1can not be overstated. Again, before MEN-1 can be diagnosed it must be suspected. Suspicion should be raised in any patient with a family history of endocrine tumors of the pancreas, family members with pituitary or parathyroid disease or a family history of endocrinopathy; in patients with renal colic with NETs; in any patient with ZES (20–25% have it as part of the MEN-1 syndrome); with a young age onset of a functional pNET; with multiple pNETs; with hyperparathyroidism with multiple gland involvement or with hyperplasia or with a pNET associated with hypercalcemia or another endocrinopathy (1,2). Genetic screening for MEN-1 is recommended when: an individual has 2 or more MEN-1 related tumors, multiple abnormal parathyroid glands before age 30 years, recurrent HPT at a young age, gastrinoma and hyperparathyroidism (HPT) or multiple pNETs at any age, plus a family history of kidney stones or endocrine tumors that are part of the syndrome (39,40).
Patients with MEN-1 are living longer and less frequently dying from the hormonal effects of tumors. However, the potentially malignant nature of some tumors like pancreatic NETs, bronchial NETs and thymic NETs are accounting for an increasingly high proportion of deaths. Additionally, there is a possibility that these patients may have a higher incidence of other malignant non-endocrine tumors (10). Awareness of these possibilities and the use of improved imaging modalities like CT, MRI, EUS, SRS and Ga 68 DOTATOC PET/CT should diagnose these tumors earlier and in a more treatable state.
MEN-4
Recently a new MEN-1 related syndrome has been defined and recognized, entitled MEN-4 (174,175). It has long been known that in patients with the MEN-1 syndrome clinically that 70–85% were found to have mutations in the MEN-1 gene with familial disease and 30% of patients with sporadic MEN-1 (7) (1, 10). In studies that also assess large deletions the percentage with MEN-1 mutations in patients with familial MEN-1 can rise to 90% (1,10) (176). Thus a percentage of patients with MEN-1 features appear to have the disease based on some other genetic alteration (176). Recently it has been established that germline mutations in the cyclin-dependent kinase (CDK) inhibitor gene, CDKN1B is responsible for causing MEN-X, a syndrome in rats with features of both the human MEN-1 and MEN-2 syndromes (177). Subsequently, a number of patients with MEN-1 features without a MEN-1 gene mutation has been described who have mutations in the CDKN1B gene (174) (178) (179). The CCDKN1B gene encodes for a member of the CDK inhibitor family, p27 (also called KIP1), a nuclear protein which is important in regulating the cell cycle, particularly the transition from G1 to S phase (174) (175,180). The CDKN1B gene, similar to the MEN-1 gene, functions as a tumor suppressor gene (174) (178). Alterations in other cyclin dependent kinase genes have also been reported in patients with clinical MEN-1 features but no MEN-1 gene mutations (181). In a study of 196 patients with clear MEN-1 or suspected MEN-1, but no MEN-1 gene mutation (181) the relative frequency of the various CDK mutations were p15 (1%), p18 (0.5%), p21 (0.5%) and p27 (1.5%) (181). In other studies it is estimated that 3% of patients with MEN1 fetaures have a CDK1B mutations (175).
MEN-2
Multiple endocrine neoplasia type 2 is composed of three distinct clinical subtypes, MEN-2a, MEN-2b, and familial medullary thyroid carcinoma (FMTC). MEN-2 is a rare syndrome with an incidence of 1 in 200,000 live births (182). Each subtype is an autosomal dominant familial cancer syndrome associated with a germline mutation of variable penetrance in the RET proto-oncogene (183). Because 50% of children of an affected parent will manifest MEN 2, the syndrome occurs in every generation of a family. The principle feature of all MEN-2 subtypes is medullary thyroid carcinoma (MTC), a cancer of the parafollicular calcitonin secreting C cells. Calcitonin and CEA are sensitive and, in the case of calcitonin, specific blood markers for MTC. Patients with MEN-2 have a 100% risk of developing MTC by age 70 years, and MTC is the most important determinant of mortality in these patients. If MTC has spread beyond the thyroid, the prognosis is poor. Further, patient survival is greatly improved when MTC is treated surgically early in the course of the disease. Therefore, the emphasis on treating patients with MEN-2 is screening and early detection of MTC (183). MEN-2 is subgrouped into two variants called MEN-2a and MEN-2b (sometimes called MEN-3) (184). Common features of the two groups are multicentric, bilateral MTC, occurring in all patients, and bilateral pheochromocytomas occurring in 50% of patients (185). MEN-2a is the most common manifestation of MEN-2, accounting for 55% of cases. MTC is often the first manifestation of MEN-2a, usually occurring between the ages of 20–30 years. MEN-2a is characterized by MTC and pheochromocytomas plus primary hyperparathyroidism apparent in 20–30% of patients, and a normal physical appearance and body habitus (183).
MEN-2b is a more rare form, accounting for approximately 5–10% of all cases. MEN-2b is marked by an early onset of a more aggressive form of MTC. MTC develops within the first year of life, and patients die before the age of 30 (186). Patients with MEN-2b do not have parathyroid disease, but do have a characteristic appearance including Marfanoid habitus, pectus abnormalities, mesodermal abnormalities, corneal nerve hypertrophy, labial and mucosal neuromas, and intestinal ganglioneuromatosis (183–185,187). Familial medullary thyroid carcinoma (FMTC) is another hereditary endocrine syndrome. It is the second most common variant. It accounts for 35% of cases. FMTC is the mildest subtype of MTC and patients usually do not die from MTC. Patients only have MTC without any of the other features seen in MEN-2a or -2b (188). The diagnosis of FMTC should be considered when at least four family members develop MTC without other endocrine findings.
Discovery of the MTC syndromes
In 1961, Sipple first described an association among malignant tumors of the thyroid, bilateral adrenal pheochromocytomas, and parathyroid hyperplasia (189). Later Steiner further characterized a kindred with pheochromocytomas, MTC, and parathyroid hyperplasia. He noted that the syndrome was transmitted in an autosomal dominant fashion with high penetrance similar to MEN-1. However, because MTC and pheochromocytomas were defining features, he reasoned that MEN-2 is genetically different from MEN-1 (190). Wells developed a method for early diagnosis of MTC based on a radioimmunoassay for calcitonin which was an excellent blood marker for MTC. Further, studies demonstrated that both calcium and pentagastrin stimulated C-cells to rapidly secrete calcitonin and this could be used for the early diagnosis of MTC. This allowed curative thyroidecomy based on stimulated plasma calcitonin levels in patients from families with MEN-2a.
In 1966, Williams and Pollock described two patients with neuromatosis, pheochromocytomas, and medullary thyroid carcinoma who died at a young age (162). Both had pheochromocytomas, multiple mucosal and ocular neuromas, ganglioneuromatosis of the intestine, and metastatic medullary carcinoma of the thyroid. In 1968, Schimke noted that marfanoid body habitus, coarse facial features, and characteristic skeletal abnormalities (pectus carinatum, saber shin) were part of this syndrome. He postulated that the entire syndrome was explained by a inheritable defect in neural crest derived tissues (191). This syndrome is now called MEN-2b and importantly, primary hyperparathyroidism is not part of this disease.
FMTC is the least malignant form of MEN-2 and patients seldom die from this form of MTC. These patients do not have other clinical manifestations of MEN-2 (157). It was first described by Farndon and Wells (188). The clinical diagnosis and onset of MTC in FMTC usually occurs later in life in the fifth or sixth decade (192).
RET proto-oncogene
RET is a proto-oncogene composed of 21 exons located on chromosome 10 (10q11.2) encoding a transmembrane receptor tyrosine kinase for members of the glial cell line-derived neurotrophic factor family (GDNF) and associated ligands (artemin, neuturin, persephin) (193–196). The RET protein is composed of three functional domains: an extracellular ligand-binding domain, a transmembrane domain, and a cytoplasmic tyrosine kinase domain. The extracellular domain contains four cadherin-like repeats and a cysteine-rich region. The cysteine-rich region is important for disulfide bond formation needed for maintaining the native tertiary structure allowing for receptor dimerization. The intracellular domain contains two tyrosine kinase subdomains that are involved in several intracellular signal transduction pathways (197). Coupling RET, its co-receptors, GNDF-family receptor alpha (GFRα 1–4) (198), and GNDF-family ligands leads to RET dimerization to form a heterohexamer complex that results in transcellular kinase activation and signaling. RET is involved in a number of cellular signaling pathways during development regulating the enteric nervous system progenitor cells, and neural crest and kidney progenitor cells (199–206). Basic studies suggest that RET is a component of the signaling pathway required for renal organogenesis and enteric neurogenesis (207,208). Inactivating mutations in RET associated with Hirschprung’s disease (209–214) and renal agenesis have been identified (207,208,215–219).
Genetic linkage analysis has mapped the MEN-2a locus to chromosome 10 (220,221). The RET gene has been found to be expressed in both familial and sporadic human pheochromocytomas and MTC (195). RET mutations have been identified in exons 7 and 8 in patients with MEN-2a and FMTC (222). Mulligan identified specific missense mutations in the RET gene at codons encoding for cysteine residues within the transition point between the extracellular and transmembrane domains (223). Subsequently, MEN-2b was shown to be associated with a T644M mutation affecting the intracellular tyrosine kinase domain of the RET proto-oncogene (224). In MEN-2a and -2b, RET mutations act by constitutively activating the kinase. MEN-2a mutations alter the disulfide bond between receptor monomers creating an activating homodimer. On the other hand, the MEN-2b mutation altered the substrate specificity for the receptor (225).
Genetic testing and risk stratification
Since the identification of mutations in the RET gene as the causative agents in MEN-2, genetic tests have been developed and refined to clinically detect these defects (226–229). Mutations vary among kindreds but are consistently inherited within kindreds. In addition, investigators determined that new direct genetic analysis supplanted established linkage-based tests, since the latter was precluded by recombination events and required the selection of informative genetic markers. There was an invariable correlation between mutation and disease. Importantly, two affected individuals that were presymtomatic were identified by genetic testing. Thus this new direct genetic analysis offered an important early diagnostic tool for the disorder. Today, genetic testing can detect nearly 100% of mutation carriers. It is the standard of care for all first-degree relatives of patients with newly diagnosed MTC. However, due to the varying clinical effects of RET mutations, strategies based on clinical phenotype, age of onset, and aggressiveness of MTC are still needed to guide therapy. Guidelines for the age of genetic testing and prophylactic thyroidectomy based on the inherited syndome have been identified (230) and revised. Recommendations on the diagnostic workup and timing of prophylactic thyroidectomy and extent of surgery are based on a classification into four risk levels utilizing the genotype-phenotype correlation (192). The American Thyroid Association classified mutations according to the risk of developing early, more aggressive MTC from A to D in increasing levels (231).
Genotype and phenotype
Since the initial discovery of RET mutations responsible for MEN-2, as many as 50 different point mutations across 7 exons (exons 8, 10, 11, 13–16) have been identified (232). Different mutations in the RET gene produce varying phenotypes for the disease, including age of onset and aggressiveness of MTC, and the presence or absence of other endocrine neoplasms, such as pheochromocytoma or hyperparathyroidism. Approximately 85% of patients with MEN-2 have a mutation at exon 11 codon 634, while mutations in codons 609, 611, 618, and 620 account for 10–15% of cases (192). Particularly early aggressive behavior and metastases in MEN-2a and MEN-2b are associated with C634 and M918T mutations, respectively, requiring early intervention (204). On the other hand, A883F mutation displays a more indolent form of MTC compared with a M918T mutation for MEN-2b. (233) In addition, polymorphism at codon 836 is associated with early metastases in patients with hereditary or sporadic MTC (234).
Prophylactic thyroidectomy
Untreated, patients with MEN-2 ultimately develop MTC and succumb to their disease. Thus, diagnostic tools for detecting MTC in patients at risk for MEN-2 were developed even before the genetic origins were known. Parafollicular cells secrete calcitonin, and blood levels of this hormone serve as a sensitive tumor marker as well as an indicator of the cancer. Intravenously administered calcium and pentagastrin are potent calcitonin secretagogues that markedly enhance the sensitivity of the calcitonin assay. Although the pentagastrin-stimulated calcitonin test provided a sensitive diagnostic test for C cell hyperplasia or carcinoma, it lacked the capacity to predict which patients would develop particularly aggressive forms of MTC and require early intervention and which patients would have a more indolent course and could be followed with biochemical surveillance. Because early detection and intervention are paramount to patient survival, genetic tests were needed to prospectively identify MEN-2 patients at-risk for accelerated MTC and requiring immediate treatment.
In 1995, shortly after mutations in RET were found to cause MEN-2, thyroidectomies were performed solely on identification of these gene mutations in patients. Using PCR-based testing (confirmed by haplotype analysis) for 19 known RET mutations, Wells studied 132 members of seven kindreds with MEN-2a. Twenty-one of the 58 subjects at risk for the disease had germline mutations in the RET oncogene associated with MEN-2a. Plasma calcitonin levels were elevated in 9 of the 21 subjects. However, 12 subjects had normal levels of calcitonin. After undergoing genetic counseling, 13 of the 21 subjects with detected germline mutations in RET, including 6 with normal serum calcitonin levels, underwent total thyroidectomies with central lymph node dissections. All of the surgically resected thyroid glands demonstrated medullary hyperplasia, many with evidence of MTC. Further, there was no evidence of lymph node metastasis in any patient at the time of surgery. In addition, postoperative plasma calcitonin levels normalized in patients who had elevated levels preoperatively. The study showed that for family members who have germline mutations in the RET proto-oncogene, total thyroidectomy is indicated irrespective of plasma calcitonin levels (235). Genetic screening and timely thyroidectomy in kindred members who have germline mutated RET alleles characteristic of MEN-2 can prevent MTC, the most common cause of death in these syndromes (197).
A major issue is the age to perform screening and prophylactic thyroidectomy. In patients from a family with MEN-2 and a RET mutation, most clinicians recommend that the individual should be screened at age 5 and the thyroid gland be removed, if affected. In this setting central compartment lymph node dissection is unnecessary as it can increase the occurence of postoperative hypoparathyroidism. In patients with MEN-2b, the surgery should be done at an early age as soon as the diagnosis is established. Since these patients commonly have a more aggressive tumor, central lymph node dissection should always be done. In patients with FMTC, screening should be done at age 21 and thyroidectomy without central lymph node dissection in patients with inherited mutations. Postoperatively at 6 months, physical examination and serum levels of calcitonin and CEA are measured. If calcitonin and CEA levels are undetectable or normal for 5 years, no additional studies are necessary.
MEN-2
MEN-2 (also termed MEN-2a) accounts for 80% of the familial MTC syndromes (Figure 7). In MEN-2, beside MTC, 50% of patients develop pheochromocytomas, and 30% develop primary hyperparathyroidism depending on the RET codon mutation (236). Patients with MEN-2 may also develop cutaneous lichen amyloidosis, Hirschsprung’s disease and prominent corneal nerves. C-cells secrete calcitonin and carcinoembryonic antigen (CEA). Either calcitonin or CEA levels in the blood serve as an excellent marker for MTC. Pentagastrin or calcium stimulated serum calcitonin levels may serve as a more sensitive marker for MTC and have been used for early diagnosis and to document surgical cure. Unlike sporadic MTC that is usually unilateral, familial forms of MTC are always bilateral. The tumor is multicentric and occupies the superior and central portion of each lobe. MTC initially remains confined to the thyroid gland but subsequently it spreads to central followed by other regional lymph nodes. Then it spreads to distant sites including the liver, lung, bone and brain. The tumor is very firm and has a fibrous acellular stroma that has staining properties similar to amyloid. On immunohistochemistry, it stains positive for calcitonin.
Figure 7. Metastatic medullary thyroid carcinoma (MTC) in MEN-2a patient. MEN-2a patient who presented with bilateral neck nodules that were found to be primary.

MTC (left panel). Mass in left lobe was 4 cm and right was 2 cm. Coronal CT of the abdomen shows multiple diffuse bilateral liver metastases (right panel).
Pheochromocytomas develop in 50% of patients with MEN-2a and -b (236) (Figure 8). The clinical findings and behavior is the same in both syndromes. The mean age at presentation is 36 years and the diagnosis is made after MTC in 40% of cases and before MTC in 10%. In this setting the pheochromocytomas are benign and confined to the adrenal gland. 65% of the time these tumors are bilateral and with 10-year follow-up patients with a surgically excised unilateral pheochromocytoma will develop a contralateral tumor. It is critical to exclude the diagnosis of pheochromocytoma in MEN-2 patients before doing any invasive procedures, because sudden death may occur if a pheochromocytoma is not detected and the patient is not appropriately prepared with an alpha-adrenergic blocking drug. Deaths have been reported during surgical procedures and child birth. Patients suspected of having a pheochromocytoma should have measurement of plasma-free metanephrine and normetanephrine levels or a 24-hour urine for VMA, metanephrines and total catecholamines. CT and MRI are used to image pheochromocytomas. The sensitivity and specificity are similar for the two procedures, 90–100% and 70–80%, respectively.
Figure 8.
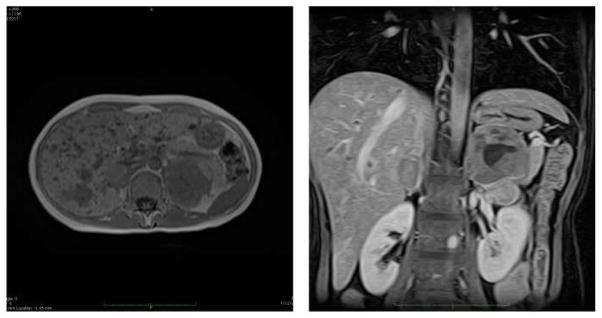
MRI of bilateral pheochromocytomas in same MEN-2a patient with metastatic MTC to the liver. Axial MRI (left panel) shows bilateral pheochromocytomas. Left adrenal tumor is larger than the right and measures 6 cm compared to 3 cm on right. Patient’s liver also shows multiple bilateral metastases that were proven to be from MTC. Coronal image (right panel also demonstrates both adrenal pheochromocytomas as axial. Left adrenal has some cystic areas.
The pheochromocytoma has priority and should be removed before the thyroid surgery. Preoperative preparation with an alpha-adrenergic blocking drug like phenoxybenzamine is done and when the patient is well blocked, a beta-adrenergic drug is added to keep the heart rate <100 beats per minute. Despite the fact that the pheochromocytomas are usually bilateral, if only one adrenal appears to contain tumor, unilateral adrenalectomy is recommended (236). This is because there have been a very high risk of Addisonian crisis in these patients following bilateral adrenalectomy. Some other surgeons have recommended bilateral subtotal adrenalectomy for these patients in an attempt to preserve cortical function. Although this approach may have merit, there has been limited follow-up and documentation of long-term adrenal function or recurrent pheochromocytomas with this procedure. Adrenalectomy can usually be accomplished laparoscopically and this approach greatly reduces morbidity. In patients who undergo bilateral adrenalectomy, corticosteroid and mineralocorticoid replacement is necessary.
Primary hyperparathyroidism develops in 20–30% of patients with MEN-2a. The mean age of onset is 36 years. It may occur prior to any other manifestation of MTC in 5% of patients. The hypercalcemia is minimal and most patients are without symptoms. The parathyroid glands are asymmetrically enlarged and contain hyperplastic nodules. Pathologically it is labelled pseudonodular hyperplasia. The operation of choice is subtotal parathyroidectomy or 3 and ½ gland parathyroidectomy.
MEN-2b
MEN-2b accounts for 5% of hereditary MTC. These patients all have MTC, bilateral pheochromocytomas, a characteristic phenotype, but they do not develop primary HPT. Patients with MEN-2b have this characteristic appearance that includes marfanoid habitus, prolonged facies, muscular skeletal abnormalities (pectus, saber shins, bowing of the extremities), ocular abnormalities (inability to cry tears and corneal nerve hypertrophy), mucosal neuromas usually on the tip of the tongue and intestinal ganglioneuromatosis. Most have gastrointestinal symptoms characterized by pain, diarrhea, constipation, bloating and even megacolon. These symptoms may occur in children and young adults with this syndrome.
The MTC is uniformly aggressive and spreads to distant sites early in these patients. It occurs during infancy and early diagnosis is critical. In approximately 50% of MEN-2b cases, de novo germ line RET mutations give rise to the disease. In patients with de novo MEN-2b, the mutated allele generally comes from the father. Babies are at a high risk in this setting because the parents are asymptomatic and the disease is not expected. This is unfortunate because there is only a narrow window during which thyroidectomy may be curative. Even in the most advantageous setting where thyroidectomy was performed during the neonatal period, most babies are not cured.
Familial medullary thyroid carcinoma (FMTC)
Familial medullary thyroid carcinoma (FMTC) accounts for 15% of hereditary MTC. These patients only have MTC that occurs at a late age and is less aggressive than the other familial forms. The diagnosis is made when an individual can identify 10 affected individuals occurring in a kindred over the age of 50 years with an adequate history and biochemical data to exclude pheochromocytoma and primary hyperparathyroidism in affected individuals.
The strongest predictor of survival for patients with MTC is the stage of disease at the time of thyroidectomy. The 10-year disease specific survival of MTC is 90% with localized disease, 78% with lymph node metastases and 40% with distant metastases. Only 10% of patients with metastases to cervical lymph nodes are cured with extensive lymph node dissection. The prognosis is excellent for familial MTC patients who have a preoperative calcitonin level <150 pg/ml, a MTC smaller than 1 cm and no lymph node metastases. The 10-year survival approaches 100%, if basal and stimulated calcitonin levels are undetectable after thyroidectomy. In patients with MTC confined to the thyroid gland, the standard operation is total thyroidectomy and resection of all lymph nodes in the central zone of the neck. The neck dissection is more extensive in patients with advanced nodal disease.
A reliable indicator of progression of MTC is the calcitonin or CEA doubling time. A calcitonin doubling time between 6 months and 2 years is associated with a 5 year survival of 92% and a 10 year survival of 37%; whereas a doubling rate of less than 6 months is associated with a 25 and 8% survival, respectively. In patients with MTC the calcitonin level is most predictive of prognosis, but in some patients the CEA level is more correlative so both should be measured. If the postoperative serum calcitonin level rises above 150 to 200 pg/ml, total body CT scan should be performed. In the absence of distant metastases and the presence of cervical lymph node disease, a neck dissection should be performed. Prior studies suggested that this strategy may cure approximately 30% of patients, but more recent long-term follow-up indicate that these patients will eventually recur. Some studies suggest that external beam radiation should be administered after neck dissection; however, this treatment has not improved outcomes.
Summary of MEN-2
MEN-2 is caused by a mutation in the RET proto-oncogene. It is inherited as an autosomal dominantt trait. It is broken down into 3 clinical syndromes: MEN-2a, MEN-2b and FMTC. Each of the three syndromes has medullary thryoid carcinoma (MTC) with complete penetrance. The MTC is the most lethal in MEN-2b, intermediate in 2a and least virulent in FMTC. Patients with MEN-2a and 2b also have a 50% chance of developing pheocromocytomas that may be bilateral and patients with MEN-2a may also get primary hyperparathryoidism caused by parathryoid hyperplasia. In children from kindreds with MEN-2a or 2b, total thyoidectomy is indicated once the RET gene mutation is detected and pheochromocutoma is excluded.
Management of Metastatic Disease in MEN-2, MEN-3 and FMTC
Development of distant metastatic disease in indivuals who were not diagnosed by screening is common in MTC. The survival of patients with distant metastases from MTC is 51% at 1 year, 26% at 5 years, and 10% at 10 years. When patients develop distant metastases from MTC and calcitonin levels increase, they may also develop secretory diarrhea that is especially difficult to control. Management of metastatic disease in patients with inherited forms of MTC is similar to management in sporadic MTC. Loperamide, codeine or sandostatin may help control the diarrhea in some cases. Tumor debulking or selective arterial embolization may provide symptomatic improvement in others. Two new compounds have recently been FDA-approved for the treatment of metastatic MTC and include vandetanib and cabozantinibz, and were shown to prolong progression-free survival in phase III clinical trials.
Key Points.
Early diagnosis of the MEN syndromes is critical for optimal clinical outcomes; before the MEN syndromes can be diagnosed, they must be suspected.
Genetic testing for germline alterations in both the MEN-1 gene and RET proto-oncogene is crucial to identifying those at risk in affected kindreds and directing timely surveillance and surgical therapy to those at greatest risk of potentially life threatening neoplasia.
Pancreatic, thymic and bronchial NETs are the leading cause of death in patients with MEN-1, and should be aggressively considered by at least biannual CT imaging.
Patients with MEN-2a or 2b who are from a kindred and have a RET gene mutation identified should undergo total thryoidectomy after a pheochromocytoma is excluded.
Footnotes
The Authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, Melmed S, Sakurai A, Tonelli F, Brandi ML. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1) J Clin Endocrinol Metab. 2012;97(9):2990–3011. doi: 10.1210/jc.2012-1230. [DOI] [PubMed] [Google Scholar]
- 2.Jensen RTBM, Bingham MD, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management and controversies. Cancer. 2008;113 (7 suppl):1807–1843. doi: 10.1002/cncr.23648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giusti FCL, Cavalli T, Brandi ML. Hereditary hyperparathyroidism syndromes. J Clin Densitom. 2013;16(1):69–74. doi: 10.1016/j.jocd.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) Best Pract Res Clin Endocrinol Metab. 2010;24(3):355–370. doi: 10.1016/j.beem.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Brandi ML, Marx SJ, Aurbach GD, Fitzpatrick LA. Familial multiple endocrine neoplasia type I: a new look at pathophysiology. Endocr Rev. 1987;8(4):391–405. doi: 10.1210/edrv-8-4-391. [DOI] [PubMed] [Google Scholar]
- 6.Marx SJ, Agarwal SK, Kester MB, Heppner C, Kim YS, Emmert-Buck MR, Debelenko LV, Lubensky IA, Zhuang Z, Guru SC, Manickam P, Olufemi SE, Skarulis MC, Doppman JL, Alexander RH, Liotta LA, Collins FS, Chandrasekharappa SC, Spiegel AM, Burns AL. Germline and somatic mutation of the gene for multiple endocrine neoplasia type 1 (MEN1) J Intern Med. 1998;243(6):447–453. doi: 10.1046/j.1365-2796.1998.00348.x. [DOI] [PubMed] [Google Scholar]
- 7.Lemos MCTR. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. 2008;29:22–32. doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- 8.SKA Exploring the tumors of multiple endocrine neoplasia type 1 in mouse models for basic and preclinical studies. Int J Endocr Oncol. 2014;1:153–161. doi: 10.2217/ije.14.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang JGB, Wan B, Matkar S, Veniaminova NA, Wan K, Merchant JL, Hua X, Lei M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature. 2012;482 (7386):542–546. doi: 10.1038/nature10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matkar STA, Hua X. Menin: a scaffold protein that controls gene expression and cell signaling. Trends Biochem Sci. 2013;38(8):394–402. doi: 10.1016/j.tibs.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito T, Igarashi H, Uehara H, Berna MJ, Jensen RT. Causes of death and prognostic factors in multiple endocrine neoplasia type 1: a prospective study: comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Medicine (Baltimore) 2013;92(3):135–181. doi: 10.1097/MD.0b013e3182954af1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore) 2004;83(1):43–83. doi: 10.1097/01.md.0000112297.72510.32. [DOI] [PubMed] [Google Scholar]
- 13.Benya RV, Metz DC, Venzon DJ, Fishbeyn VA, Strader DB, Orbuch M, Jensen RT. Zollinger-Ellison syndrome can be the initial endocrine manifestation in patients with multiple endocrine neoplasia-type I. Am J Med. 1994;97(5):436–444. doi: 10.1016/0002-9343(94)90323-9. [DOI] [PubMed] [Google Scholar]
- 14.Burgess JR, David R, Parameswaran V, Greenaway TM, Shepherd JJ. The outcome of subtotal parathyroidectomy for the treatment of hyperparathyroidism in multiple endocrine neoplasia type 1. Arch Surg. 1998;133(2):126–129. doi: 10.1001/archsurg.133.2.126. [DOI] [PubMed] [Google Scholar]
- 15.Skogseid B, Rastad J, Gobl A, Larsson C, Backlin K, Juhlin C, Akerstrom G, Oberg K. Adrenal lesion in multiple endocrine neoplasia type 1. Surgery. 1995;118(6):1077–1082. doi: 10.1016/s0039-6060(05)80117-5. [DOI] [PubMed] [Google Scholar]
- 16.Burgess JR, Harle RA, Tucker P, Parameswaran V, Davies P, Greenaway TM, Shepherd JJ. Adrenal lesions in a large kindred with multiple endocrine neoplasia type 1. Arch Surg. 1996;131(7):699–702. doi: 10.1001/archsurg.1996.01430190021006. [DOI] [PubMed] [Google Scholar]
- 17.Darling TN, Skarulis MC, Steinberg SM, Marx SJ, Spiegel AM, Turner M. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133(7):853–857. [PubMed] [Google Scholar]
- 18.Gibril F, Chen YJ, Schrump DS, Vortmeyer A, Zhuang Z, Lubensky IA, Reynolds JC, Louie A, Entsuah LK, Huang K, Asgharian B, Jensen RT. Prospective study of thymic carcinoids in patients with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. 2003;88(3):1066–1081. doi: 10.1210/jc.2002-021314. [DOI] [PubMed] [Google Scholar]
- 19.Hofmann M, Schilling T, Heilmann P, Haisken O, Wuster C, Brandi ML, Ziegler R, Nawroth PP. Multiple endocrine neoplasia associated with multiple lipomas. Med Klin (Munich) 1998;93(9):546–549. doi: 10.1007/BF03042664. [DOI] [PubMed] [Google Scholar]
- 20.Marx S, Spiegel AM, Skarulis MC, Doppman JL, Collins FS, Liotta LA. Multiple endocrine neoplasia type 1: clinical and genetic topics. Ann Intern Med. 1998;129(6):484–494. doi: 10.7326/0003-4819-129-6-199809150-00011. [DOI] [PubMed] [Google Scholar]
- 21.Raef H, Zou M, Baitei EY, Al-Rijjal RA, Kaya N, Al-Hamed M, Monies D, Abu-Dheim NN, Al-Hindi H, Al-Ghamdi MH, Meyer BF, Shi Y. A novel deletion of the MEN1 gene in a large family of multiple endocrine neoplasia type 1 (MEN1) with aggressive phenotype. Clin Endocrinol (Oxf) 2011;75(6):791–800. doi: 10.1111/j.1365-2265.2011.04134.x. [DOI] [PubMed] [Google Scholar]
- 22.Teh BT, Zedenius J, Kytola S, Skogseid B, Trotter J, Choplin H, Twigg S, Farnebo F, Giraud S, Cameron D, Robinson B, Calender A, Larsson C, Salmela P. Thymic carcinoids in multiple endocrine neoplasia type 1. Ann Surg. 1998;228(1):99–105. doi: 10.1097/00000658-199807000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilkinson S, Teh BT, Davey KR, McArdle JP, Young M, Shepherd JJ. Cause of death in multiple endocrine neoplasia type 1. Arch Surg. 1993;128(6):683–690. doi: 10.1001/archsurg.1993.01420180085016. [DOI] [PubMed] [Google Scholar]
- 24.Asgharian B, Chen YJ, Patronas NJ, Peghini PL, Reynolds JC, Vortmeyer A, Zhuang Z, Venzon DJ, Gibril F, Jensen RT. Meningiomas may be a component tumor of multiple endocrine neoplasia type 1. Clin Cancer Res. 2004;10(3):869–880. doi: 10.1158/1078-0432.ccr-0938-3. [DOI] [PubMed] [Google Scholar]
- 25.Asgharian BTM, Gibril F, Entsuah LK, Serrano J, Jensen RT. Cutaneous tumors in patients with Multiple Endocrine Neoplasm Type 1 (MEN1) and gastrinomas: Prospective study of frequency and development of criteria with high sensitivity and speecificity for MEM-1. J Clin Endocrinol Metab. 2004;89:6328–5336. doi: 10.1210/jc.2004-0218. [DOI] [PubMed] [Google Scholar]
- 26.Berna MJ, Annibale B, Marignani M, Luong TV, Corleto V, Pace A, Ito T, Liewehr D, Venzon DJ, Delle Fave G, Bordi C, Jensen RT. A prospective study of gastric carcinoids and enterochromaffin-like cell changes in multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: identification of risk factors. J Clin Endocrinol Metab. 2008;93(5):1582–1591. doi: 10.1210/jc.2007-2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lodewijk LBP, Kist JW, Conemans EB, de Laat JM, Pieterman CR, van der Horst-Schrivers AN, Jorna C, Hermus AR, Dekkers OM, de Herder WW, Drent ML, Bisschop PH, Havekes B, Rinkes IH, Vriens MR, Valk GD. Thyroid incidentalomas in patients with multiple endocrine neoplasia type 1. Eur J Endocrinol. 2015;172:337–342. doi: 10.1530/EJE-14-0897. [DOI] [PubMed] [Google Scholar]
- 28.Papillon ERA, Calender A, Chabre O, Barnoud R, Fournet J. A malignant gastrointestinal stromal tumour in a patient with multiple endocrine neoplasia type 1. Eur J Gastroenterol Hepatol. 2001;13:207–211. doi: 10.1097/00042737-200102000-00021. [DOI] [PubMed] [Google Scholar]
- 29.De Toma G, Plocco M, Nicolanti V, Brozzetti S, Letizia C, Cavallaro A. Type B1 thymoma in multiple endocrine neoplasia type 1 (MEN-1) syndrome. Tumori. 2001;87(4):266–268. doi: 10.1177/030089160108700411. [DOI] [PubMed] [Google Scholar]
- 30.Denker PS, Wright D, Hilscher JR, Saba SR, Ramirez G. Hypernephroma associated with multiple endocrine neoplasia type I: a case report. J Urol. 1986;136(4):896–898. doi: 10.1016/s0022-5347(17)45120-2. [DOI] [PubMed] [Google Scholar]
- 31.Dong Q, Debelenko LV, Chandrasekharappa SC, Emmert-Buck MR, Zhuang Z, Guru SC, Manickam P, Skarulis M, Lubensky IA, Liotta LA, Collins FS, Marx SJ, Spiegel AM. Loss of heterozygosity at 11q13: analysis of pituitary tumors, lung carcinoids, lipomas, and other uncommon tumors in subjects with familial multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. 1997;82(5):1416–1420. doi: 10.1210/jcem.82.5.3944. [DOI] [PubMed] [Google Scholar]
- 32.Goudet P, Murat A, Cardot-Bauters C, Emy P, Baudin E, du Boullay Choplin H, Chapuis Y, Kraimps JL, Sadoul JL, Tabarin A, Verges B, Carnaille B, Niccoli-Sire P, Costa A, Calender A. Thymic neuroendocrine tumors in multiple endocrine neoplasia type 1: a comparative study on 21 cases among a series of 761 MEN1 from the GTE (Groupe des Tumeurs Endocrines) World J Surg. 2009;33(6):1197–1207. doi: 10.1007/s00268-009-9980-y. [DOI] [PubMed] [Google Scholar]
- 33.Kojima Y, Ito H, Hasegawa S, Sasaki T, Inui K. Resected invasive thymoma with multiple endocrine neoplasia type 1. Jpn J Thorac Cardiovasc Surg. 2006;54(4):171–173. doi: 10.1007/BF02662474. [DOI] [PubMed] [Google Scholar]
- 34.Mallek R, Mostbeck G, Walter RM, Herold CH, Imhof H, Tscholakoff D. Contrast MRI in multiple endocrine neoplasia type 1 (MEN) associated with renal cell carcinoma. Eur J Radiol. 1990;10(2):105–108. doi: 10.1016/0720-048x(90)90116-s. [DOI] [PubMed] [Google Scholar]
- 35.Nishimura Y, Yamashita K, Yumita W, Yamazaki M, Katai M, Sakurai A, Hashizume K. Multiple endocrine neoplasia type 1 with unusual concomitance of various neoplastic disorders. Endocr J. 2004;51(1):75–81. doi: 10.1507/endocrj.51.75. [DOI] [PubMed] [Google Scholar]
- 36.Tanabe T, Yasuo M, Tsushima K, Urushihata K, Yamamoto H, Hanaoka M, Koizumi T, Fujimoto K, Yamazaki Y, Hirose Y, Hamano H, Sakurai A, Kubo K. Mediastinal seminoma in a patient with multiple endocrine neoplasia type 1. Intern Med. 2008;47(18):1615–1619. doi: 10.2169/internalmedicine.47.1226. [DOI] [PubMed] [Google Scholar]
- 37.Toshimori H, Okamoto M, Nakatsuru K, Hidaka H, Yamaguchi H, Ushiroda Y, Kisanuki A, Tsubouchi H, Sumiyoshi A, Matsukura S. Multiple endocrine neoplasia type 1 associated with malignant lymphoma and other complications. Intern Med. 1996;35(11):849–854. doi: 10.2169/internalmedicine.35.849. [DOI] [PubMed] [Google Scholar]
- 38.Goudet P1DA, Le Bras A, Cardot-Bauters C, Niccoli P, Lévy-Bohbot N, du Boullay H, Bertagna X, Ruszniewski P, Borson-Chazot F, Vergès B, Sadoul J, Ménégaux F, Tabarin A, Kühn J, d’Anella P, Chabre O, Christin-Maitre S, Cadiot G, Binquet C, Delemer B. MEN1 disease occurring before 21 years old. A 160-patient cohort study from the GTE (Groupe d’étude des Tumeurs Endocrines) J Clin Endocrinol Metab. 2015;(Jan 16):jc20143659. doi: 10.1210/jc.2014-3659. [DOI] [PubMed] [Google Scholar]
- 39.Vasen HF, Lamers CB, Lips CJ. Screening for the multiple endocrine neoplasia syndrome type I. A study of 11 kindreds in The Netherlands. Arch Intern Med. 1989;149(12):2717–2722. [PubMed] [Google Scholar]
- 40.Marx SJ, Vinik AI, Santen RJ, Floyd JC, Jr, Mills JL, Green J., 3rd Multiple endocrine neoplasia type I: assessment of laboratory tests to screen for the gene in a large kindred. Medicine (Baltimore) 1986;65(4):226–241. [PubMed] [Google Scholar]
- 41.Skogseid B, Oberg K. Experience with multiple endocrine neoplasia type 1 screening. J Intern Med. 1995;238(3):255–261. doi: 10.1111/j.1365-2796.1995.tb00932.x. [DOI] [PubMed] [Google Scholar]
- 42.Waldmann J, Fendrich V, Habbe N, Bartsch DK, Slater EP, Kann PH, Rothmund M, Langer P. Screening of patients with multiple endocrine neoplasia type 1 (MEN-1): a critical analysis of its value. World J Surg. 2009;33(6):1208–1218. doi: 10.1007/s00268-009-9983-8. [DOI] [PubMed] [Google Scholar]
- 43.Macleod AF, Ayers B, Young AE, Medd WE, Sonksen PH. Resolution of hypergastrinaemia after parathyroidectomy in multiple endocrine neoplasia syndrome type I (MEN type I) Clin Endocrinol (Oxf) 1987;26(6):693–698. doi: 10.1111/j.1365-2265.1987.tb00827.x. [DOI] [PubMed] [Google Scholar]
- 44.Norton JACM, Doppman JL, Maton PN, Gardner JD, Jensen RT. Effect of parathyoidectomy in patients with hyperparathryoidism in patients wth hyperparathyroidism, Zollinger-Ellison syndrome and multiple endocrine neoplasia type 1: a prospective study. Surgery. 1987;102:958–966. [PubMed] [Google Scholar]
- 45.Norton JA, Venzon DJ, Berna MJ, Alexander HR, Fraker DL, Libutti SK, Marx SJ, Gibril F, Jensen RT. Prospective study of surgery for primary hyperparathyroidism (HPT) in multiple endocrine neoplasia-type 1 and Zollinger-Ellison syndrome: long-term outcome of a more virulent form of HPT. Ann Surg. 2008;247(3):501–510. doi: 10.1097/SLA.0b013e31815efda5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marx SJ. Multiplicity of hormone-secreting tumors: common themes about cause, expression, and management. J Clin Endocrinol Metab. 2013;98(8):3139–3148. doi: 10.1210/jc.2013-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marx SJ, Menczel J, Campbell G, Aurbach GD, Spiegel AM, Norton JA. Heterogeneous size of the parathyroid glands in familial multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf) 1991;35(6):521–526. doi: 10.1111/j.1365-2265.1991.tb00938.x. [DOI] [PubMed] [Google Scholar]
- 48.Nilubol NWA, Weinstein LS, Simonds WF, Jensen RT, Phan GQ, Hughes MS, Libutti SK, Marx S, Kebebew E. Utility of intraoperative parathyroid hormone monitoring in patients with multiple endocrine neoplasia type 1-associated primary hyperparathyroidism undergoing initial parathyroidectomy. World J Surg. 2013;37:1966–1972. doi: 10.1007/s00268-013-2054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lairmore TCGC, Quinn CE, Sigmond BR, Lee CY, Jupiter DC. A randomized, prospective trial of operative treatments for hyperparathyroidism in patients with multiple endocrine neoplasia type 1. Surgery. 2014;156(6):1326–1334. doi: 10.1016/j.surg.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 50.Versnick M, Popadich A, Sidhu S, Sywak M, Robinson B, Delbridge L. Minimally invasive parathyroidectomy provides a conservative surgical option for multiple endocrine neoplasia type 1-primary hyperparathyroidism. Surgery. 2013;154(1):101–105. doi: 10.1016/j.surg.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 51.Filopanti MVU, Ermetici F, Olgiati L, Eller-Vainicher C, Corbetta S, Persani L, Beck-Peccoz P, Spada A. MEN1-related hyperparathyroidism: response to cinacalcet and its relationship with the calcium-sensing receptor gene variant Arg990Gly. Eur J Endocrinol. 2012;167:157–164. doi: 10.1530/EJE-12-0117. [DOI] [PubMed] [Google Scholar]
- 52.Del Prete MMV, Ramundo V, Marciello F, Di Sarno A, Esposito R, Carratù AC, De Luca Di Roseto C, Di Somma C, Colao A, Faggiano A. Impact of cinacalcet hydrochloride in clinical management of primary hyperparathyroidism in multiple endocrine neoplasia type 1. Minerva Endocrinol. 2013;38:389–394. [PubMed] [Google Scholar]
- 53.Giusti F, Tonelli F, Brandi ML. Primary hyperparathyroidism in multiple endocrine neoplasia type 1: when to perform surgery? Clinics (Sao Paulo) 2012;67 (Suppl 1):141–144. doi: 10.6061/clinics/2012(Sup01)23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.del Pozo CG-PL, Balsells M, Barahona MJ, Veloso E, González C, Anglada-Barceló J. Parathyroid carcinoma in multiple endocrine neoplasia type 1. Case report and review of the literature. Hormones. 2011;4:326–331. doi: 10.14310/horm.2002.1325. [DOI] [PubMed] [Google Scholar]
- 55.Singh Ospina NST, Thompson GB, Clarke BL, Young WF., Jr Prevalence of parathyroid carcinoma in 348 patients with multiple endocrine neoplasia type 1 - case report and review of the literature. Clin Endocrinol. 2014;(Dec 31) doi: 10.1111/cen.12714. [DOI] [PubMed] [Google Scholar]
- 56.Burgess JRDR, Greenaway TM, Parameswaran V, Shepherd JJ. Osteoporosis in multiple endocrine neoplasia type 1: severity, clinical significance, relationship to primary hyperparathyroidism, and response to parathyroidectomy. Arch Surg. 1999;134:1119–1123. doi: 10.1001/archsurg.134.10.1119. [DOI] [PubMed] [Google Scholar]
- 57.Kann PHBD, Langer P, Waldmann J, Hadji P, Pfützner A, Klüsener J. Peripheral bone mineral density in correlation to disease-related predisposing conditions in patients with multiple endocrine neoplasia type 1. J Endocrinol Invest. 2012;35:573–579. doi: 10.3275/7880. [DOI] [PubMed] [Google Scholar]
- 58.Kanazawa ICL, Abi Rafeh J, Angrula A, Li J, Riddle RC, Boraschi-Diaz I, Komarova SV, Clemens TL, Murshed M, Hendy GN. Osteoblast menin regulates bone mass in vivo. J Biol Chem. 2015;290:3910–3924. doi: 10.1074/jbc.M114.629899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Singh Ospina NTG, Lee RA, Reading CC, Young WF., Jr Safety and efficacy of percutaneous parathyroid ethanol ablation in patients with recurrent primary hyperparathyroidism and multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. 2015;100:E87–90. doi: 10.1210/jc.2014-3255. [DOI] [PubMed] [Google Scholar]
- 60.Nunes VSSG, Perone D, Conde SJ, Nogueira CR. Frequency of multiple endocrine neoplasia type 1 in a group of patients with pituitary adenoma: genetic study and familial screening. Pituitary. 2014;17:30–37. doi: 10.1007/s11102-013-0462-8. [DOI] [PubMed] [Google Scholar]
- 61.Thakker RV. Multiple endocrine neoplasia type 1. Indian J Endocrinol Metab. 2012;16(Suppl 2):S272–274. doi: 10.4103/2230-8210.104058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de Laat JMPC, Weijmans M, Hermus AR, Dekkers OM, de Herder WW, van der Horst-Schrivers AN, Drent ML, Bisschop PH, Havekes B, Vriens MR, Valk GD. Low accuracy of tumor markers for diagnosing pancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1 patients. J Clin Endocrinol Metab. 2013;98:4143–4151. doi: 10.1210/jc.2013-1800. [DOI] [PubMed] [Google Scholar]
- 63.Alexander HRN, Bartlett DL, Tio L, Benjamin SB, Doppman JL, Goebel SU, Serrano J, Gibril F, Jensen RT. Prospective study of somatostatin receptor scinigraphy and its effect on operative outcome in patients with Zollinger-Ellison syndrome. Ann Surg. 1998;228:228–2238. doi: 10.1097/00000658-199808000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langer P, Kann PH, Fendrich V, Richter G, Diehl S, Rothmund M, Bartsch DK. Prospective evaluation of imaging procedures for the detection of pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. World J Surg. 2004;28(12):1317–1322. doi: 10.1007/s00268-004-7642-7. [DOI] [PubMed] [Google Scholar]
- 65.van Asselt SJBA, van Dullemen HM, van der Jagt EJ, Bongaerts AH, Kema IP, Koopmans KP, Valk GD, Timmers HJ, de Herder WW, Feelders RA, Fockens P, Sluiter WJ, de Vries EG, Links TP. EUS is superior for detection of pancreatic lesions compared with standard imaging in patients with multiple endocrine neoplasia type 1. Gastrointestinal Endosc. 2015;81:159–167. doi: 10.1016/j.gie.2014.09.037. [DOI] [PubMed] [Google Scholar]
- 66.Barbe C, Murat A, Dupas B, Ruszniewski P, Tabarin A, Vullierme MP, Penfornis A, Rohmer V, Baudin E, Le Rhun M, Gaye D, Marcus C, Cadiot G. Magnetic resonance imaging versus endoscopic ultrasonography for the detection of pancreatic tumours in multiple endocrine neoplasia type 1. Dig Liver Dis. 2012;44(3):228–234. doi: 10.1016/j.dld.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 67.Thompson NWLR, Nishiyama RH, Vinik AI, Strodel WE, Allo MD, Eckhauser FE, Talpos G, Mervak T. MEN I pancreas: a histological and immunohistochemical study. World J Surg. 1984;8:561–574. doi: 10.1007/BF01654938. [DOI] [PubMed] [Google Scholar]
- 68.Anlauf M, Schlenger R, Perren A, Bauersfeld J, Koch CA, Dralle H, Raffel A, Knoefel WT, Weihe E, Ruszniewski P, Couvelard A, Komminoth P, Heitz PU, Kloppel G. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol. 2006;30(5):560–574. doi: 10.1097/01.pas.0000194044.01104.25. [DOI] [PubMed] [Google Scholar]
- 69.Kloppel G, Willemer S, Stamm B, Hacki WH, Heitz PU. Pancreatic lesions and hormonal profile of pancreatic tumors in multiple endocrine neoplasia type I. An immunocytochemical study of nine patients. Cancer. 1986;57(9):1824–1832. doi: 10.1002/1097-0142(19860501)57:9<1824::aid-cncr2820570920>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 70.Debelenko LV, Zhuang Z, Emmert-Buck MR, Chandrasekharappa SC, Manickam P, Guru SC, Marx SJ, Skarulis MC, Spiegel AM, Collins FS, Jensen RT, Liotta LA, Lubensky IA. Allelic deletions on chromosome 11q13 in multiple endocrine neoplasia type 1-associated and sporadic gastrinomas and pancreatic endocrine tumors. Cancer Res. 1997;57(11):2238–2243. [PubMed] [Google Scholar]
- 71.Thomas-Marques L, Murat A, Delemer B, Penfornis A, Cardot-Bauters C, Baudin E, Niccoli-Sire P, Levoir D, du Choplin HB, Chabre O, Jovenin N, Cadiot G. Prospective endoscopic ultrasonographic evaluation of the frequency of nonfunctioning pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. Am J Gastroenterol. 2006;101(2):266–273. doi: 10.1111/j.1572-0241.2006.00367.x. [DOI] [PubMed] [Google Scholar]
- 72.Wautot V, Vercherat C, Lespinasse J, Chambe B, Lenoir GM, Zhang CX, Porchet N, Cordier M, Beroud C, Calender A. Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: search for correlation between phenotype and the functional domains of the MEN1 protein. Hum Mutat. 2002;20(1):35–47. doi: 10.1002/humu.10092. [DOI] [PubMed] [Google Scholar]
- 73.Giraud S, Zhang CX, Serova-Sinilnikova O, Wautot V, Salandre J, Buisson N, Waterlot C, Bauters C, Porchet N, Aubert JP, Emy P, Cadiot G, Delemer B, Chabre O, Niccoli P, Leprat F, Duron F, Emperauger B, Cougard P, Goudet P, Sarfati E, Riou JP, Guichard S, Rodier M, Meyrier A, Caron P, Vantyghem MC, Assayag M, Peix JL, Pugeat M, Rohmer V, Vallotton M, Lenoir G, Gaudray P, Proye C, Conte-Devolx B, Chanson P, Shugart YY, Goldgar D, Murat A, Calender A. Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am J Hum Genet. 1998;63(2):455–467. doi: 10.1086/301953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kouvaraki MASS, Cote GJ, Lee JE, Yao JC, Waguespack SG, Gagel RF, Evans DB, Perrier ND. Management of pancreatic endocrine tumors in multiple endocrine neoplasia type 1. World J Surg. 2006;30:643–653. doi: 10.1007/s00268-006-0360-y. [DOI] [PubMed] [Google Scholar]
- 75.Thevenon J1BA, Faivre L, Cardot-Bauters C, Calender A, Murat A, Giraud S, Niccoli P, Odou MF, Borson-Chazot F, Barlier A, Lombard-Bohas C, Clauser E, Tabarin A, Parfait B, Chabre O, Castermans E, Beckers A, Ruszniewski P, Le Bras M, Delemer B, Bouchard P, Guilhem I, Rohmer V, Goichot B, Caron P, Baudin E, Chanson P, Groussin L, Du Boullay H, Weryha G, Lecomte P, Penfornis A, Bihan H, Archambeaud F, Kerlan V, Duron F, Kuhn JM, Vergès B, Rodier M, Renard M, Sadoul JL, Binquet C, Goudet P. Higher risk of death among MEN1 patients with mutations in the JunD interacting domain: a Groupe d’etude des Tumeurs Endocrines (GTE) cohort study. Hum Mol Genet. 2013;22:1940–1948. doi: 10.1093/hmg/ddt039. [DOI] [PubMed] [Google Scholar]
- 76.Bartsch DK1SE, Albers M, Knoop R, Chaloupka B, Lopez CL, Fendrich V, Kann PH, Waldmann J. Higher risk of aggressive pancreatic neuroendocrine tumors in MEN1 patients with MEN1 mutations affecting the CHES1 interacting MENIN domain. J Clin Endocrinol Metab. 2014;99:E2387–2391. doi: 10.1210/jc.2013-4432. [DOI] [PubMed] [Google Scholar]
- 77.Tonelli F, Fratini G, Falchetti A, Nesi G, Brandi ML. Surgery for gastroenteropancreatic tumours in multiple endocrine neoplasia type 1: review and personal experience. J Intern Med. 2005;257(1):38–49. doi: 10.1111/j.1365-2796.2004.01424.x. [DOI] [PubMed] [Google Scholar]
- 78.MacFarlane MP, Fraker DL, Alexander HR, Norton JA, Lubensky I, Jensen RT. Prospective study of surgical resection of duodenal and pancreatic gastrinomas in multiple endocrine neoplasia type 1. Surgery. 1995;118(6):973–979. doi: 10.1016/s0039-6060(05)80102-3. discussion 979–980. [DOI] [PubMed] [Google Scholar]
- 79.Anlauf MGN, Henopp T, Schmitt A, Schlenger R, Raffel A, et al. Sporadic versus hereditary gastrinomas of the duodenum and pancreas: distinct clinicopathological and epidemiological features. World J Surg. 2006;12(34):5440–5446. doi: 10.3748/wjg.v12.i34.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lopez CLFM, Waldmann J, Boninsegna L, Fendrich V, Goretzki PK, Langer P, Kann PH, Partelli S, Bartsch DK. Partial pancreaticoduodenectomy can provide cure for duodenal gastrinoma associated with multiple endocrine neoplasia type 1. Ann Surg. 2013;257(2):308–314. doi: 10.1097/SLA.0b013e3182536339. [DOI] [PubMed] [Google Scholar]
- 81.Pipeleers-Marichal M, Somers G, Willems G, Foulis A, Imrie C, Bishop AE, Polak JM, Hacki WH, Stamm B, Heitz PU, et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med. 1990;322(11):723–727. doi: 10.1056/NEJM199003153221103. [DOI] [PubMed] [Google Scholar]
- 82.Thom AKNJ, Axiotis CA, Jensen RT. Location, incidence and malignant potential of duodenal gastrinomas. Surgery. 1991;110:1086–1093. [PubMed] [Google Scholar]
- 83.Norton JAAH, Fraker DL, Venzon DJ, Gibril F, Jensen RT. Comparison of surgical results in patients with advanced and limited disease with multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome. Ann Surg. 2001;234(4):495–506. doi: 10.1097/00000658-200110000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.LeBodic MFHM, Lecomte M, Berger N, Berger F, Louvel A, et al. Immunohistochemical study of 100 pancreatic tumors in 28 patients with Multiple Endocrine Neoplasia, Type 1. Am J Surg Pathol. 1996;20(11):1378–1384. doi: 10.1097/00000478-199611000-00009. [DOI] [PubMed] [Google Scholar]
- 85.Tonelli F, Giudici F, Fratini G, Brandi ML. Pancreatic endocrine tumors in multiple endocrine neoplasia type 1 syndrome: review of literature. Endocr Pract. 2011;17 (Suppl 3):33–40. doi: 10.4158/EP10376.RA. [DOI] [PubMed] [Google Scholar]
- 86.Ito TIH, Jensen RT. Pancreatic neuroendocrine tumors: clinical features, diagnosis and medical treatment: Advances. Best Pract Res Clin Gastroenterol. 2012;26:737–753. doi: 10.1016/j.bpg.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jensen RTCG, Brandi ML, de Herder WW, Kaltsas G, Komminoth P, Scoazec JY, Salazar R, Sauvanet A, Kianmanesh R. ENETS Consensus Guidelines for the Management of Patients with Digestive Neuroendocrine Neoplasms: Functional Pancreatic Endocrine Tumor Syndromes. Neuroendocrinology. 2012;95(2):98–119. doi: 10.1159/000335591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gibril FVD, Ojeaburu JV, Bashir S, Jensen RT. Prospective study of the natural history of gastrinoma in patients with MEN1: Definition of an aggressive and a nonaggressive form. J Clin Endocrinol Metab. 2001;86(11):5282–5293. doi: 10.1210/jcem.86.11.8011. [DOI] [PubMed] [Google Scholar]
- 89.Weber HCVD, Lin JT, Fishbein VA, Orbuch M, Strader DB, Gibril F, Metz DC, Fraker DL, Norton JA, Jensen RT. Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: a prospective long-term study. Gastroenterology. 1995;108:1637–1649. doi: 10.1016/0016-5085(95)90124-8. [DOI] [PubMed] [Google Scholar]
- 90.Triponez FDD, Goudet P, Cougard P, Bauters C, Murat A, Cadiot G, Niccoli-Sire P, Chayvialle JA, Calender A, Proye CA. Epidemiology Data on 108 MEN 1 Patients From the GTE With Isolated Nonfunctioning Tumors of the Pancreas. Ann Surg. 2006;243(2):265–272. doi: 10.1097/01.sla.0000197715.96762.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.You YN, Thompson GB, Young WF, Jr, Larson D, Farley DR, Richards M, Grant CS. Pancreatoduodenal surgery in patients with multiple endocrine neoplasia type 1: Operative outcomes, long-term function, and quality of life. Surgery. 2007;142(6):829–836. doi: 10.1016/j.surg.2007.09.010. discussion 836 e821. [DOI] [PubMed] [Google Scholar]
- 92.Norton JAFD, Alexander HR, Venzon DJ, Doppman JL, Serrano J, Goebel SU, Peghini PL, Roy PK, Gibril F, Jensen RT. Surgery to cure the Zollinger-Ellison syndrome. N Engl J Med. 1999;341(9):635–644. doi: 10.1056/NEJM199908263410902. [DOI] [PubMed] [Google Scholar]
- 93.Akerstrom GHO, Skogseid B. Timing and extent of surgery in symptomatic and asymptomatic neuroendocrine tumors of the pancreas in MEN1. Lagenbecks Arch Surg. 2002;386:558–569. doi: 10.1007/s00423-001-0274-6. [DOI] [PubMed] [Google Scholar]
- 94.Singh MH, Fraker DL, Metz DC. Importance of surveillance for multiple endocrine neoplasia-1 and surgery in patients with sporadic Zollinger-Ellison syndrome. Clin Gastroenterol Hepatol. 2012;10(11):1262–1269. doi: 10.1016/j.cgh.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 95.Adkisson CDSJ, Bowers SP, Raimondo M, Wallace MB, Riegert-Johnson DL, et al. What Extent of Pancreatic Resection Do Patients with MEN-1 Require? JOP. 2012;13(4):402–408. doi: 10.6092/1590-8577/657. [DOI] [PubMed] [Google Scholar]
- 96.Tonelli FGF, Fratini G, Brandi ML. Pancreatic endocrine tumors in multiple endocrine neoplasia type 1 syndrome: review of literature. Endocr Pract. 2011;17 (suppl 3):33–40. doi: 10.4158/EP10376.RA. [DOI] [PubMed] [Google Scholar]
- 97.Lopez CLWJ, Fendrich V, Langer P, Kann PH, Bartsch DK. Long-term results of surgery for pancreatic neuroendocrine neoplasms in patients with MEN1. Langenbecks Arch Surg. 2011;396(8):1187–1197. doi: 10.1007/s00423-011-0828-1. [DOI] [PubMed] [Google Scholar]
- 98.Jensen RT. Management of the Zollinger-Ellison syndrome in patients with multiple endocrine neoplasia type 1. J Intern Med. 1998;243(6):477–488. doi: 10.1046/j.1365-2796.1998.00281.x. [DOI] [PubMed] [Google Scholar]
- 99.Triponez FGP, Dosseh D, Cougard P, Bauters C, Murat A, Cadiot G, Niccoli-Sire P, Calender A, Proye CA. Is surgery beneficial for MEN1 patients with small (< or = 2 cm), nonfunctioning pancreaticoduodenal endocrine tumor? An analysis of 65 patients from the GTE. World J Surg. 2006;30(5):654–662. doi: 10.1007/s00268-005-0354-9. [DOI] [PubMed] [Google Scholar]
- 100.Giudici F, Nesi G, Brandi ML, Tonelli F. Surgical management of insulinomas in multiple endocrine neoplasia type 1. Pancreas. 2012;41(4):547–553. doi: 10.1097/MPA.0b013e3182374e08. [DOI] [PubMed] [Google Scholar]
- 101.Norton JA, Fang TD, Jensen RT. Surgery for gastrinoma and insulinoma in multiple endocrine neoplasia type 1. J Natl Compr Canc Netw. 2006;4(2):148–153. doi: 10.6004/jnccn.2006.0015. [DOI] [PubMed] [Google Scholar]
- 102.Ballard HSFB, Harstock RT. Familial endocrine adenoma-peptic ulcer disease. Medicine (Baltimore) 1964;43:481–515. [PubMed] [Google Scholar]
- 103.Majewski JTWS. The MEN-I syndrome: an all or none phenomenon? Surgery. 1979;86(3):475–484. [PubMed] [Google Scholar]
- 104.Ito TIH, Uehara H, Jensen RT. Pharmacotherapy of Zollinger-Ellison syndrome. Expert Opin Pharmacotherapy. 2013;14(3):307–321. doi: 10.1517/14656566.2013.767332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vierimaa O, Ebeling TM, Kytola S, Bloigu R, Eloranta E, Salmi J, Korpi-Hyovalti E, Niskanen L, Orvola A, Elovaara E, Gynther A, Sane T, Valimaki M, Ignatius J, Leisti J, Salmela PI. Multiple endocrine neoplasia type 1 in Northern Finland; clinical features and genotype phenotype correlation. Eur J Endocrinol. 2007;157(3):285–294. doi: 10.1530/EJE-07-0195. [DOI] [PubMed] [Google Scholar]
- 106.Machens A, Schaaf L, Karges W, Frank-Raue K, Bartsch DK, Rothmund M, Schneyer U, Goretzki P, Raue F, Dralle H. Age-related penetrance of endocrine tumours in multiple endocrine neoplasia type 1 (MEN1): a multicentre study of 258 gene carriers. Clin Endocrinol (Oxf) 2007;67(4):613–622. doi: 10.1111/j.1365-2265.2007.02934.x. [DOI] [PubMed] [Google Scholar]
- 107.Norton JA, Melcher ML, Gibril F, Jensen RT. Gastric carcinoid tumors in multiple endocrine neoplasia-1 patients with Zollinger-Ellison syndrome can be symptomatic, demonstrate aggressive growth, and require surgical treatment. Surgery. 2004;136(6):1267–1274. doi: 10.1016/j.surg.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 108.Teh BTHN, Walters MK, Shepherd JJ, Wilkinson S, Nordenskjold M, Larsson C. Genetic studies of thymic carcinoids in multiple endocrine neoplasia type 1. J Med Genet. 1994;31:261–262. doi: 10.1136/jmg.31.3.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wickenhauser C, Aichelmann E, Neuhaus H, Holscher AH, Dienes HP. Glucagon-secreting malignant neuroendocrine tumor of the pancreas. Med Klin (Munich) 2000;95(8):466–469. doi: 10.1007/s000630050010. [DOI] [PubMed] [Google Scholar]
- 110.Jadoul M, Koppeschaar HP, Bax MA, Mali WP, Wit JM, Huber J, Vasen HF, Van der Sluys Veer J, Struyvenberg A, Lips CJ. Insulinomas in MEN-I patients: early detection and treatment of insulinomas in patients with the multiple endocrine neoplasia syndrome type-I. Neth J Med. 1990;37(3–4):95–102. [PubMed] [Google Scholar]
- 111.McCallum RW, Parameswaran V, Burgess JR. Multiple endocrine neoplasia type 1 (MEN 1) is associated with an increased prevalence of diabetes mellitus and impaired fasting glucose. Clin Endocrinol (Oxf) 2006;65(2):163–168. doi: 10.1111/j.1365-2265.2006.02563.x. [DOI] [PubMed] [Google Scholar]
- 112.van Wijk JPDK, Pieterman CR, Lips CJ, Zelissen PM, Valk GD. Increased prevalence of impaired fasting glucose in MEN1 gene mutation carriers. Clin Endocrinol (Oxf) 2012;76(1):67–71. doi: 10.1111/j.1365-2265.2011.04166.x. [DOI] [PubMed] [Google Scholar]
- 113.King JKK, Matsumoto J, Reber HA, Yeh MW, Hines OJ, Eibi G. Distal pancreatectomy: incidence of postoperative diabetes. J Gastrointest Surg. 2008;12(9):1548–1553. doi: 10.1007/s11605-008-0560-5. [DOI] [PubMed] [Google Scholar]
- 114.Worhunsky DJKG, Poullos PD, Visser BC, Kunz PL, Fisher GA, Norton JA, Poultsides GA. Pancreatic neuroendocrine tumours: hypoenhancement on arterial phase computed tomography predicts biological aggressiveness. HPB (Oxford) 2013 doi: 10.111/hpb:12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Poultsides GAHL, Chen Y, Visser BC, Pai RK, Jeffrey RB, Park WG, Chen AM, Kunz PL, Fisher GA, Norton JA. Pancreatic neuroendocrine tumors: radiographic calcifications correlate with grade and metastasis. Ann Surg Oncol. 2012;19(7):2295–2303. doi: 10.1245/s10434-012-2305-7. [DOI] [PubMed] [Google Scholar]
- 116.Anlauf MPA, Kloppel G. Endocrine precursor lesions and microadenomas of the duodenum and pancreas with and without MEN1: criteria, molecular concepts and clinical significance. Pathobiology. 2007;74(5):279–284. doi: 10.1159/000105810. [DOI] [PubMed] [Google Scholar]
- 117.Imamura M. Treatment for pancreatic endocrine tumors with or without multiple endocrine neoplasia type 1. Nihon Geka Gakkai Zasshi. 2005;106(8):472–478. [PubMed] [Google Scholar]
- 118.Imamura M, Komoto I, Ota S, Hiratsuka T, Kosugi S, Doi R, Awane M, Inoue N. Biochemically curative surgery for gastrinoma in multiple endocrine neoplasia type 1 patients. World J Gastroenterol. 2011;17(10):1343–1353. doi: 10.3748/wjg.v17.i10.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Norton JA, Jensen RT. Current surgical management of Zollinger-Ellison syndrome (ZES) in patients without multiple endocrine neoplasia-type 1 (MEN1) Surg Oncol. 2003;12(2):145–151. doi: 10.1016/s0960-7404(03)00035-5. [DOI] [PubMed] [Google Scholar]
- 120.O’Riordain DS, O’Brien T, van Heerden JA, Service FJ, Grant CS. Surgical management of insulinoma associated with multiple endocrine neoplasia type I. World J Surg. 1994;18(4):488–493. doi: 10.1007/BF00353743. discussion 493–484. [DOI] [PubMed] [Google Scholar]
- 121.Grama D, Skogseid B, Wilander E, Eriksson B, Martensson H, Cedermark B, Ahren B, Kristofferson A, Oberg K, Rastad J, et al. Pancreatic tumors in multiple endocrine neoplasia type 1: clinical presentation and surgical treatment. World J Surg. 1992;16(4):611–618. doi: 10.1007/BF02067335. discussion 618–619. [DOI] [PubMed] [Google Scholar]
- 122.Mignon M, Ruszniewski P, Podevin P, Sabbagh L, Cadiot G, Rigaud D, Bonfils S. Current approach to the management of gastrinoma and insulinoma in adults with multiple endocrine neoplasia type I. World J Surg. 1993;17(4):489–497. doi: 10.1007/BF01655108. [DOI] [PubMed] [Google Scholar]
- 123.Thompson NW. Management of pancreatic endocrine tumors in patients with multiple endocrine neoplasia type 1. Surg Oncol Clin N Am. 1998;7(4):881–891. [PubMed] [Google Scholar]
- 124.Gauger PG, Thompson NW. Early surgical intervention and strategy in patients with multiple endocrine neoplasia type 1. Best Pract Res Clin Endocrinol Metab. 2001;15(2):213–223. doi: 10.1053/beem.2001.0136. [DOI] [PubMed] [Google Scholar]
- 125.Lopez CL, Falconi M, Waldmann J, Boninsegna L, Fendrich V, Goretzki PK, Langer P, Kann PH, Partelli S, Bartsch DK. Partial pancreaticoduodenectomy can provide cure for duodenal gastrinoma associated with multiple endocrine neoplasia type 1. Ann Surg. 2013;257(2):308–314. doi: 10.1097/SLA.0b013e3182536339. [DOI] [PubMed] [Google Scholar]
- 126.Bartsch DK, Langer P, Wild A, Schilling T, Celik I, Rothmund M, Nies C. Pancreaticoduodenal endocrine tumors in multiple endocrine neoplasia type 1: surgery or surveillance? Surgery. 2000;128(6):958–966. doi: 10.1067/msy.2000.109727. [DOI] [PubMed] [Google Scholar]
- 127.Kulke MHAL, Bushnell DL, de Herder WW, Goldsmith SJ, Klimstra DS, Marx SJ, Pasieka JL, Pommier RF, Yao JC, Jensen RT. NANETS Treatment Guidelines: Well-Differentiated Neuroendocrine Tumors of the Stomach and Pancreas. Pancreas. 2010;39(6):735–752. doi: 10.1097/MPA.0b013e3181ebb168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Krampitz GWN, Poultsides GA, Visser BC, Sun L, Jensen RT. Lymph nodes and survival in pancreatic neuroendocrine tumors. Arch Surg. 2012;147(9):820–827. doi: 10.1001/archsurg.2012.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bartsch DKFV, Boninsegna L, Lopez CL, Partelli S, Falconi M. Impact of lymphadenectomy on survival after surgery for sporadic gastrinoma. Br J Surg. 2012;99:1234–1240. doi: 10.1002/bjs.8843. [DOI] [PubMed] [Google Scholar]
- 130.Hashim YMLD, Strasberg SS, Fields RC, Cao D, Hawkins WG. Regional lymphadenectomy is indicated in the surgical treatment of pancreatic neuroendocrine tumors (PNETS) Ann Surg. 2014;259:197–203. doi: 10.1097/SLA.0000000000000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Norton JAKG, Zemek A, Longacre T, Jensen RT. Surgical Perspectives: Changing Causes of Death in Patients with MEN-1. Ann Surg. 2015 doi: 10.1097/SLA.0000000000001211. in press. [DOI] [PubMed] [Google Scholar]
- 132.Kunz PLR-LD, Anthony LB, Bertino EM, Brendtro K, Chan JA, Chen H, Jensen RT, Kim MK, Klimstra DS, Kulke MH, Liu EH, Metz DC, Phan AT, Sippel RS, Strosberg JR, Yao JC. Consensus Guidelines for the Management and Treatment of Neuroendocrine Tumors. Pancreas. 2013;42(4):557–577. doi: 10.1097/MPA.0b013e31828e34a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Thakker RV. Multiple endocrine neoplasia type 1. Endocrinol Metab Clin North Am. 2000;29(3):541–567. doi: 10.1016/s0889-8529(05)70150-x. [DOI] [PubMed] [Google Scholar]
- 134.Goudet PMA, Binquet C, Cardot-Bauters C, Costa A, Ruszniewski P, Niccoli P, Menegaux F, Chabrier G, Borson-Chazot F, Tabarin A, Bouchard P, Delemer B, Beckers A, Bonithon-Kopp C. Risk factors and causes of death in MEN1 disease. A GTE cohort study among 758 patients. World J Surg. 2010;34:249–255. doi: 10.1007/s00268-009-0290-1. [DOI] [PubMed] [Google Scholar]
- 135.Bartsch DKKR, Kann PH, Fendrich V, Waldmann J. Enucleation and limited pancreatic resection provide long-term cure for insulinoma in Multiple Endocrine Neoplasia type 1. Neuroendocrinology. 2014;98(4):290–298. doi: 10.1159/000357779. [DOI] [PubMed] [Google Scholar]
- 136.Tonelli F, Fratini G, Nesi G, Tommasi MS, Batignani G, Falchetti A, Brandi ML. Pancreatectomy in multiple endocrine neoplasia type 1-related gastrinomas and pancreatic endocrine neoplasias. Ann Surg. 2006;244(1):61–70. doi: 10.1097/01.sla.0000218073.77254.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sakurai A, Yamazaki M, Suzuki S, Fukushima T, Imai T, Kikumori T, Okamoto T, Horiuchi K, Uchino S, Kosugi S, Yamada M, Komoto I, Hanazaki K, Itoh M, Kondo T, Mihara M, Imamura M. Clinical features of insulinoma in patients with multiple endocrine neoplasia type 1: analysis of the database of the MEN Consortium of Japan. Endocr J. 2012;59(10):859–866. doi: 10.1507/endocrj.ej12-0173. [DOI] [PubMed] [Google Scholar]
- 138.Cougard P, Goudet P, Peix JL, Henry JF, Sarfati E, Proye C, Calender A. Insulinomas in multiple endocrine neoplasia type 1. Report of a series of 44 cases by the multiple endocrine neoplasia study group. Ann Chir. 2000;125(2):118–123. doi: 10.1016/s0001-4001(00)00112-4. [DOI] [PubMed] [Google Scholar]
- 139.Hiramoto JSFV, LaBerge JM, Norton JA. Intraoperative ultrasound and preoperative localization detects all occult insulinomas; discussion 1025–6. Arch Surg. 2001;136(9):1020–1025. doi: 10.1001/archsurg.136.9.1020. [DOI] [PubMed] [Google Scholar]
- 140.Doppman JLMD, Chang R, Maton PN, London JF, Gardner JD, Jensen RT, Norton JA. Gastrinomas: localization by means of selective intraarterial injection of secretin. Radiology. 1990;(174):25–29. doi: 10.1148/radiology.174.1.2294556. [DOI] [PubMed] [Google Scholar]
- 141.Vezzosi DC-BC, Bouscaren N, Lebras M, Bertholon-Grégoire M, Niccoli P, Levy-Bohbot N, Groussin L, Bouchard P, Tabarin A, Chanson P, Lecomte P, Guilhem I, Carrere N, Mirallié E, Pattou F, Peix JL, Goere D, Borson-Chazot F, Caron P, Bongard V, Carnaille B, Goudet P, Baudin E. Long-term results of the surgical management of insulinoma patients with MEN1: a Groupe d’étude des Tumeurs Endocrines (GTE) retrospective study. Eur J Endocrinol. 2015;172:309–319. doi: 10.1530/EJE-14-0878. [DOI] [PubMed] [Google Scholar]
- 142.Pipeleers-Marichal M, Donow C, Heitz PU, Kloppel G. Pathologic aspects of gastrinomas in patients with Zollinger-Ellison syndrome with and without multiple endocrine neoplasia type I. World J Surg. 1993;17(4):481–488. doi: 10.1007/BF01655107. [DOI] [PubMed] [Google Scholar]
- 143.Bartsch DK, Langer P, Rothmund M. Surgical aspects of gastrinoma in multiple endocrine neoplasia type 1. Wien Klin Wochenschr. 2007;119(19–20):602–608. doi: 10.1007/s00508-007-0883-3. [DOI] [PubMed] [Google Scholar]
- 144.Skogseid B, Oberg K, Eriksson B, Juhlin C, Granberg D, Akerstrom G, Rastad J. Surgery for asymptomatic pancreatic lesion in multiple endocrine neoplasia type I. World J Surg. 1996;20(7):872–876. doi: 10.1007/s002689900133. discussion 877. [DOI] [PubMed] [Google Scholar]
- 145.Kann PH, Balakina E, Ivan D, Bartsch DK, Meyer S, Klose KJ, Behr T, Langer P. Natural course of small, asymptomatic neuroendocrine pancreatic tumours in multiple endocrine neoplasia type 1: an endoscopic ultrasound imaging study. Endocr Relat Cancer. 2006;13(4):1195–1202. doi: 10.1677/erc.1.01220. [DOI] [PubMed] [Google Scholar]
- 146.Kann PH, Kann B, Fassbender WJ, Forst T, Bartsch DK, Langer P. Small neuroendocrine pancreatic tumors in multiple endocrine neoplasia type 1 (MEN1): least significant change of tumor diameter as determined by endoscopic ultrasound (EUS) imaging. Exp Clin Endocrinol Diabetes. 2006;114(7):361–365. doi: 10.1055/s-2006-924322. [DOI] [PubMed] [Google Scholar]
- 147.D’souza SLEB, Scheiman JM. Long-term follow-up of asymptomatic pancreatic neuroendocrine tumors in multiple endocrine neoplasia type I syndrome. J Clin Gastroenterol. 2014;48:458–461. doi: 10.1097/MCG.0000000000000062. [DOI] [PubMed] [Google Scholar]
- 148.FT How to follow-up and when to operate asymptomatic pancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1. J Clin Gastroenterol. 2014;48:387–389. doi: 10.1097/MCG.0000000000000087. [DOI] [PubMed] [Google Scholar]
- 149.Gauger PG, Scheiman JM, Wamsteker EJ, Richards ML, Doherty GM, Thompson NW. Role of endoscopic ultrasonography in screening and treatment of pancreatic endocrine tumours in asymptomatic patients with multiple endocrine neoplasia type 1. Br J Surg. 2003;90(6):748–754. doi: 10.1002/bjs.4142. [DOI] [PubMed] [Google Scholar]
- 150.Froeling V, Elgeti F, Maurer MH, Scheurig-Muenkler C, Beck A, Kroencke TJ, Pape UF, Hamm B, Brenner W, Schreiter NF. Impact of Ga-68 DOTATOC PET/CT on the diagnosis and treatment of patients with multiple endocrine neoplasia. Ann Nucl Med. 2012;26(9):738–743. doi: 10.1007/s12149-012-0634-z. [DOI] [PubMed] [Google Scholar]
- 151.Sabet A, Nagarajah J, Dogan AS, Biersack HJ, Sabet A, Guhlke S, Ezziddin S. Does PRRT with standard activities of 177Lu-octreotate really achieve relevant somatostatin receptor saturation in target tumor lesions?: insights from intra-therapeutic receptor imaging in patients with metastatic gastroenteropancreatic neuroendocrine tumors. EJNMMI Res. 2013;3(1):82. doi: 10.1186/2191-219X-3-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Stoeltzing O, Loss M, Huber E, Gross V, Eilles C, Mueller-Brand J, Schlitt HJ. Staged surgery with neoadjuvant 90Y-DOTATOC therapy for down-sizing synchronous bilobular hepatic metastases from a neuroendocrine pancreatic tumor. Langenbecks Arch Surg. 2010;395(2):185–192. doi: 10.1007/s00423-009-0520-x. [DOI] [PubMed] [Google Scholar]
- 153.Levy-Bohbot NMC, Goudet P, Delemer B, Calender A, Jolly D, Thiéfin G, Cadiot G Groupe des Tumeurs Endocrines. Prevalence, characteristics and prognosis of MEN 1-associated glucagonomas, VIPomas, and somatostatinomas: study from the GTE (Groupe des Tumeurs Endocrines) registry. Gastroenterol Clin Biol. 2004;28(11):1075–1081. doi: 10.1016/s0399-8320(04)95184-6. [DOI] [PubMed] [Google Scholar]
- 154.Gatta-Cherifi BMA, Niccoli P, Cardot-Bauters C, Rohmer V, Young J, Delemer B, DuBoullay H, Verger MF, Kuhn JM, Sadoul JL, Ruszniewski, Beckers A, Monsaingeon M, Baudin E, Goudet P, Tabarin A. Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d’etude des tumeurs endocrines database. European journal of endocrinology. 2012;166:269–272. doi: 10.1530/EJE-11-0679. [DOI] [PubMed] [Google Scholar]
- 155.Sachithanandan N, Harle RA, Burgess JR. Bronchopulmonary carcinoid in multiple endocrine neoplasia type 1. Cancer. 2005;103(3):509–515. doi: 10.1002/cncr.20825. [DOI] [PubMed] [Google Scholar]
- 156.Teh BT, McArdle J, Chan SP, Menon J, Hartley L, Pullan P, Ho J, Khir A, Wilkinson S, Larsson C, Cameron D, Shepherd J. Clinicopathologic studies of thymic carcinoids in multiple endocrine neoplasia type 1. Medicine (Baltimore) 1997;76(1):21–29. doi: 10.1097/00005792-199701000-00002. [DOI] [PubMed] [Google Scholar]
- 157.Scherubl HCG, Jensen RT, Rosch T, Stolzel U, Kloppel G. Neuroendocrine tumors of the stomach (gastric carcinoids) are on the rise: small tumors, small problems? Endoscopy. 2010;42(8):664–671. doi: 10.1055/s-0030-1255564. [DOI] [PubMed] [Google Scholar]
- 158.Debelenko LV, Emmert-Buck MR, Zhuang Z, Epshteyn E, Moskaluk CA, Jensen RT, Liotta LA, Lubensky IA. The multiple endocrine neoplasia type I gene locus is involved in the pathogenesis of type II gastric carcinoids. Gastroenterology. 1997;113(3):773–781. doi: 10.1016/s0016-5085(97)70171-9. [DOI] [PubMed] [Google Scholar]
- 159.Fossmark R1SØ, Jianu CS, Qvigstad G, Nordrum IS, Boyce M, Waldum HL. Treatment of gastric carcinoids type 1 with the gastrin receptor antagonist netazepide (YF476) results in regression of tumours and normalisation of serum chromogranin A. Aliment Pharmacol Ther. 2012;36(11–12):1067–1075. doi: 10.1111/apt.12090. [DOI] [PubMed] [Google Scholar]
- 160.Manfredi SPM, de Lajarte-Thirouard AS, Bretagne JF. Type 1 and 2 gastric carcinoid tumors: long-term follow-up of the efficacy of treatment with a slow-release somatostatin analogue. Eur J Gastroenterol Hepatol. 2007;19(11):1021–1025. doi: 10.1097/MEG.0b013e328220eae0. [DOI] [PubMed] [Google Scholar]
- 161.Teh BTMJ, Chan SP, Menon J, Hartley L, Pullan P, Ho J, Khir A, Wilkinson S, Larsson C, Cameron D, Shepherd J. Clinicopathologic studies of thymic carcinoids in multiple endocrine neoplasia type 1. Medicine. 1997;76:21–29. doi: 10.1097/00005792-199701000-00002. [DOI] [PubMed] [Google Scholar]
- 162.Berglund G, Liden A, Hansson MG, Oberg K, Sjoden PO, Nordin K. Quality of life in patients with multiple endocrine neoplasia type 1 (MEN 1) Fam Cancer. 2003;2(1):27–33. doi: 10.1023/a:1023252107120. [DOI] [PubMed] [Google Scholar]
- 163.Bordi CFA, Azzoni C, D’Adda T, Canavese G, Guariglia A, et al. Aggressive forms of gastric neuroendocrine tumors in multiple endocrine neoplasia type I. Am J Surg Pathol. 1997;21(9):1075–1082. doi: 10.1097/00000478-199709000-00012. [DOI] [PubMed] [Google Scholar]
- 164.Grieco A, Bianco A, Alfei B, Rufini V, Vecchio FM, Marcoccia S, Gasbarrini G. Liver metastases of endocrine tumour associated with multiple endocrine neoplasia type 1: a sustained response to interferon therapy or a peculiar benign course? Hepatogastroenterology. 2000;47(35):1269–1272. [PubMed] [Google Scholar]
- 165.Belizzi AM. Assigning site of origin in metastatic neuroendocrine neoplasms: a clinically significant application of diagnostic immunohistochemistry. Adv Anat Pathol. 2013;20(5):285–314. doi: 10.1097/PAP.0b013e3182a2dc67. [DOI] [PubMed] [Google Scholar]
- 166.AMB Assigning site of origin in metastatic neuroendocrine neoplasms: a clinically significant application of diagnostic immunohistochemistry. Adv Anat Pathol. 2013;20:285–314. doi: 10.1097/PAP.0b013e3182a2dc67. [DOI] [PubMed] [Google Scholar]
- 167.Ito TIH, Jensen RT. Therapy of metastatic pancreatic neuroendocrine tumors (pNETs): recent insights and advances. J Gastroenterol. 2012;47(9):941–960. doi: 10.1007/s00535-012-0642-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pavel MBE, Couvelard A, Krenning E, Oberg K, Steinmuller T, Anlauf M, Wiedenmann B, Salazar R. ENETS Consensus Guidelines for the Management of Patients with Liver and Other Distant Metastases from Neuroendocrine Neoplasms of Foregut, Midgut, Hindgut, and Unknown Primary. Neuroendocrinology. 2012;95(2):157–176. doi: 10.1159/000335597. [DOI] [PubMed] [Google Scholar]
- 169.Yao JCSM, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, Tomassetti P, Pavel ME, Hoosen S, Haas T, Lincy J, Lebwohl D, Öberg K. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N Engl J Med. 2011;364(6):514–523. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A, Chen JS, Horsch D, Hammel P, Wiedenmann B, Van Cutsem E, Patyna S, Lu DR, Blanckmeister C, Chao R, Ruszniewski P. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- 171.Rinke AMH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, Mayer C, Aminossadati B, Pape UF, Bläker M, Harder J, Arnold C, Gress T, Arnold R. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656–4663. doi: 10.1200/JCO.2009.22.8510. [DOI] [PubMed] [Google Scholar]
- 172.Blumberg JCM the UK and Ireland NET Society/NETS. The CLARINET Study-Assessing the effect of Lantreotide Autogel on Tumor progression-free survival in patients with non-functioning gastroenteropancreatic neuroendocrine tumors (GEP-NETS) AGA. 2012:C4. [Google Scholar]
- 173.Bergsma HvVE, Teunissen JJ, Kam BL, de Herder WW, Peeters RP, Krenning EP, Kwekkeboom DJ. Peptide receptor radionuclide therapy (PRRT) for GEP-NETs. Best Pract Res Clin Gastroenterol. 2012;26(6):867–881. doi: 10.1016/j.bpg.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 174.Lee MPN. Multiple endocrine neoplasia type 4. Front Horm Res. 2013;41:63–78. doi: 10.1159/000345670. [DOI] [PubMed] [Google Scholar]
- 175.RVT Multiple endocrine neoplasia type ! (MEN1) and type 4 (MEN4) Mol Cell Endocrinol. 2014;386:2–15. doi: 10.1016/j.mce.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tham EGU, Lindgren E, Toss G, Skogseid B, Nordenskjöld M. Clinical testing for mutations in the MEN1 gene in Sweden: a report on 200 unrelated cases. J Clin Endocrinol Metab. 2007;92:3389–3395. doi: 10.1210/jc.2007-0476. [DOI] [PubMed] [Google Scholar]
- 177.Pellegata NSQ-ML, Siggelkow H, Samson E, Bink K, Höfler H, Fend F, Graw J, Atkinson MJ. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA. 2006;103:15558–15563. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4) Mol Cell Endocrinol. 2013 doi: 10.1016/j.mce.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pardi EMS, Pellegata NS, Benfini K, Borsari S, Saponaro F, Torregrossa L, Cappai A, Satta C, Mastinu M, Marcocci C, Cetani F. Functional characterization of a CDKN1B mutation in a Sardinian kindred with multiple endocrine neoplasia type 4 (MEN4) Endocr Connect. 2014:EC-14-0116. doi: 10.1530/EC-14-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Marinoni IPN. p27kip1: a new multiple endocrine neoplasia gene? Neuroendocrinology. 2011;93:19–28. doi: 10.1159/000320366. [DOI] [PubMed] [Google Scholar]
- 181.Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. 2009;94(5):1826–1834. doi: 10.1210/jc.2008-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.PL NJaK. Cancer: Principles and Practice of Oncology. 10 2015. Multiple Endocrine Neoplasias. [Google Scholar]
- 183.Moline J, Eng C. Multiple endocrine neoplasia type 2: An overview. Genet Med. 2011 doi: 10.1097/GIM.0b013e318216cc6d. [DOI] [PubMed] [Google Scholar]
- 184.Sizemore GW, Health H, 3rd, Carney JA. Multiple endocrine neoplasia type 2. Clin Endocrinol Metab. 1980;9(2):299–315. doi: 10.1016/s0300-595x(80)80035-1. [DOI] [PubMed] [Google Scholar]
- 185.Howe JR, Norton JA, Wells SA., Jr Prevalence of pheochromocytoma and hyperparathyroidism in multiple endocrine neoplasia type 2A: results of long-term follow-up. Surgery. 1993;114(6):1070–1077. [PubMed] [Google Scholar]
- 186.Norton JA, Froome LC, Farrell RE, Wells SA., Jr Multiple endocrine neoplasia type IIb: the most aggressive form of medullary thyroid carcinoma. Surg Clin North Am. 1979;59(1):109–118. doi: 10.1016/s0039-6109(16)41737-8. [DOI] [PubMed] [Google Scholar]
- 187.Carney JA, Go VL, Sizemore GW, Hayles AB. Alimentary-tract ganglioneuromatosis. A major component of the syndrome of multiple endocrine neoplasia, type 2b. N Engl J Med. 1976;295(23):1287–1291. doi: 10.1056/NEJM197612022952304. [DOI] [PubMed] [Google Scholar]
- 188.Farndon JR, Leight GS, Dilley WG, Baylin SB, Smallridge RC, Harrison TS, Wells SA., Jr Familial medullary thyroid carcinoma without associated endocrinopathies: a distinct clinical entity. Br J Surg. 1986;73(4):278–281. doi: 10.1002/bjs.1800730411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sipple J. The association of pheochromocytoma with carcinoma of the thyroid gland. The American Journal of Medicine. 1961;31(1):163–166. [Google Scholar]
- 190.Steiner AL, Goodman AD, Powers SR. Study of a kindred with pheochromocytoma, medullary thyroid carcinoma, hyperparathyroidism and Cushing’s disease: multiple endocrine neoplasia, type 2. Medicine (Baltimore) 1968;47(5):371–409. doi: 10.1097/00005792-196809000-00001. [DOI] [PubMed] [Google Scholar]
- 191.Schimke RN, Hartmann WH, Prout TE, Rimoin DL. Syndrome of bilateral pheochromocytoma, medullary thyroid carcinoma and multiple neuromas. A possible regulatory defect in the differentiation of chromaffin tissue. N Engl J Med. 1968;279(1):1–7. doi: 10.1056/NEJM196807042790101. [DOI] [PubMed] [Google Scholar]
- 192.Raue F, Frank-Raue K. Update multiple endocrine neoplasia type 2. Fam Cancer. 2010;9(3):449–457. doi: 10.1007/s10689-010-9320-2. [DOI] [PubMed] [Google Scholar]
- 193.Durbec P, Marcos-Gutierrez CV, Kilkenny C, Grigoriou M, Wartiowaara K, Suvanto P, Smith D, Ponder B, Costantini F, Saarma M, et al. GDNF signalling through the Ret receptor tyrosine kinase. Nature. 1996;381(6585):789–793. doi: 10.1038/381789a0. [DOI] [PubMed] [Google Scholar]
- 194.Robertson K, Mason I. The GDNF-RET signalling partnership. Trends Genet. 1997;13(1):1–3. doi: 10.1016/s0168-9525(96)30113-3. [DOI] [PubMed] [Google Scholar]
- 195.Nosrat CA, Tomac A, Hoffer BJ, Olson L. Cellular and developmental patterns of expression of Ret and glial cell line-derived neurotrophic factor receptor alpha mRNAs. Exp Brain Res. 1997;115(3):410–422. doi: 10.1007/pl00005711. [DOI] [PubMed] [Google Scholar]
- 196.Trupp M, Arenas E, Fainzilber M, Nilsson AS, Sieber BA, Grigoriou M, Kilkenny C, Salazar-Grueso E, Pachnis V, Arumae U. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature. 1996;381(6585):785–789. doi: 10.1038/381785a0. [DOI] [PubMed] [Google Scholar]
- 197.Wells SA, Jr, Santoro M. Targeting the RET pathway in thyroid cancer. Clin Cancer Res. 2009;15(23):7119–7123. doi: 10.1158/1078-0432.CCR-08-2742. [DOI] [PubMed] [Google Scholar]
- 198.Jing S, Wen D, Yu Y, Holst PL, Luo Y, Fang M, Tamir R, Antonio L, Hu Z, Cupples R, Louis JC, Hu S, Altrock BW, Fox GM. GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell. 1996;85(7):1113–1124. doi: 10.1016/s0092-8674(00)81311-2. [DOI] [PubMed] [Google Scholar]
- 199.Golden JP, Hoshi M, Nassar MA, Enomoto H, Wood JN, Milbrandt J, Gereau RWt, Johnson EM, Jr, Jain S. RET signaling is required for survival and normal function of nonpeptidergic nociceptors. J Neurosci. 2010;30(11):3983–3994. doi: 10.1523/JNEUROSCI.5930-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tee JB, Choi Y, Shah MM, Dnyanmote A, Sweeney DE, Gallegos TF, Johkura K, Ito C, Bush KT, Nigam SK. Protein kinase A regulates GDNF/RET-dependent but not GDNF/Ret-independent ureteric bud outgrowth from the Wolffian duct. Dev Biol. 2010;347(2):337–347. doi: 10.1016/j.ydbio.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ryu H, Jeon GS, Cashman NR, Kowall NW, Lee J. Differential expression of c-Ret in motor neurons versus non-neuronal cells is linked to the pathogenesis of ALS. Lab Invest. 2011;91(3):342–352. doi: 10.1038/labinvest.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ohgami N, Ida-Eto M, Sakashita N, Sone M, Nakashima T, Tabuchi K, Hoshino T, Shimada A, Tsuzuki T, Yamamoto M, Sobue G, Jijiwa M, Asai N, Hara A, Takahashi M, Kato M. Partial impairment of c-Ret at tyrosine 1062 accelerates age-related hearing loss in mice. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 203.Pachnis V, Mankoo B, Costantini F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development. 1993;119(4):1005–1017. doi: 10.1242/dev.119.4.1005. [DOI] [PubMed] [Google Scholar]
- 204.Moore SW, Zaahl MG. Multiple endocrine neoplasia syndromes, children, Hirschsprung’s disease and RET. Pediatr Surg Int. 2008;24(5):521–530. doi: 10.1007/s00383-008-2137-5. [DOI] [PubMed] [Google Scholar]
- 205.Reginensi A, Clarkson M, Neirijnck Y, Lu B, Ohyama T, Groves AK, Sock E, Wegner M, Costantini F, Chaboissier MC, Schedl A. SOX9 controls epithelial branching by activating RET effector genes during kidney development. Hum Mol Genet. 2011;20(6):1143–1153. doi: 10.1093/hmg/ddq558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Brantley MA, Jr, Jain S, Barr EE, Johnson EM, Jr, Milbrandt J. Neurturin-mediated ret activation is required for retinal function. J Neurosci. 2008;28(16):4123–4135. doi: 10.1523/JNEUROSCI.0249-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Schuchardt A, D’Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367(6461):380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- 208.Schuchardt A, D’Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. RET-deficient mice: an animal model for Hirschsprung’s disease and renal agenesis. J Intern Med. 1995;238(4):327–332. doi: 10.1111/j.1365-2796.1995.tb01206.x. [DOI] [PubMed] [Google Scholar]
- 209.Rungby J. RET, a gene responsible for familial endocrine neoplasias and Hirschsprung disease. Ugeskr Laeger. 1994;156(21):3194. [PubMed] [Google Scholar]
- 210.Lyonnet S, Edery P, Mulligan LM, Pelet A, Dow E, Abel L, Holder S, Nihoul-Fekete C, Ponder BA, Munnich A. Mutations of RET proto-oncogene in Hirschsprung disease. C R Acad Sci III. 1994;317(4):358–362. [PubMed] [Google Scholar]
- 211.Edery P, Lyonnet S, Mulligan LM, Pelet A, Dow E, Abel L, Holder S, Nihoul-Fekete C, Ponder BA, Munnich A. Mutations of the RET proto-oncogene in Hirschsprung’s disease. Nature. 1994;367(6461):378–380. doi: 10.1038/367378a0. [DOI] [PubMed] [Google Scholar]
- 212.Romeo G, Ronchetto P, Luo Y, Barone V, Seri M, Ceccherini I, Pasini B, Bocciardi R, Lerone M, Kaariainen H, et al. Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung’s disease. Nature. 1994;367(6461):377–378. doi: 10.1038/367377a0. [DOI] [PubMed] [Google Scholar]
- 213.Smith DP, Eng C, Ponder BA. Mutations of the RET proto-oncogene in the multiple endocrine neoplasia type 2 syndromes and Hirschsprung disease. J Cell Sci Suppl. 1994;18:43–49. doi: 10.1242/jcs.1994.supplement_18.6. [DOI] [PubMed] [Google Scholar]
- 214.Attie T, Edery P, Lyonnet S, Nihoul-Fekete C, Munnich A. Identification of mutation of RET proto-oncogene in Hirschsprung disease. C R Seances Soc Biol Fil. 1994;188(5–6):499–504. [PubMed] [Google Scholar]
- 215.Jeanpierre C, Mace G, Parisot M, Moriniere V, Pawtowsky A, Benabou M, Martinovic J, Amiel J, Attie-Bitach T, Delezoide AL, Loget P, Blanchet P, Gaillard D, Gonzales M, Carpentier W, Nitschke P, Tores F, Heidet L, Antignac C, Salomon R. RET and GDNF mutations are rare in fetuses with renal agenesis or other severe kidney development defects. J Med Genet. 2011;48(7):497–504. doi: 10.1136/jmg.2010.088526. [DOI] [PubMed] [Google Scholar]
- 216.Jain S. The many faces of RET dysfunction in kidney. Organogenesis. 2009;5(4):177–190. doi: 10.4161/org.5.4.10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Skinner MA, Safford SD, Reeves JG, Jackson ME, Freemerman AJ. Renal aplasia in humans is associated with RET mutations. Am J Hum Genet. 2008;82(2):344–351. doi: 10.1016/j.ajhg.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Gestblom C, Sweetser DA, Doggett B, Kapur RP. Sympathoadrenal hyperplasia causes renal malformations in Ret(MEN2B)-transgenic mice. Am J Pathol. 1999;155(6):2167–2179. doi: 10.1016/S0002-9440(10)65534-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Schuchardt A, D’Agati V, Pachnis V, Costantini F. Renal agenesis and hypodysplasia in ret-k- mutant mice result from defects in ureteric bud development. Development. 1996;122(6):1919–1929. doi: 10.1242/dev.122.6.1919. [DOI] [PubMed] [Google Scholar]
- 220.Mathew CG, Chin KS, Easton DF, Thorpe K, Carter C, Liou GI, Fong SL, Bridges CD, Haak H, Kruseman AC, et al. A linked genetic marker for multiple endocrine neoplasia type 2A on chromosome 10. Nature. 1987;328(6130):527–528. doi: 10.1038/328527a0. [DOI] [PubMed] [Google Scholar]
- 221.Simpson NE, Kidd KK, Goodfellow PJ, McDermid H, Myers S, Kidd JR, Jackson CE, Duncan AM, Farrer LA, Brasch K, et al. Assignment of multiple endocrine neoplasia type 2A to chromosome 10 by linkage. Nature. 1987;328(6130):528–530. doi: 10.1038/328528a0. [DOI] [PubMed] [Google Scholar]
- 222.Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, Howe JR, Moley JF, Goodfellow P, Wells SA., Jr Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet. 1993;2(7):851–856. doi: 10.1093/hmg/2.7.851. [DOI] [PubMed] [Google Scholar]
- 223.Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, Love DR, Mole SE, Moore JK, Papi L, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363(6428):458–460. doi: 10.1038/363458a0. [DOI] [PubMed] [Google Scholar]
- 224.Hofstra RM, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Luo Y, Pasini B, Hoppener JW, van Amstel HK, Romeo G, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. 1994;367(6461):375–376. doi: 10.1038/367375a0. [DOI] [PubMed] [Google Scholar]
- 225.Santoro M, Carlomagno F, Romano A, Bottaro DP, Dathan NA, Grieco M, Fusco A, Vecchio G, Matoskova B, Kraus MH, et al. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science. 1995;267(5196):381–383. doi: 10.1126/science.7824936. [DOI] [PubMed] [Google Scholar]
- 226.Unger K, Malisch E, Thomas G, Braselmann H, Walch A, Jackl G, Lewis P, Lengfelder E, Bogdanova T, Wienberg J, Zitzelsberger H. Array CGH demonstrates characteristic aberration signatures in human papillary thyroid carcinomas governed by RET/PTC. Oncogene. 2008;27(33):4592–4602. doi: 10.1038/onc.2008.99. [DOI] [PubMed] [Google Scholar]
- 227.Xue F, Yu H, Maurer LH, Memoli VA, Nutile-McMenemy N, Schuster MK, Bowden DW, Mao J, Noll WW. Germline RET mutations in MEN 2A and FMTC and their detection by simple DNA diagnostic tests. Hum Mol Genet. 1994;3(4):635–638. doi: 10.1093/hmg/3.4.635. [DOI] [PubMed] [Google Scholar]
- 228.Chi DD, Toshima K, Donis-Keller H, Wells SA., Jr Predictive testing for multiple endocrine neoplasia type 2A (MEN 2A) based on the detection of mutations in the RET protooncogene. Surgery. 1994;116(2):124–132. discussion 132–123. [PubMed] [Google Scholar]
- 229.Pazaitou-Panayiotou K, Kaprara A, Sarika L, Zovoilis T, Belogianni I, Vainas I, Nasioulas G. Efficient testing of the RET gene by DHPLC analysis for MEN 2 syndrome in a cohort of patients. Anticancer Res. 2005;25(3B):2091–2095. [PubMed] [Google Scholar]
- 230.Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, Libroia A, Lips CJ, Lombardi G, Mannelli M, Pacini F, Ponder BA, Raue F, Skogseid B, Tamburrano G, Thakker RV, Thompson NW, Tomassetti P, Tonelli F, Wells SA, Jr, Marx SJ. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86(12):5658–5671. doi: 10.1210/jcem.86.12.8070. [DOI] [PubMed] [Google Scholar]
- 231.Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H, Moley JF, Pacini F, Ringel MD, Schlumberger M, Wells SA., Jr Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid. 2009;19(6):565–612. doi: 10.1089/thy.2008.0403. [DOI] [PubMed] [Google Scholar]
- 232.Frank-Raue K, Rondot S, Schulze E, Raue F. Change in the spectrum of RET mutations diagnosed between 1994 and 2006. Clin Lab. 2007;53(5–6):273–282. [PubMed] [Google Scholar]
- 233.Jasim S, Ying AK, Waguespack SG, Rich TA, Grubbs EG, Jimenez C, Hu MI, Cote G, Habra MA. Multiple endocrine neoplasia type 2B with a RET proto-oncogene A883F mutation displays a more indolent form of medullary thyroid carcinoma compared with a RET M918T mutation. Thyroid. 2011;21(2):189–192. doi: 10.1089/thy.2010.0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Siqueira DR, Romitti M, da Rocha AP, Ceolin L, Meotti C, Estivalet A, Punales MK, Maia AL. The RET polymorphic allele S836S is associated with early metastatic disease in patients with hereditary or sporadic medullary thyroid carcinoma. Endocr Relat Cancer. 2010;17(4):953–963. doi: 10.1677/ERC-09-0312. [DOI] [PubMed] [Google Scholar]
- 235.Wells SA, Jr, Chi DD, Toshima K, Dehner LP, Coffin CM, Dowton SB, Ivanovich JL, DeBenedetti MK, Dilley WG, Moley JF, et al. Predictive DNA testing and prophylactic thyroidectomy in patients at risk for multiple endocrine neoplasia type 2A. Ann Surg. 1994;220(3):237–247. doi: 10.1097/00000658-199409000-00002. discussion 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Scholten AVG, Ulfman, et al. Unilateral adrenalectomy for pheochromocytoma in Multiple Endocrine Neoplasia Type 2 patients. Ann Surg. 2011;254:1022–1027. doi: 10.1097/SLA.0b013e318237480c. [DOI] [PubMed] [Google Scholar]
- 237.Lamers CB. Familial multiple endocrine neoplasia type I (Wermer’s syndrome) Neth J Med. 1978;21(6):270–274. [PubMed] [Google Scholar]
- 238.Doherty GM, Olson JA, Frisella MM, Lairmore TC, Wells SA, Jr, Norton JA. Lethality of multiple endocrine neoplasia type I. World J Surg. 1998;22(6):581–586. doi: 10.1007/s002689900438. discussion 586–587. [DOI] [PubMed] [Google Scholar]
- 239.Geerdink EA, Van der Luijt RB, Lips CJ. Do patients with multiple endocrine neoplasia syndrome type 1 benefit from periodical screening? Eur J Endocrinol. 2003;149(6):577–582. doi: 10.1530/eje.0.1490577. [DOI] [PubMed] [Google Scholar]
- 240.Kouvaraki MA, Shapiro SE, Cote GJ, Lee JE, Yao JC, Waguespack SG, Gagel RF, Evans DB, Perrier ND. Management of pancreatic endocrine tumors in multiple endocrine neoplasia type 1. World J Surg. 2006;30(5):643–653. doi: 10.1007/s00268-006-0360-y. [DOI] [PubMed] [Google Scholar]