

Abstract
Memory/effector T cells recirculate through extralymphoid tissues by entering from blood and egressing via afferent lymph. While T cell entry into effector sites is key to inflammation, the relevance of T cell egress to this process is unknown. Here we found that antigen recognition at the effector site reduced the tissue egress of pro-inflammatory Th1 cells in a mouse model of delayed hypersensitivity. Transgenic expression of ‘tissue exit receptor’ CCR7 enhanced lymphatic egress of antigen-sequestered Th1 cells from the inflamed site and ameliorated inflammation. In contrast, lack of CCR7 on Th1 cells diminished their tissue egress while enhancing inflammation. Lymph-borne Th1 and Th17 cells draining the inflamed skin of sheep migrated toward the CCR7 ligand CCL21, suggesting the CCR7-CCL21 axis as a physiological target in regulating inflammation. In conclusion, exit receptors can be targeted to modulate T cell dwell time and inflammation at effector sites, revealing T cell tissue egress as a novel control point of inflammation.
Introduction
During immunosurveillance and inflammation, effector/memory T cells, unlike naïve T cells, recirculate through extralymphoid tissues, entering from the blood and exiting via the afferent lymph (1). From the afferent lymph, T cells reach regional lymph nodes, which they leave via the efferent lymph, returning them back into blood. T cell migration into tissues is regulated by a multistep cascade involving adhesion and chemoattractant receptors on T cells interacting with their respective endothelial ligands (2). Similarly, T cell egress from lymph nodes is controlled by chemoattractant sphingosine-1 phosphate and its receptors (3). In the absence of inflammation, CD4+ and CD8+ T cells require expression of CCR7 to egress from extralymphoid tissues (4, 5). Congruently, lymphatic endothelial cells constitutively express the CCR7 ligand CCL21 in many organs (6).
T cell entry into effector sites is critical for inflammation and a target of anti-inflammatory therapy (7). Given the large number of lymphocytes, including pro-inflammatory Th1 and Th17 cells, that egress from chronically inflamed sites (8, 9); T cell egress potentially determines effector T cell accumulation and their downstream effector functions in situ. During inflammation and infection, the CCR7-CCL21 receptor-ligand pair guides T cells out of affected tissues via afferent lymph (5, 9, 10), and CCR7 deficiency promotes the development of long-term tissue-resident memory T cells (TRM)4 key to site-specific immunity (11). However, inflammation also facilitates CCR7-independent T cell tissue egress (9, 12) with contributions from additional chemoattractants, such as sphingosine-1 phosphate (9, 13).
Ccr7−/− mice demonstrate exacerbated inflammation in various models, such as cutaneous hypersensitivity (14), chronic arthritis (15), gastrointestinal inflammation (16, 17), and autoimmunity (reviewed in (18)), suggesting a role of T cell egress in regulating tissue inflammation. Conversely, mice with transgenic expression of Ccr7 (Ccr7TG) exhibit reduced effector responses (19). Nevertheless, in these models it is impossible to dissect the role of T cell egress in inflammation, in particular as other anomalies contribute to the observed phenotypes, e.g. regulatory T cell defects in Ccr7−/− mice (14, 20) or retention of Ccr7TG T cells in splenic white pulp (19). As a result, although discussed (9, 16, 17, 21), the role of T cell egress in the modulation of tissue inflammation remains unresolved.
In this study, we revisited this question and demonstrate that targeting the tissue egress capacity of pro-inflammatory T cells modulates the local inflammatory response, enhancing our understanding of the pathogenesis of inflammation and laying the foundation for future therapies for inflammatory diseases.
Materials and Methods
Animals and surgical procedures in sheep
All mice were on C57BL/6 background, bred in our animal facility, and used between 8 and 16 weeks of age in male or female sex- and age-matched groups. CD45.1 and CD45.2 congenic mice, and OTII mice were obtained from The Jackson Laboratory, Ccr7−/− mice (22) were from Martin Lipp (Max Delbrück Center), Ccr7TG mice (23) from Nigel Killeen (UCSF), and plt mice (24) from Avinash Bhandoola. OTII mice were crossed with Ccr7−/− mice and with Ccr7TG mice to obtain Ccr7−/− OTII mice and Ccr7TG OTII mice, respectively. Ccr7−/− OTII and Ccr7TG OTII breeders, but not their offspring, were maintained on an antibiotic diet (Mouse Helicobacter MD’s™ 4 Drug Combo, Bio-Serv). For sheep experiments, 5–10 month-old female mixed breed or Dorset sheep were purchased from Animal Biotech Industries or Pine Ridge Dorsets, respectively. Skin draining ‘pseudoafferent’ lymph vessels were induced by lymphectomy of the subiliac (prefemoral) lymph nodes as detailed (25). Pseudoafferent (prenodal) skin draining lymph vessels were cannulated with heparin-coated sterile catheters (Carmeda) in a surgical procedure under isoflurane anesthesia as described (9, 25). Lymph was collected from unanesthetized animals into sterile collection bottles containing heparin (APP Pharmaceuticals, LLC). All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Cell isolation, culture, and labeling, and chemotaxis assay
Lymphocytes were isolated from lymph nodes and spleens by passage through 40 μm cell strainers (BD Biosciences), as described (10). Red blood cells were lysed with 160 mM ammonium chloride and cells washed in RPMI 1640 with 10% fetal bovine serum. Th1 cells were generated from microbead-sorted (Miltenyi Biotec) CD4+ T cells cultured on plate-bound anti-CD3 (145-2C11; UCSF Monoclonal Antibody Core (MAC)) and anti-CD28 (37.51; UCSF MAC) in the presence of IL-12 (R&D Systems), IFN-γ (R&D Systems), and anti-IL-4 (11B11, BioXCell) as described (9). On day 5 of culture, dead cells were removed using a Nycodenz gradient (Axis-Shield). Th1 cells were labeled with 0.5 mM CFSE (Life Sciences) or 5 mM eFluor670 (eF670; eBioscience) in HBSS with 25 mM HEPES (Gibco) for 10 minutes at 37°C. Bovine serum was added to stop the reaction, and cells were washed 3 times. Femurs and tibias were flushed to isolate bone marrow (BM) cells. BM-derived dendritic cells (BMDCs) were generated as a source of APCs by culture of BM cells with 20 ng/ml of GM-CSF (PeproTech) for 8–9 days. BMDCs were pulsed overnight with 500 ng/ml LPS (Sigma-Aldrich), 10 ng/ml mouse TNFα (R&D Systems) and 1 mg/ml of BSA (Sigma-Aldrich) or OVA protein (Sigma-Aldrich). Cells were isolated from footpad skin by mincing mouse feet followed by two 30-minute enzymatic digestion steps in HBSS at 37°C with 0.1 mg/ml DNase I (Roche) and 12.5 μg/ml Liberase TM (Roche). Subsequently, samples were passed through a 100-μm cell strainer. The chemotaxis assay was carried out and analyzed as described (26). Briefly, 5×105 lymph-borne cells in RPMI 1640 containing 0.5% BSA were added to the upper chamber of 5-μm pore size Transwell inserts (Corning). Mouse CCL21 (R&D Systems) was added to the lower chamber at 100 nM, its optimal concentration to attract ovine CD4+ T cells (4, 27). CD4+ T cells that migrated to the lower chamber during the 90 minute incubation at 37°C were quantified by flow cytometry using a bead standard (15 μm polystyrene beads, Polysciences Inc). Frequencies of cytokine producing CD4+ T cells were determined in input and migrated wells by intracellular staining (described below) to calculate percent migration of total IFN-γ+ and IL-17+ CD4+ T cells.
Induction of skin inflammation in sheep and mice, and T cell egress assay
Chronic skin inflammation was induced in sheep by subcutaneous injection of 0.3–0.5 ml of CFA (Sigma-Aldrich), emulsified at 1:1 with saline, into two sites in the area of the flank (9). ≥ 3 weeks later, lymph vessels draining the inflamed skin and the control side were cannulated and lymph-borne CD4+ T cells analyzed. To elicit inflammation in mice and to test T cell egress from the site, we modified a model of delayed hypersensitivity by Streilein (28). Unless otherwise indicated, responder (OTII) Th1 cells and antigen-pulsed BMDCs generated from wild-type (WT) or CCL19-deficient plt mice were co-cultured at a 2:1 ratio, keeping APCs constant at 2×105 cells per recipient. After 1 h, the APC-T cell mixture was injected subcutaneously in 10 μl of PBS into the footpads of recipient mice, and tissue swelling was blindly measured over time using an engineer’s caliper (Fisher Scientific). Δ Swelling was defined as the footpad thickness after injection of cells subtracted by pre-injection values for the same foot. In T cell egress assays, 20–70 h post-injection of 4–6×106 responder Th1 cell subsets and 1×106 antigen-loaded APCs, the draining and non-draining (control) popliteal lymph nodes and spleens of recipient mice were analyzed for donor cells based on fluorescent and/or congenic labels as well as CD4 expression as described (9). Enumeration of migrated Th1 cell subsets was done by flow cytometry using a fixed number of beads (15 μm polystyrene beads) added to each sample. Cell counts and ratios were corrected for differences in input populations.
Flow cytometry
To reduce non-specific staining, mouse cells were pre-incubated with rat IgG (Jackson ImmunoResearch), anti-CD16/CD32 (2.4G2; UCSF MAC), and if necessary, with donkey and mouse IgG (Jackson ImmunoResearch). Sheep cells were pre-incubated with mouse and sheep IgG. Cells were stained for surface markers and analyzed as described (9), using fluorochrome-conjugated (fluorescein isothiocyanate, Pacific Blue, eFluor450, phycoerythrin, and/or allophycocyanin) anti-mouse antibodies from eBioscience: CD4 (RM4-5), CD45.1 (A20), and CD45.2 (104) or anti-ovine CD4 (44.38) from Serotec. CCR7 was stained with a CCL19-human Ig chimeric protein (provided by Daniel Campbell) followed by biotinylated donkey anti-human IgG F(ab′)2 (Jackson ImmunoResearch) and phycoerythrin-conjugated streptavidin (BD Bioscience). Dead cells were stained using the LIVE/DEAD Fixable Aqua Dead Cell Stain Kit and excluded from standard analysis or used in combination with annexin V binding to quantify dead and apoptotic cells according to the manufacturer’s instructions (Life Technologies). To detect intracellular cytokines, mouse Th1 cells were stimulated with antigen-pulsed BMDCs for 5 hours. Additionally, mouse and sheep T cells were stimulated with ionomycin and PMA, and stained as described (9) using monoclonal antibodies recognizing mouse IFN-γ (XMG1.2; eBioscience), IL-4 (11B11; eBioscience), and IL-10 (JES5-16E3; BD Bioscience) or ovine IFN-γ (CC302; Serotec) and IL-17A (eBio64DEC17; eBioscience). Samples were acquired on a BD LSRII or LSRFortessa using FACSDiVa software (BD Biosciences), and analyzed with FlowJo software (Tree Star).
Statistical analysis
The Mann Whitney U test (GraphPad Prism) was used for statistical analyses, and P < 0.05 was considered statistically significant.
Results
Antigen recognition at the effector site decreases tissue egress of effector Th1 cells
T cell-mediated inflammation involves antigenic activation of pro-inflammatory T cells at the target site. To develop a model of T cell-mediated inflammation in which the expression of T cell exit receptors is the sole variable and that is independent of cell recruitment from the blood, we modified Streilein’s “local adoptive transfer assay of delayed hypersensitivity” (28). Using OVA-specific CD4+ T cells from TCR-transgenic (OTII) mice, pro-inflammatory Th1 cells were generated, co-incubated for one hour with OVA-pulsed APCs and transferred into the footpads of mice. The induced local tissue inflammation was transient and measurable as footpad swelling that depended on the number of transferred Th1 cells (Fig. 1A).
FIGURE 1. Antigen recognition reduces the tissue egress of CD4+ effector T cells.
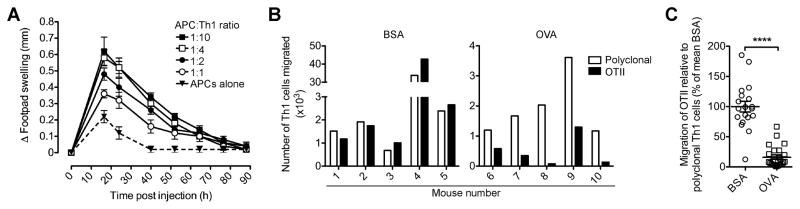
(A) Different ratios of OVA-specific OTII Th1 cells and OVA-pulsed APCs (constant per recipient) were co-incubated and subcutaneously injected into the footpads of WT recipient mice. Footpad thickness was measured and corrected for pre-injection values (Δ swelling). Data points show mean ± SEM (5 mice per group) in one representative of 3 performed experiments. (B–C) A mixture of CFSE-labeled polyclonal CD45.1+ and OTII CD45.2+ Th1 cells were co-incubated with BSA- or OVA-pulsed APCs before injection into the footpads of recipient mice. 20 h later, donor cells that had egressed from the effector site and reached the draining lymph node (dLN) were enumerated by flow cytometry. Migration of OTII Th1 cells relative to polyclonal Th1 cells (B) in one representative experiment (5 mice per group) or (C) in all (n=4) independent experiments combined and expressed relative to the mean ratio of migrated OTII Th1 cells to polyclonal Th1 cells in the presence of BSA (set as 100%). Data points indicate individual recipients and the mean ± SEM of each group. ****P < 0.0001.
To test the influence of antigen recognition on egress from the inflamed site, CFSE-labeled CD45.1+ polyclonal and CD45.2+ (OVA-specific) OTII Th1 cells were mixed, and co-incubated with APCs pulsed with OVA or a control protein (BSA) before transfer into the footpads of recipient mice. As previously (4, 9, 27), we assessed T cell tissue egress by enumerating donor T cells that had left the skin and migrated into the draining popliteal lymph node (dLN) (Fig. 1B, 1C). While at 20 h after transfer many donor Th1 cells had egressed from the inflamed site and reached the dLN, transferred cells in the contralateral lymph node or the spleen were below the level of detection (Fig. 1B, 1C, and data not shown), confirming migration via afferent lymph before transferred cells enter the blood circulation. In the presence of OVA-pulsed APCs, the number of OTII Th1 cells that left the site of inflammation and reached the dLN was drastically reduced relative to that of polyclonal Th1 cells (~75% reduction on average; P < 0.0001; Fig. 1C). We conclude that antigen recognition leads to down-regulation of effector CD4+ T cell egress from inflamed tissue.
Transgenic expression of CCR7 enhances effector T cell egress from inflamed tissue and accelerates resolution of inflammation
To address whether tissue exit receptors can be targeted to modulate tissue egress of antigen-responding pro-inflammatory T cells, we generated Th1 cells from OTII mice and OTII mice whose T cells are Ccr7-transgenic (Ccr7TG). Ccr7TG OTII Th1 cells showed a more uniformly high CCR7 expression relative to Ccr7WT OTII Th1 cells (Fig. 2A). These Th1 cell subsets were differentially labeled with fluorescent dyes and, after co-incubation with OVA-pulsed APCs, co-injected into the footpads of recipient mice. Strikingly, Ccr7TG OTII Th1 cells showed a significantly enhanced capacity to egress from the inflamed skin and enter the dLN 20 h after transfer relative to Ccr7WT OTII Th1 cells (P = 0.0002; Fig. 2B, 2C). At this time point after cell transfer, both Ccr7TG and Ccr7WT Th1 cells were below the level of detection in spleens and contralateral lymph nodes (data not shown), confirming that cells had reached the dLN through the afferent lymph. Migratory differences were independent of the cell labels, as there were no differences in the migration of differentially labeled Ccr7WT OTII Th1 cells (Fig. 2C). At this time point, we could not detect differences in cell proliferation or survival between Ccr7WT and Ccr7TG OTII Th1 cells in skin or dLN (Supplemental Fig. 1A–C). The CCR7 transgene did not affect differentiation into Th1 cells as indicated by similar cytokine expression profiles after stimulation with OVA-pulsed APCs or PMA and ionomycin prior to injection (Fig. 2D, and data not shown) and after re-stimulation of transferred Th1 cells recovered from skin (Supplemental Fig. 1D). Strikingly, although both Th1 cell subsets initially induced a similar magnitude of inflammation, resolution of inflammation was accelerated in mice that had received Ccr7TG OTII Th1 cells (Fig. 2E). We conclude that enhancing exit receptor expression is able to dislodge antigen-sequestered effector T cells from inflamed tissue, translating into faster resolution of inflammation.
FIGURE 2. Transgenic expression of CCR7 enhances effector T cell egress from inflamed tissue and accelerates resolution of inflammation.

(A–C) CFSE-labeled Ccr7WT or Ccr7TG OTII Th1 cells were mixed with eF670-labeled Ccr7WT OTII Th1 cells and incubated with OVA-pulsed APCs, before injection into the footpads of WT recipients. 20 h later, dLN were analyzed for donor cells. (A) Representative CCR7 staining of Th1 cells prior to injection. (B) Quantification of donor Th1 cells that migrated to the dLN. (C) Tissue egress of CFSE-labeled Ccr7WT and Ccr7TG OTII Th1 cells relative to eF670-labeled Ccr7WT OTII Th1 cells. Data are expressed as a percentage of the mean ratio of CFSE+ to eF670+ Ccr7WT Th1 cells that migrated to the dLN (set as 100%) combined from 2 experiments. (D) Intracellular cytokine profile of in vitro generated Ccr7WT OTII and Ccr7TG OTII Th1 cells prior to injection with and without stimulation with OVA-pulsed APCs. (E) Ccr7WT OTII or Ccr7TG OTII Th1 cells were co-incubated with OVA-pulsed APCs, injected into the footpads of separate groups of mice and inflammation was measured over time. Data points depict individual recipients (B and C) and/or the mean ± SEM of each group (C and E). One experiment of ≥ 2 with similar results (A, B, D, E) or the combined analysis of all (N=2) experiments (C) using 5 mice per group is shown. *P < 0.05; ***P < 0.005.
Lack of T cell CCR7 enhances local tissue inflammation
Next, we analyzed in vitro polarized Th1 cells from Ccr7−/− OTII and Ccr7WT OTII mice (Fig. 3A), which produced cytokines at similar frequency after stimulation with OVA-pulsed APCs or PMA and ionomycin (Fig. 3B, and data not shown). Consistent with CCR7 as a major exit receptor (4, 5, 9), Ccr7−/− Th1 cells were drastically reduced in the dLN relative to Ccr7WT Th1 cells (P = 0.0159) and below the level of detection in the spleens of most recipients even 3 days after transfer together with OVA-loaded APCs into footpad skin (Fig. 3C, and data not shown). These results are in line with data showing that lack of T cell CCR7 promotes T cell retention at effector sites (11, 16, 17, 29). Importantly, the reduced egress of Ccr7−/− OTII Th1 cells from the site of inflammation translated into enhanced and prolonged inflammation in our hypersensitivity model (Fig. 3D). Together with our findings that enhancing T cell egress ameliorates the inflammatory response (Fig. 2E), we conclude that the tissue egress capacity of effector T cells is a critical determinant of the course of inflammation and that tissue exit receptors can be targeted to modulate inflammation.
FIGURE 3. Lack of CCR7 impairs effector T cell egress and enhances local inflammation.

Ccr7WT and Ccr7−/− (CD45.2+) OTII Th1 cells were separately co-incubated with OVA-pulsed DCs, before subcutaneous injection into the footpads of two groups of CD45.1+ congenic WT recipients. (A) Representative CCR7 staining of Th1 cells prior to injection. (B) Intracellular cytokine profile of Ccr7WT OTII and Ccr7−/− OTII Th1 cells prior to injection after stimulation with OVA-pulsed APCs. (C) 70 h after transfer, recovered donor Th1 cells were quantified in the dLN. (D) Footpad swelling induced by Ccr7WT and Ccr7−/− OTII Th1 cells was measured over time. Data points depict individual recipients (C) and/or the mean ± SEM of each group (C and D). One experiment of ≥ 2 with similar results using 5 recipients per group. *P < 0.05.
The CCR7-CCL21 axis is active in tissue egressing pro-inflammatory T cells
If the CCR7-CCL21 axis is a physiological target relevant to the egress of pro-inflammatory T cells, then effector T cells that have entered the afferent lymph draining inflamed sites should be responsive to CCR7 ligand. Because analysis of afferent lymph T cells is not feasible in mice and humans, the sheep has emerged as a classical model of afferent lymph cannulation, allowing for the analysis of tissue-recirculating lymphocytes (1, 4, 8, 30). Afferent lymph vessel cannulation in sheep revealed that lymph-borne IFN-γ+ Th1 and IL-17+ Th17 cells egressing from uninflamed or chronically inflamed skin migrated toward CCL21 in an ex vivo chemotaxis assay (Fig. 4). We conclude that the CCR7-CCL21 axis is not only active in adoptively transferred effector T cells but is also a physiological target in pro-inflammatory T cells that recirculate through extralymphoid sites.
FIGURE 4. Inflammation exiting Th1 and Th17 cells chemotax to CCR7 ligand.

Chronic skin inflammation was induced in sheep by subcutaneous injection of CFA. ≥ 3 weeks later afferent lymph vessels draining the inflamed and uninflamed control skin were cannulated and skin-egressing T cells collected. Chemotaxis of lymph-borne CD4+ T cells toward CCL21 was tested ex vivo in a Transwell chemotaxis assay. (A) Representative intracellular cytokine staining of inflammation-draining CD4+ T cells in input and migrated populations after polyclonal stimulation. (B) Chemotaxis of CD4+ T cell subsets. Bars indicate the mean ± SD for triplicate wells for one representative animal of ≥ 3 analyzed for each condition.
Discussion
In this paper we demonstrate that the magnitude and duration of T cell-mediated inflammation is a function of the capacity of pro-inflammatory T cells to leave the inflamed tissue via the afferent lymph. This fills an important gap in our knowledge regarding the regulation of T cell-driven inflammation and highlights the importance of studies delineating the mechanisms that regulate T cell egress versus retention.
We found that CD4+ effector T cells that recognize antigen at the inflamed site greatly reduce their egress from the tissue (Fig. 1). This is in agreement with data demonstrating that upon antigenic stimulation in situ, virus-specific CD8+ T cells decrease their egress from the lung during influenza virus infection (10) and/or develop into long-term TRM cells in the vesicular stomatitis virus-infected brain (31). As T cell entry into tissues is independent of antigen specificity (32), enhancing the retention of antigen-specific T cells by decreased egress from sites of inflammation and infection ensures continued T cell effector functions, while allowing bystander T cells to egress and redistribute. Notably, also long-term retention of CD4+ T cells as TRM cells at effector sites was proposed to involve antigenic stimulation (33). In contrast, formation of CD8+ TRM cells requires additional or alternative antigen-independent cytokine-dependent transcriptional reprograming (reviewed in (34, 35)).
Importantly, we show that tissue exit receptors can be targeted to modulate inflammation: transgenic expression of T cell CCR7 enhanced egress and accelerated resolution of inflammation, while lack of T cell CCR7 exacerbated inflammation. These data are consistent with reduced delayed hypersensitivity responses in Ccr7TG mice, a phenotype that was attributed to effector T cell redistribution to the splenic white pulp (19). We demonstrate that an additional or alternative explanation is that Ccr7TG effector T cells may enter inflamed tissues but egress prematurely via afferent lymph, thereby dampening local inflammation. This is likely accomplished by enhanced infiltrate clearance and/or limited access to antigen bearing APCs at the effector site. Thus, enhancing T cell egress or preventing reduced egress from the effector site represents a novel potential target of anti-inflammatory therapy.
The finding that the CCR7-CCL21 axis is active in physiologically recirculating effector T cells that have entered the inflammation-draining afferent lymph (Fig. 4) highlights the potential importance of this receptor-ligand pair as a therapeutic target. While many inflammatory signals upregulate lymphatic CCL21 at effector sites, its expression levels as well as that of other lymphatically expressed chemokines and adhesion molecules are strongly stimulus-dependent during inflammation (reviewed in (36)). Since we show that diminished T cell egress exacerbates inflammation, future studies will be critical to reveal the regulation of CCL21 on lymphatic endothelial cells and CCR7 on tissue-infiltrating T cells in addition to identifying alternative tissue exit receptors active in different types of inflammation.
Dampening inflammation by guiding pro-inflammatory T cells out of the inflamed site adds to the various anti-inflammatory functions of lymphatic endothelial cells (reviewed in (21)). One example of counter-regulation of inflammation by lymphatic endothelial cells is the expression of atypical chemokine receptor 2 (ACKR2, D6), which scavenges inflammatory chemokines while preserving CCL21 (37, 38). Other examples demonstrate that lymphatic vessel function and activation (e.g. by VEGF-C and VEGF-D) decrease inflammation by expanding the lymphatic network and enhancing the drainage of fluid and inflammatory mediators from the tissue (21, 36). Thus, afferent lymphatics fulfill a key anti-inflammatory role by counteracting the function of blood vascular endothelial cells, which allow for the entry of pro-inflammatory cells, fluid and mediators from the blood into the tissue.
In conclusion, our data show a proof-of-principle that the capacity of effector T cells to egress from the inflamed site via the afferent lymph modulates the local inflammatory response, revealing a novel checkpoint of tissue inflammation with relevance to autoimmune and inflammatory diseases.
Supplementary Material
Acknowledgments
The authors thank Michael Lee for technical support, Eugene Butcher for helpful discussion and Dan Campbell for critical comments on the manuscript. We are indebted to Nigel Killeen, Martin Lipp, and Avinash Bhandoola for providing mice.
Footnotes
This work was supported by NIH/NIAMS grant R01-AR056730 to GFD, an AAI Careers in Immunology Fellowship to DG and GFD, and NIH/NIAID grant F32-AI114080 to TW.
Abbreviations used: dLN, draining lymph node; eF670, eFluor670; MAC, monoclonal antibody core; Ccr7TG, Ccr7-transgenic; TRM, resident memory T cells; WT, wild-type.
References
- 1.Mackay CR, Marston WL, Dudler L. Naive and memory T cells show distinct pathways of lymphocyte recirculation. J Exp Med. 1990;171:801–817. doi: 10.1084/jem.171.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butcher EC. The multistep model of leukocyte trafficking: a personal perspective from 15 years later. In: Hamann A, Engelhardt B, editors. Leukocyte Trafficking: Molecular Mechanisms, Therapeutic Targets, and Methods. Wiley-VCH; Weinheim, Germany: 2005. pp. 3–13. [Google Scholar]
- 3.Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. 2007;8:1295–1301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- 4.Debes GF, Arnold CN, Young AJ, Krautwald S, Lipp M, Hay JB, Butcher EC. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat Immunol. 2005;6:889–894. doi: 10.1038/ni1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bromley SK, Thomas SY, Luster AD. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat Immunol. 2005;6:895–901. doi: 10.1038/ni1240. [DOI] [PubMed] [Google Scholar]
- 6.Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, Williams LT. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc Natl Acad Sci U S A. 1998;95:258–263. doi: 10.1073/pnas.95.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griffith JW, Luster AD. Targeting cells in motion: migrating toward improved therapies. Eur J Immunol. 2013;43:1430–1435. doi: 10.1002/eji.201243183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith JB, McIntosh GH, Morris B. The migration of cells through chronically inflamed tissues. J Pathol. 1970;100:21–29. doi: 10.1002/path.1711000104. [DOI] [PubMed] [Google Scholar]
- 9.Brown MN, Fintushel SR, Lee MH, Jennrich S, Geherin SA, Hay JB, Butcher EC, Debes GF. Chemoattractant receptors and lymphocyte egress from extralymphoid tissue: changing requirements during the course of inflammation. J Immunol. 2010;185:4873–4882. doi: 10.4049/jimmunol.1000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jennrich S, Lee MH, Lynn RC, Dewberry K, Debes GF. Tissue exit: a novel control point in the accumulation of antigen-specific CD8 T cells in the influenza a virus-infected lung. J Virol. 2012;86:3436–3445. doi: 10.1128/JVI.07025-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, Gebhardt T. The developmental pathway for CD103(+)CD8(+) tissue-resident memory T cells of skin. Nat Immunol. 2013;14:1294–1301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 12.Vander Lugt B, Tubo NJ, Nizza ST, Boes M, Malissen B, Fuhlbrigge RC, Kupper TS, Campbell JJ. CCR7 plays no appreciable role in trafficking of central memory CD4 T cells to lymph nodes. J Immunol. 2013;191:3119–3127. doi: 10.4049/jimmunol.1200938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ledgerwood LG, Lal G, Zhang N, Garin A, Esses SJ, Ginhoux F, Merad M, Peche H, Lira SA, Ding Y, Yang Y, He X, Schuchman EH, Allende ML, Ochando JC, Bromberg JS. The sphingosine 1-phosphate receptor 1 causes tissue retention by inhibiting the entry of peripheral tissue T lymphocytes into afferent lymphatics. Nat Immunol. 2008;9:42–53. doi: 10.1038/ni1534. [DOI] [PubMed] [Google Scholar]
- 14.Schneider MA, Meingassner JG, Lipp M, Moore HD, Rot A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J Exp Med. 2007;204:735–745. doi: 10.1084/jem.20061405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wengner AM, Hopken UE, Petrow PK, Hartmann S, Schurigt U, Brauer R, Lipp M. CXCR5- and CCR7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis Rheum. 2007;56:3271–3283. doi: 10.1002/art.22939. [DOI] [PubMed] [Google Scholar]
- 16.Höpken UE, Wengner AM, Loddenkemper C, Stein H, Heimesaat MM, Rehm A, Lipp M. CCR7 deficiency causes ectopic lymphoid neogenesis and disturbed mucosal tissue integrity. Blood. 2007;109:886–895. doi: 10.1182/blood-2006-03-013532. [DOI] [PubMed] [Google Scholar]
- 17.McNamee EN, Masterson JC, Veny M, Collins CB, Jedlicka P, Byrne FR, Ng GY, Rivera-Nieves J. Chemokine receptor CCR7 regulates the intestinal TH1/TH17/Treg balance during Crohn’s-like murine ileitis. J Leukoc Biol. 2015;97:1011–1022. doi: 10.1189/jlb.3HI0614-303R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Förster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol. 2008;8:362–371. doi: 10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- 19.Unsoeld H, Voehringer D, Krautwald S, Pircher H. Constitutive expression of CCR7 directs effector CD8 T cells into the splenic white pulp and impairs functional activity. J Immunol. 2004;173:3013–3019. doi: 10.4049/jimmunol.173.5.3013. [DOI] [PubMed] [Google Scholar]
- 20.Kocks JR, Davalos-Misslitz AC, Hintzen G, Ohl L, Forster R. Regulatory T cells interfere with the development of bronchus-associated lymphoid tissue. J Exp Med. 2007;204:723–734. doi: 10.1084/jem.20061424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dieterich LC, Seidel CD, Detmar M. Lymphatic vessels: new targets for the treatment of inflammatory diseases. Angiogenesis. 2014;17:359–371. doi: 10.1007/s10456-013-9406-1. [DOI] [PubMed] [Google Scholar]
- 22.Förster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 23.Kwan J, Killeen N. CCR7 directs the migration of thymocytes into the thymic medulla. J Immunol. 2004;172:3999–4007. doi: 10.4049/jimmunol.172.7.3999. [DOI] [PubMed] [Google Scholar]
- 24.Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, Nakano H. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young AJ, Hein WR, Hay JB. Cannulation of lymphatic vessels and its use in the study of lymphocyte traffic. In: Levkovits I, editor. Manual of immunological methods: the comprehensive source book of techniques. Academic Press; San Diego: 1997. pp. 2039–2059. [Google Scholar]
- 26.Geherin SA, Lee MH, Wilson RP, Debes GF. Ovine skin-recirculating gammadelta T cells express IFN-gamma and IL-17 and exit tissue independently of CCR7. Vet Immunol Immunopathol. 2013;155:87–97. doi: 10.1016/j.vetimm.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geherin SA, Wilson RP, Jennrich S, Debes GF. CXCR4 Is dispensable for T Cell egress from chronically inflamed skin via the afferent lymph. PLoS One. 2014;9:e95626. doi: 10.1371/journal.pone.0095626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kezuka T, Streilein JW. Analysis of in vivo regulatory properties of T cells activated in vitro by TGFbeta2-treated antigen presenting cells. Invest Ophthalmol Vis Sci. 2000;41:1410–1421. [PubMed] [Google Scholar]
- 29.Doebis C, Menning A, Neumann K, Ghani S, Schlawe K, Lauer U, Hamann A, Huehn J, Syrbe U. Accumulation and local proliferation of antigen-specific CD4+ T cells in antigen-bearing tissue. Immunol Cell Biol. 2011;89:566–572. doi: 10.1038/icb.2010.128. [DOI] [PubMed] [Google Scholar]
- 30.Hein WR, Griebel PJ. A road less travelled: large animal models in immunological research. Nat Rev Immunol. 2003;3:79–84. doi: 10.1038/nri977. [DOI] [PubMed] [Google Scholar]
- 31.Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A. 2010;107:17872–17879. doi: 10.1073/pnas.1010201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ager A, Drayson MT. Lymphocyte migration in the rat. In: Husband AJ, editor. Migration and homing of lymphoid cells. CRC Press; Boca Raton: 1988. pp. 19–49. [Google Scholar]
- 33.Iijima N, Iwasaki A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science. 2014;346:93–98. doi: 10.1126/science.1257530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gebhardt T, Mueller SN, Heath WR, Carbone FR. Peripheral tissue surveillance and residency by memory T cells. Trends Immunol. 2013;34:27–32. doi: 10.1016/j.it.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity. 2014;41:886–897. doi: 10.1016/j.immuni.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aebischer D, Iolyeva M, Halin C. The inflammatory response of lymphatic endothelium. Angiogenesis. 2014;17:383–393. doi: 10.1007/s10456-013-9404-3. [DOI] [PubMed] [Google Scholar]
- 37.Jamieson T, Cook DN, Nibbs RJ, Rot A, Nixon C, McLean P, Alcami A, Lira SA, Wiekowski M, Graham GJ. The chemokine receptor D6 limits the inflammatory response in vivo. Nat Immunol. 2005;6:403–411. doi: 10.1038/ni1182. [DOI] [PubMed] [Google Scholar]
- 38.Lee KM, McKimmie CS, Gilchrist DS, Pallas KJ, Nibbs RJ, Garside P, McDonald V, Jenkins C, Ransohoff R, Liu L, Milling S, Cerovic V, Graham GJ. D6 facilitates cellular migration and fluid flow to lymph nodes by suppressing lymphatic congestion. Blood. 2011;118:6220–6229. doi: 10.1182/blood-2011-03-344044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.