

Endoplasmic reticulum (ER) stress induces the degradation of mRNAs by Ire1. In mammalian cells, this process depends on specific stem-loop structures in the target mRNAs and on Perk, a second sensor of ER stress. Perk appears to be required to translationally attenuate the stem-loop regions of target mRNAs.
Abstract
Endoplasmic reticulum (ER) stress occurs when misfolded proteins overwhelm the capacity of the ER, resulting in activation of the unfolded protein response (UPR). Ire1, an ER transmembrane nuclease and conserved transducer of the UPR, cleaves the mRNA encoding the transcription factor Xbp1 at a dual stem-loop (SL) structure, leading to Xbp1 splicing and activation. Ire1 also cleaves other mRNAs localized to the ER membrane through regulated Ire1-dependent decay (RIDD). We find that during acute ER stress in mammalian cells, Xbp1-like SLs within the target mRNAs are necessary for RIDD. Furthermore, depletion of Perk, a UPR transducer that attenuates translation during ER stress, inhibits RIDD in a substrate-specific manner. Artificially blocking translation of the SL region of target mRNAs fully restores RIDD in cells depleted of Perk, suggesting that ribosomes disrupt SL formation and/or Ire1 binding. This coordination between Perk and Ire1 may serve to spatially and temporally regulate RIDD.
INTRODUCTION
The endoplasmic reticulum (ER) is the entry point for proteins targeted to the secretory pathway. Secreted proteins are translated from mRNAs localized to the cytosolic face of the ER membrane and enter the ER as nascent chains that are folded and modified before exiting the organelle. The flux of proteins through the ER varies extensively among cell types and environments. Changes in this flux can result in ER stress, an imbalance between the load of unfolded proteins entering the ER and the capacity of the organelle to fold and modify them efficiently. In metazoans, ER stress activates three ER transmembrane proteins: inositol-requiring 1 (Ire1), PKR-like endoplasmic reticulum kinase (Perk), and activating transcription factor 6 (Atf6), which coordinate a signaling network known as the unfolded protein response (UPR; Walter and Ron, 2011). Although ER stress results from a variety of pathological conditions, loss of individual UPR sensors also affects normal development and physiology in several model organisms (Moore and Hollien, 2012).
Perk directly phosphorylates eukaryotic translation initiation factor 2 α (eIF2α), which leads to the attenuation of translation initiation and limits the protein-folding load on the ER (Harding et al., 1999). This phosphorylation event also leads to translational up-regulation of certain proteins, including activating transcription factor 4 (Atf4) (Harding et al., 2000). Concurrently, Ire1 oligomerizes in response to ER stress, activating its cytosolic nuclease domain (Li et al., 2010), and cleaves the mRNA encoding X-box binding protein 1 (Xbp1). This cleavage occurs at two specific sites in a dual stem-loop (SL) structure (Yoshida et al., 2001; Calfon et al., 2002). The resulting 5′ and 3′ fragments are then ligated, forming a spliced transcript encoding the active transcription factor, which, together with other UPR transcription factors, up-regulates numerous genes that increase the capacity of the secretory pathway (Travers et al., 2000; Harding et al., 2003).
Ire1 is also responsible for the cleavage of other ER-localized mRNAs, leading to their degradation through regulated Ire1-dependent decay (RIDD; Hollien and Weissman, 2006; Hollien et al., 2009). RIDD was originally observed in Drosophila melanogaster S2 cells, where a large number of mRNAs associated with the ER are degraded during ER stress (Hollien and Weissman, 2006). RIDD is important for Drosophila eye development, confirming a physiological role for this pathway in vivo (Coelho et al., 2013). In S2 cells, ER localization of an mRNA is both necessary and sufficient for its degradation by RIDD (Gaddam et al., 2013). However, exceptions to this rule exist. For example, the Drosophila transcript encoding small ubiquitin-modifier (Sumo) is targeted to RIDD despite localizing to the cytosol. This mRNA requires an Xbp1-like SL in its coding region to be degraded by Ire1 (Moore et al., 2013). In addition, for unknown reasons, RIDD of Sumo requires Perk (Moore et al., 2013), even though Perk depletion does not appear to generally affect RIDD in S2 cells (Hollien and Weissman, 2006).
RIDD also occurs in mammalian cells (Han et al., 2009; Hollien et al., 2009). Activation of Ire1 through overexpression in cultured cells or tissue-specific Xbp1 mutations in mice, which result in hyperactivation of Ire1, induces broad cleavage of ER- localized mRNAs (Han et al., 2009; So et al., 2012). However, during chemical induction of ER stress in both mammalian cell culture and mice, the magnitude of degradation and number of mRNAs targeted to the pathway are more limited than in S2 cells (Hollien et al., 2009; So et al., 2012). This restriction of RIDD substrates suggests a dependence on additional factors or sequence elements beyond mRNA localization to the ER. One likely requirement is the presence an Xbp1-like SL within the target mRNA sequence. These SLs are more prevalent in mammalian RIDD targets than in those of D. melanogaster (Gaddam et al., 2013). Furthermore, mutation of a conserved guanine (G) within the loop blocks mRNA cleavage by human Ire1 in vitro (Hur et al., 2012) and also affects the regulation of at least one RIDD target in human cells (Bright et al., 2015).
In this study, we investigate the mechanism and substrate selectivity of RIDD during acute, chemically induced ER stress in mammalian cells and describe an unexpected role for Perk in the RIDD pathway.
RESULTS
RIDD targeting in different cell types
Previous work in mammalian cells suggested that the extent of degradation of RIDD targets in the absence of Ire1 overexpression is fairly small, on the order of twofold (Hollien et al., 2009). We first asked whether this result was cell-line dependent. We treated several different mammalian cell lines with chemical inducers of ER stress: dithiothreitol (DTT), which blocks disulfide bonding; thapsigargin (Tg), which depletes ER calcium reserves; and tunicamycin (Tm), an inhibitor of N-linked glycosylation. We then used quantitative real-time PCR (qPCR) to measure the stress-dependent changes in the relative levels of mRNAs that were previously identified as RIDD targets in mouse fibroblasts (Hollien et al., 2009; Figure 1, A and B). Note that because our qPCR expression data are inherently ratiometric, we use a log 2 scale throughout this article, meaning that a unit of 1.0 refers to a twofold change in expression.
FIGURE 1:

The RIDD pathway varies across mammalian cell lines. For all abundance measurements, mRNA was reverse transcribed and measured by qPCR and data were normalized to the housekeeping control mRNA Rpl19. The legend in A applies to bar graphs in A, B, D, and F. (A) Relative mRNA levels of RIDD targets in mouse MC3T3-E1 cells treated with either DTT (2 mM) or Tg (2 μM) for 4 h to induce ER stress. (B) Relative mRNA levels of Blos1 (black) and Scara3 (gray) in the indicated cell lines treated with DTT (2 mM), Tm (2.5 μg/ml), or Tg (2 μM) for 4 h. Note that Scara3 was not expressed strongly enough in Min6 cells to measure mRNA levels. (C) Samples from A and B were amplified by PCR using primers surrounding the Xbp1 splice sites. Shown are representative agarose gels with the spliced and unspliced products and averages and SDs of the percentage spliced Xbp1 for three independent experiments. (D) Relative mRNA levels of RIDD targets in MC3T3-E1 cells transfected with either Neg (negative control) or Ire1 siRNAs and then treated with or without DTT (2 mM, 4 h). (E) Xbp1 splicing in samples from D. (F, G) Relative mRNA levels of RIDD targets (F) and Xbp1 (G) in MC3T3-E1 cells transfected with Neg or Xbp1 siRNAs and then treated with or without DTT (2 mM, 4 h). Shown in all panels are the averages and SDs from two (Hek293 cells, Tm treatment) or three (all other panels) independent experiments. Ut, untreated.
Xbp1 splicing was nearly complete in all stress conditions tested (Figure 1C). However, the extent of RIDD targeting varied among individual mRNAs and among the different cell types (Figure 1, A and B). In both human cell lines tested (Hek293 and Hep G2), Blos1 was degraded during ER stress, but other mouse RIDD targets were either not degraded (Scara3) or not expressed to detectable levels (Col6a1 and Hgsnat; Figure 1B). Of note, the mouse Scara3 transcript contains an Xbp1-like SL, but the human transcript does not.
We observed the most robust RIDD in MC3T3-E1 cells, a preosteoblast cell line derived from mouse calvaria (Kodama et al., 1981), and therefore used these cells for further study. Using small interfering RNA (siRNA)–mediated silencing, we verified that the down-regulation of RIDD targets was Ire1 dependent and Xbp1 independent (Figure 1, D–G).
Xbp1-like stem loops are necessary for RIDD in mammalian cells
To test the importance of Xbp1-like SLs for RIDD in a cellular context, we used a reporter-based approach. We created plasmids expressing the coding sequences (CDSs) of the mouse RIDD targets Hgsnat and Blos1 with vector-derived 5′ and 3′ untranslated regions (UTRs) and stably transfected them into MC3T3-E1 cells. After treatment of cells with or without DTT (2 mM, 4 h), we measured the relative abundance of the reporter mRNAs by qPCR, using primers that spanned the CDSs and reporter UTRs and therefore did not amplify the endogenous transcripts. As expected, the mRNAs expressed from both of these plasmids were down-regulated during ER stress (Figure 2, B and C), indicating that the CDS is sufficient for RIDD of these transcripts.
FIGURE 2:

An Xbp1-like stem loop is necessary for RIDD and sufficient to induce GFP mRNA degradation in mammalian cells during ER stress. (A) RNA SLs from mouse Hgsnat, Blos1, and Xbp1. Red lettering indicates Xbp1 loop residues conserved across species, and arrows indicate putative Ire1 cleavage sites. Numbering is relative to mRNA translation start sites. (B–F) We stably transfected MC3T3-E1 cells with plasmids expressing reporter mRNAs, incubated cells with or without DTT (2 mM, 4 h), and measured relative abundances of the mRNA reporters by qPCR relative to the housekeeping control Rpl19. (B, C) Reporters expressing the mHgsnat (B) or mBlos1 (C) coding sequences (CDSs) with and without mutations in the Xbp1-like loop. (D) Reporters expressing the Blos1 CDS with and without mutations in the stem region of the Xbp1-like SL. Blue lettering indicates mutated residues. (E) Changes in mRNA abundance for the WT Blos1 reporter in two independent cell lines (WT1 and WT2) after DTT treatment. The cell lines differ only in their levels of reporter expression (Ex), either low (4-fold above endogenous levels) or high (32-fold above endogenous levels). (F) Reporters expressing GFP or an ER-targeted GFP (ssGFP) with and without the mBlos1 SL inserted 15 nt downstream of the stop codon. (G) Fraction membrane (membrane/total) of mRNAs from MC3T3-E1 cells stably expressing different GFP reporters measured by digitonin fractionation followed by qPCR. (H) We depleted Ire1 from stably transfected cells and then measured reporter mRNA levels as in B–F. Shown are averages and SDs from three or more independent experiments. *p < 0.05, two-tailed unpaired t test. Ut, untreated.
The CDSs of Blos1 and Hgsnat contain Xbp1-like SLs (Figure 2A), as defined by a seven-nucleotide (nt) loop with the four conserved residues essential for Xbp1 splicing (Calfon et al., 2002) and a stem of at least four consecutive base pairs (allowing for AU, GC, and GU pairs). To test whether these sites were important for RIDD, we mutated the putative Ire1 cleavage site G to cytosine (C) and measured reporter degradation. For Blos1, this mutation, as well as mutation of a second conserved loop residue, completely ablated degradation (Figure 2C). For Hgsnat, mutation of the putative cleavage site in one of the two SLs (Hgsnat SL #1) blocked RIDD (Figure 2B), whereas the corresponding mutation in a second SL (Hgsnat SL #2) did not affect its degradation during ER stress (Figure 2B). The stem of Hgsnat SL #2 is shorter and has fewer GC pairs than Hgsnat SL #1 (Figure 2A), suggesting that the stability of the stem is important for RIDD. To test this, we made mutations that disrupted the base-pairing of the Xbp1-like SL of our Blos1 reporter. These mutations blocked RIDD targeting (Figure 2D). Restoring base-pairing within the putative stem region with complementary mutations that preserved the GC content of the SL restored RIDD. However, mutations that replaced GC pairs with AU pairs prevented RIDD (Figure 2D). Together these results indicate that both the sequence and stability of Xbp1-like SLs are important for RIDD in mouse cells, as suggested previously for human cells (Bright et al., 2015).
To ensure that reporter expression levels did not influence RIDD, we measured the level of overexpression of Blos1 mRNA in our reporter cell lines. Total Blos1 mRNA abundance was measured by qPCR using primers that annealed within the CDS of the Blos1 transcript and therefore amplified both endogenous and reporter mRNAs. The overexpression of the Blos1 reporter mRNAs varied from ∼4- to 32-fold above endogenous Blos1 levels, which were measured using a control cell line transfected with green fluorescent protein (GFP). However, there was no correlation between reporter expression level and degradation during ER stress. Furthermore, we created two independent cell lines that expressed WT Blos1 to different levels (4- vs. 32-fold overexpression) and observed no difference in the extent of the reporter mRNA degradation during stress (Figure 2E).
An Xbp1-like stem loop is sufficient to target GFP mRNA to RIDD
To determine whether an Xbp1-like SL is sufficient to induce degradation of a transcript not normally targeted to the RIDD pathway, we used reporters expressing either GFP or an ER-targeted GFP (ssGFP) containing the signal sequence from Drosophila Hsp70-3. In S2 cells, this ssGFP mRNA reporter (but not the cytosolic GFP mRNA) is degraded by RIDD (Gaddam et al., 2013). Similarly, rat cells overexpressing Ire1 degrade an ER-targeted GFP mRNA (Han et al., 2009). However, in MC3T3-E1 cells, neither GFP nor ssGFP transcripts were down-regulated during ER stress (Figure 2F), supporting the idea that mRNA membrane association is not sufficient for RIDD in mammalian cells during acute ER stress.
To confirm that reporter mRNAs were correctly localized, we used detergent fractionation to separate membrane-associated versus cytosolic mRNAs, as described previously (Stephens et al., 2008; Gaddam et al., 2013). As expected, ssGFP mRNA fractionated predominately with the membrane, along with a membrane-bound control, BiP. In contrast, GFP mRNA fractionated predominantly with the cytosol, similarly to the control glyceraldehyde 3-phosphate dehydrogenase (Gapdh; Figure 2G).
Addition of the Blos1 SL to the 3′ UTR of either GFP reporter mRNA (GFP-SLUTR or ssGFP-SLUTR) resulted in its Ire1-dependent degradation during ER stress (Figure 2, F and H), indicating that an Xbp1-like SL is sufficient to target GFP mRNA to RIDD. Addition of the SL also resulted in a partial shift of GFP mRNA localization toward the membrane fraction (Figure 2G), suggesting that the SL alone may mediate membrane association.
Xbp1-like SLs do not predict RIDD targets generally
On the basis of these results, we hypothesized that endogenous mRNAs with Xbp1-like SLs would be RIDD targets. Previous work in mammalian cells has not led to a comprehensive list of RIDD targets, in part because transcription is highly regulated during ER stress and complicates the global analysis of mRNA degradation. Therefore we carried out a limited test of our hypothesis by blocking transcription in MC3T3-E1 cells using actinomycin D (2 μg/ml) and then measuring the relative degradation of several mRNAs in the presence and absence of DTT (1 mM, 4 h). We chose mRNAs that met the following criteria: 1) they were expressed in MC3T3-E1 cells (Nabavi et al., 2012), 2) they were associated with Gene Ontology terms indicating ER, Golgi, lysosome, plasma membrane, or extracellular localization of the encoded protein, and 3) they contained strong Xbp1-like SLs with at least three GC base pairs in the stem. Surprisingly, none of the 10 mRNAs we measured was degraded more strongly during ER stress (Figure 3). These results indicate that although the presence of an Xbp1-like SL is sufficient to target GFP mRNA to RIDD, it is not generally sufficient to target endogenous mRNAs to RIDD and additional targeting features must exist.
FIGURE 3:
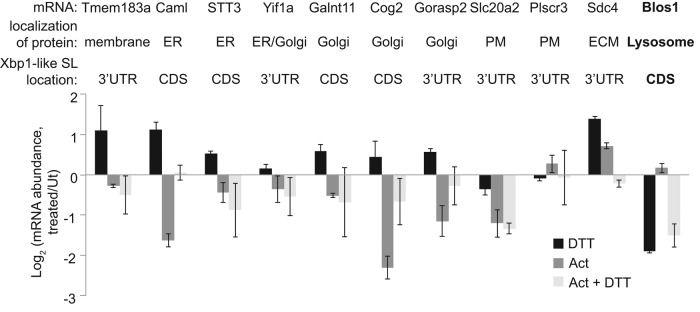
Xbp1-like SLs are not sufficient to target endogenous mRNAs to RIDD. MC3T3-E1 cells were treated with 1 mM DTT, 2 μg/ml actinomycin D (Act), or both for 4 h. We then measured relative mRNA levels of noted transcripts by qPCR. Transcripts were chosen based on their predicted localization to the ER (based on Gene Ontology term analysis) and the presence of Xbp1-like SLs, defined as 1) a seven-membered loop with the four conserved residues (as in Figure 2A), and 2) a stem of at least 5 base pairs including three GC pairs. The verified RIDD target Blos1 was also measured as a control. Shown are averages and SDs from two independent experiments. ECM, extracellular matrix; PM, plasma membrane; Ut, untreated.
Perk-mediated attenuation of translation is important for RIDD
Previously we determined that the noncanonical Drosophila RIDD target Sumo relies on both a SL and the presence of Perk to be degraded during ER stress (Moore et al., 2013). To determine whether Perk plays a role in the mammalian RIDD pathway, we transfected MC3T3-E1 cells with either a negative control (Neg) siRNA or a combination of four siRNAs targeting Perk and then induced ER stress with either DTT or Tg. Depletion of Perk strongly inhibited RIDD of both Blos1 and Col6a1 and partially inhibited RIDD of Scara3 (Figure 4, A–C). RIDD of Hgsnat, however, was not affected by Perk knockdown (Figure 4B; see next section). We saw similar effects when two distinct Perk siRNAs were transfected individually (Supplemental Figure S1, A and B). Finally, Perk knockdown also inhibited RIDD of Blos1 in Hek293 cells (Figure 4, D and E), indicating a conserved effect across species.
FIGURE 4:

Translation attenuation mediated by Perk is important for RIDD. (A–C) We transfected MC3T3-E1 cells with Neg or Perk siRNAs and then incubated them with and without 1 mM DTT (B) or 2 μM Tg (C) for 4 h. We then measured the percentage of Perk mRNA remaining (A) and RIDD target mRNA levels (B, C). The legend in B applies to bar graphs in B, C, F, and G. Asterisks represent significant differences between Neg and Perk siRNA-treated samples. (D, E) Perk (D) or human Blos1 (E) mRNA measured from Neg or Perk siRNA–treated Hek293 cells with or without DTT (2 mM, 4 h). (F) Blos1 (black bars) and Col6a1 (gray bars) mRNA levels in MC3T3-E1 cells treated with 500 nM ISRIB, 1 mM DTT, or both for 4 h. (G, H) Blos1 (black bars) and Col6a1 (gray bars) mRNA levels (G) and Xbp1 splicing (H) from control or Perk-depleted MC3T3-E1 cells treated with 1 μM harringtonine (Har), 1 mM DTT, or both for 4 h. All mRNA levels were determined by qPCR. Shown are averages and SDs from at least three independent experiments. *p < 0.05, two-tailed paired t test. Ut, untreated.
In addition to phosphorylating eIF2α and thereby attenuating translation initiation, Perk also phosphorylates other targets, including Nrf2 (Cullinan et al., 2003) and diacylglycerol (Bobrovnikova-Marjon et al., 2012). To determine which aspect of Perk signaling is important for RIDD, we used integrated stress response inhibitor (ISRIB), a chemical that blocks translation attenuation during ER stress but does not affect the phosphorylation of eIF2α or other Perk targets (Sidrauski et al., 2013). ISRIB significantly inhibited RIDD (Figure 4F and Supplemental Figure S1C). Therefore Perk's ability to attenuate translation during ER stress is important for RIDD. Accordingly, artificially attenuating translation with the initiation inhibitor harringtonine fully restored RIDD in cells depleted of Perk (Figure 4G).
Knockdown of Perk also resulted in a 25–40% decrease in Xbp1 splicing in response to ER stress (Figure 4H), an effect noted previously (Majumder et al., 2012). As with RIDD, inhibition of translation initiation by harringtonine fully restored Xbp1 splicing. Harringtonine did not cause a general increase in Ire1 activity, as harringtonine treatment alone actually led to a reduction in constitutive Xbp1 splicing in unstressed cells (Figure 4H). Overall these results indicate that attenuating translation initiation during ER stress allows for more efficient RIDD and Xbp1 splicing.
RIDD relies on the translational status of target mRNAs
There are two general possibilities for why Perk-mediated translation attenuation is important for RIDD: either halting translation allows for depletion of an unstable factor that is important for RIDD, or depletion of ribosomes from the RIDD target mRNA allows it to be degraded. The fact that Perk knockdown had varying effects on different mRNAs suggests that translation attenuation of the RIDD target itself is of primary importance. In support of this model, we noted that Hgsnat, the RIDD target that was insensitive to Perk depletion, has two large clusters of rare codons near the 5′ end of the transcript, which may act to constitutively reduce translation and allow for Hgsnat mRNA degradation during ER stress, regardless of Perk activity. Rare codon clusters were not found in the 5′ regions of Perk-sensitive RIDD targets (see later discussion of Figure 7A).
FIGURE 7:

RIDD target summary and model. (A) Summary of Perk-dependent RIDD targeting for endogenous and reporter mRNAs. RNA CDSs are shown in color; UTRs are in gray. Diagrams are the same as in Figures 5 and 6. Orange bars denote clusters of rare codon usage, defined as >10% usage of infrequent codons over multiple 18-codon groupings as calculated by the Rare Codon Calculator (Clarke and Clark, 2008). (B) Model of differential Ire1 targeting, with legend of diagrams used.
To test directly whether the translational status of mRNA targets is important for RIDD, we asked whether limiting translation of Perk-sensitive RIDD targets caused them to become Perk insensitive. We introduced translation-stalling SLs (Vattem and Wek, 2004) 6 nt upstream of the translation start site within the 5′ UTRs of two RIDD reporters, one expressing the Blos1 CDS (as in Figure 2C) and one expressing ssGFP with the Blos1 SL inserted in the CDS, 68 nt upstream of the stop codon (ssGFP-SLCDS). We then stably transfected these reporters into MC3T3-E1 cells and tested for RIDD as described. In Neg siRNA–treated cells, these reporter mRNAs were degraded similarly to their wild-type counterparts. However, unlike the wild-type reporters, degradation of the translationally stalled reporters was unaffected by depletion of Perk (Figure 5, B and C). We conclude that attenuating translation of the target itself is important for degradation by RIDD.
FIGURE 5:

RIDD relies on the translational status of target mRNAs. (A) Legend for the diagrams. (B, C) We stably transfected MC3T3-E1 cells with plasmids expressing reporter mRNAs and then transfected them with Neg or Perk siRNAs and incubated cells with or without DTT (2 mM, 4 h). Reporters express ssGFP-SLCDS (B) or mBlos1 (C) with or without upstream translation-blocking SLs inserted into their 5′ UTRs 6 nt upstream of the start codons. mRNA levels were measured by qPCR with reporter-specific primers. (D) Endogenous RIDD targets from MC3T3-E1 cells treated with 35 μM Chx, 1 mM DTT, or both for 4 h. Shown are averages and SDs from at least three independent experiments. *p < 0.05, two-tailed paired t test. Ut, untreated.
Ribosome binding to an mRNA may limit Ire1's access, thus inhibiting cleavage and subsequent degradation of the mRNA. To test this idea we used cycloheximide (Chx), a translation elongation inhibitor that stalls ribosomes along mRNAs without releasing them. Chx significantly inhibited RIDD of both Blos1 and Col6a1 but not Scara3 (Figure 5D), correlating with the relative sensitivities of these mRNAs to Perk depletion. These results indicate that attenuating translation initiation and essentially reducing the number of ribosomes on an mRNA enhances RIDD, whereas blocking translation elongation by locking ribosomes on an mRNA inhibits RIDD.
Translation attenuation of Xbp1-like SLs is important for RIDD
Based on the evidence that Ire1 directly cleaves RIDD targets in their Xbp1-like SLs, we wondered whether reduced ribosome occupancy in this specific region, rather than the entire message, is important for RIDD. We devised two strategies to test this hypothesis. First, we predicted that RIDD targets with Xbp1-like SLs in the CDS would be sensitive to Perk depletion, whereas RIDD targets with SLs in the 3′ UTR would be insensitive to Perk. As noted, degradation of the ssGFP-SLCDS reporter during ER stress was reduced when Perk was depleted (Figure 5B). In contrast, the ssGFP-SLUTR reporter, which has a stop codon 15 nt upstream of the Xbp1-like SL, was not sensitive to Perk knockdown (Figure 6B). Because these two constructs differ only in the presence of the upstream stop codon, the overall translation of the two constructs should be the same. Thus, translation of the Xbp1-like SL region appears to strongly influence whether a RIDD target will be affected by Perk.
FIGURE 6:

Translation attenuation of Xbp1-like SLs is required for RIDD. (A) Legend for the diagrams. (B–D) We stably transfected MC3T3-E1 cells with plasmids expressing reporter mRNAs and then transfected them with Neg or Perk siRNAs and incubated cells with or without DTT (2 mM, 4 h) as in Figure 5. (B) Reporters expressing ssGFP-SLCDS or ssGFP-SLUTR. (C) Reporters expressing RIDD-insensitive Blos1 containing the G360C loop mutation, with a functional Xbp1-like SL (from wild-type Blos1) added to the CDS at nt 261 (two independent experiments) or to the 3′ UTR. (D) Reporters expressing the mBlos1 CDS with or without the IBV pseudoknot (orange) or SRV pseudoknot (purple) inserted 15 nt upstream of the Xbp1-like SL. For B–D, we measured relative mRNA abundances by qPCR using reporter-specific primers. Shown are averages and SDs from at least three independent experiments except where noted. *p < 0.05, two-tailed paired t test. Ut, untreated.
We observed a similar trend for reporters expressing the Blos1 CDS. To determine whether moving Blos1's SL would affect its Perk dependence, we used the Blos1 reporter containing the G360C loop mutation, which is not degraded during ER stress (Figure 2C). We inserted a functional Xbp1-like SL (from wild-type Blos1) at an alternative position in the CDS of the mutated Blos1 reporter (position 261 relative to the translation start) or in the 3′ UTR (21 nt downstream of the stop codon). Moving the SL within the Blos1 CDS did not change the Perk sensitivity of the reporter (Figure 6C). Moving the SL to the 3′ UTR resulted in a decrease in overall degradation; however, this degradation was not affected by Perk depletion (Figure 6C).
In a second approach to test the importance of translation in the SL region, we used ribosome-stalling pseudoknots with different structures and sequences from either the infectious bronchitis virus (IBV) or simian retrovirus-1 (SRV-1; Kontos et al., 2001). We introduced these sequences 15 nt upstream of the Xbp1-like SL in the CDS of our Blos1 reporter, preserving the original reading frame. Conveniently, the endogenous Blos1 SL is located within 30 nt of the stop codon, meaning that the majority of the Blos1 mRNA was translated normally with or without the pseudoknots. Both of these reporter mRNAs were degraded during ER stress similarly to the wild-type Blos1 reporter; however, Perk knockdown did not affect degradation of either pseudoknot-containing mRNA (Figure 6D). Differences in the efficiency of Perk depletion did not account for these effects, as endogenous Blos1 measured by qPCR with primers that amplified the endogenous CDS and 3′ UTR but not the reporter 3′ UTR, was equally sensitive to Perk knockdown in all cell lines tested (Supplemental Figure S1D). These data suggest that translation attenuation of only the SL region of the RIDD target is required for degradation.
DISCUSSION
In response to ER stress, the nuclease activity of Ire1 has two outputs. One is to initiate the splicing of the Xbp1 mRNA, leading to the transcriptional regulation of a large number of target genes. The second is to initiate the degradation of RIDD targets. Although these two outputs can be uncoupled (Han et al., 2009; Hollien et al., 2009), the mRNA sequence elements important for cleavage of RIDD substrates in mammalian cells are remarkably similar to those important for cleavage of the Xbp1 mRNA, namely stable SL structures with specific, conserved loop residues. However, despite the apparent sufficiency of such a SL in targeting GFP to the RIDD pathway (Figure 2F), there are many mRNAs in the cell that possess Xbp1-like SLs but are not targeted to RIDD (Figure 3; Bright et al., 2015). We suggest that this additional specificity arises in part from the translational status of would-be target mRNAs, which we propose influences the accessibility of the Xbp1-like SLs.
A large body of evidence supports a role for translation in dictating an mRNA's susceptibility to degradation (Roy and Jacobson, 2013; Walters and Parker, 2014), and it appears that this is true for RIDD as well. We show here that RIDD in mammalian cells relies on Perk-mediated attenuation of translational initiation during ER stress, in a substrate-specific manner. Two RIDD targets with Xbp1-like SLs in their CDSs (Blos1 and Col6a1) were highly sensitive to knockdown of Perk, which blocked their degradation during ER stress. A third RIDD target (Scara3), with an Xbp1-like SL immediately upstream of its start codon, was partially sensitive to Perk depletion. The translation elongation inhibitor Chx inhibited RIDD of Blos1 and Col6a1 but not of Scara3. This is consistent with the idea that it is translation of the SL region that is important, as elongation inhibitors should not affect the small ribosomal subunit while it is scanning the 5′ UTR. Finally, Hgsnat, a RIDD target with natural clusters of rare codons (one at the 5′ end of the CDS and one immediately upstream of the Xbp1-like SL), was completely insensitive to Perk depletion, suggesting that it is normally translated at a low enough level to allow for RIDD in the absence of further translational attenuation (Figure 7A). Artificially stalling translation of RIDD reporter mRNAs extended these observations, as mRNAs containing translation-stalling SLs, pseudoknots, or stop codons upstream of their Xbp1-like SLs were insensitive to Perk depletion (Figure 7A).
Our data support a model in which attenuation of translation in mammalian cells, mediated by Perk or by natural sequence elements, leads to the formation of an accessible Xbp1-like SL in a target mRNA, which is then cleaved by Ire1 to initiate degradation. When Perk is depleted and translation is allowed to proceed, ribosomes would be expected to disrupt the secondary structure of the Xbp1-like SL as they move through this region. This effect, combined with ribosome physical occupancy of the mRNA, would limit the ability of Ire1 to access and cleave the target mRNA. These same mechanisms may also apply to Xbp1, as Xbp1 splicing was reduced when Perk was depleted and rescued by the addition of the translation initiation inhibitor harringtonine (Figure 4H). Although Perk depletion did not broadly inhibit RIDD in Drosophila S2 cells, target mRNAs are likely sensitive to translation, as continued, high levels of translation during ER stress can protect certain transcripts from RIDD (Gaddam et al., 2013). In S2 cells, however, Xbp1-like SLs are not required for RIDD, and thus ribosomes may sterically hinder Ire1 access to the mRNA in a more general manner.
This coordination between Perk-mediated translation attenuation and Ire1 cleavage of mRNAs may tailor the UPR to specific types of stress (Figure 7B). UPR sensors are activated differentially under distinct forms of ER stress, and the involvement of Perk may limit RIDD to cases of ER stress in which both Ire1 and Perk are activated, such as hypoxia (Koumenis et al., 2002; Drogat et al., 2007). Furthermore, this requirement would ensure that inappropriate activation of the RIDD pathway does not occur in cases of stress in which only Ire1 is activated, such as plasma cell differentiation (Ma et al., 2010). In addition, Perk initiates a negative feedback loop via induction of the phosphatase GADD34, which dephosphorylates eIF2α and restores protein translation (Novoa et al., 2001; Ma and Hendershot, 2003). Through this mechanism, Perk may temporally limit robust degradation of RIDD targets.
Our data indicate that in mammals, RIDD is much more selective than in flies and suggest that the specific targeting of particular mRNAs is important for ER stress recovery. However, when Ire1 is overexpressed or otherwise hyperactivated, the requirements for both Perk and Xbp1-like SLs are lost (Han et al., 2009; So et al., 2012), suggesting that mammalian Ire1 is capable of a much broader specificity in certain circumstances (Figure 7B). We speculate that broad cleavage of mRNAs may occur in mammalian cells exposed to acute ER stress as well but to a small degree such that the steady-state levels for most mRNAs do not measurably change. This activity may be important in the local control of ER load or in the response to viruses, in which Rig-I has recently been shown to be activated by the products of RIDD (Cho et al., 2013).
MATERIALS AND METHODS
Cell culture/ER stress
We cultured MC3T3-E1 cells (American Type Culture Collection, Manassas, VA) in MEMα with nucleosides and no ascorbic acid (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) and antibiotics unless otherwise stated. Min6, Hek293, and Hep G2 cells (from J. Rutter, A. V. Mariqc, and C. H. Hagedorn, respectively, University of Utah) were cultured in DMEM (Invitrogen) supplemented with 10% FBS, antibiotics, and Gln. All cell lines were maintained at 37°C and 5% CO2. We added 1 or 2 mM DTT (Sigma-Aldrich, St. Louis, MO), 2 μM Tg (Sigma-Aldrich), or 2 μg/ml Tm (EMD Millipore, Billerica, MA) to cell media for 4 h to induce ER stress. For inhibition of transcription or translation elongation, we added 2 μg/ml ActD (Sigma-Aldrich) or 35 μM Chx (Sigma-Aldrich), respectively, to cells for ∼5 min before adding 1 mM DTT. ISRIB was a kind gift from the Peter Walter lab (University of California at San Francisco, San Francisco, CA). For ISRIB experiments, we added 500 nM ISRIB to cells for ∼5 min before adding DTT.
siRNA
For Ire1, Xbp1, and Perk siRNA experiments, we cultured cells in antibiotic-free media. We followed published RNAiMAX (Invitrogen) protocols to transfect cells with organism-specific siRNAs (Qiagen, Valencia, CA). The following siRNA reagents were used: Ire1 (SI00995890, 897, 904), Xbp1 (GS22433), Perk combined (GS1366-mouse or GS9451-human), Perk #1 (SI00991319), and Perk #3 (SI00991333). We controlled for the effects of the siRNA procedure by including Neg siRNA (Qiagen)–transfected samples in all experiments. We incubated cells for 48–72 h before replacing media and treating with or without ER stress. For harringtonine experiments, we added 1 μg/ml harringtonine (LKT Laboratories, St. Paul, MN) to media in Neg and Perk siRNA–treated cells and incubated for ∼5 min before the addition of DTT.
Plasmid reporter construction and transfection
For wild-type Blos1 and Hgsnat reporters, we amplified the Blos1 (UniGene ID Mm.30118) or Hgsnat (UniGene ID Mm.28326) CDS from MC3T3-E1 cell cDNA and subcloned downstream of the human EF-1α promoter. To create plasmids expressing our reporter mRNA and a hygromycin resistance gene, we then subcloned the promoter and CDS between the NruI and NotI sites of the pcDNA3.1-Hygro(+) vector (Invitrogen). For our GFP and ssGFP reporters, we subcloned the GFP or ssGFP sequences previously described (Gaddam et al., 2013) into our expression vector. To create GFP-SLUTR and ssGFP-SLUTR reporters, we added 33 nt from the 3′ end of the Blos1 CDS, which includes the Xbp1-like SL sequence, 15 nt downstream of the GFP or ssGFP CDS. We introduced site-directed mutations in reporters by fusion PCR and translation-stalling SLs (Vattem and Wek, 2004) or pseudoknots (Kontos et al., 2001) in reporters from Figures 5 and 6 by oligo cassette mutagenesis. We inserted pseudoknot sequences 15 nt upstream of the Blos1 SL.
For all reporters, we created polyclonal stable cell lines by transfecting 2 μg of plasmid into MC3T3-E1 cells using Lipofectamine 2000 (Invitrogen). We replaced media after 1.5–2 h and allowed cells to recover for an additional 24–36 h before passaging and adding 100 μg/ml hygromycin B. Hygromycin B–resistant cells were selected over a 2- to 3-wk period and cultured in 100 μg/ml hygromycin B thereafter. For reporter assays, cells were passaged into hygromycin-free media 48–72 h before treatment with or without 2 mM DTT for 4 h.
Digitonin fractionation
We used a modified procedure based on a protocol developed by Stephens et al. (2008) for separation of cytosolic and membrane-bound mRNAs. Briefly, we incubated MC3T3-E1 cells with 35 μM Chx for 10 min and then trypsinized and pelleted cells. We resuspended cells in cytosol buffer (150 mM KOAc, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5, 2.5 mM Mg(OAc)2, 200 U/ml RNaseOUT, 35 μM Chx). We immediately permeabilized cells with 1 mg/ml digitonin and incubated on ice for 15 min. Cell lysates were then centrifuged at 500 × g for 5 min, and supernatant was collected as the cytosolic fraction. We resuspended the pellet in cytosol buffer with 1% Triton X-100, and incubated and centrifuged as before. The supernatant was collected as the membrane-bound fraction.
mRNA isolation and analysis
We isolated mRNA using either TRIzol reagent (Invitrogen) or Zymo Research Quick-RNA MiniPrep kits. We then synthesized cDNA using 1–2 μg of total RNA as template, a T18 primer, and MMLV reverse transcriptase (NEB, Ipswich, MA). We measured relative mRNA levels by qPCR using the Masterplex ep realplex (Eppendorf, Hauppauge, NY) with SYBR green fluorescent dye. Each sample was measured in triplicate, and target mRNA levels were normalized to those of ribosomal protein 19 (Rpl19) mRNA. To ensure that signal was not due to contaminating plasmid or genomic DNA, we also measured mRNA levels from samples to which no reverse transcriptase was added.
For specifically detecting mRNA expressed from reporters, we used one primer designed to bind the reporter CDS and one primer designed to bind the 3′ or 5′ UTR derived from the vector. These primers did not amplify endogenous transcripts, which we tested using untransfected cells or cells transfected with control reporters (e.g., GFP).
We quantified Xbp1 splicing by amplifying cDNA with primers that surround the Xbp1 splice site and running the products on a 2% agarose gel. Relative band intensities for the spliced and unspliced products were quantified using ImageJ (National Institutes of Health, Bethesda, MD).
All primer sequences are shown in Table 1.
TABLE 1:
Primers used for qPCR and Xbp1 splicing measurements (5′ to 3′).
| RNA | Primer 1 | Primer 2 |
|---|---|---|
| mBlos1 (endogenous) | CAAGGAGCTGCAGGAGAAGA | CCAGGAGGGTGAAGTAAGAGG |
| mScara3 | TGCATGGATACTGACCCTGA | GCCGTGTTACCAGCTTCTTC |
| mCol6a1 | TGCTCAACATGAAGCAGACC | TTGAGGGAGAAAGCTCTGGA |
| mHgsnat (endogenous) | TCTCCGCTTTCTCCATTTTG | CGCATACACGTGGAAAGTCA |
| hBlos1 | CAAGGAGCTGCAGGAGAAGA | GCCTGGTTGAAGTTCTCCAC |
| hScara3 | AACTTCCTGCACACACTGGC | CAAACCAGTTGCACATCCAG |
| mXbp1 | AGAAGAGAACCACAAACTCCAG | GGGTCCAACTTGTCCAGAATGC |
| hXbp1 | AGCTCAGACTGCCAGAGATCG | AATCCATGGGGAGATGTTCTA |
| mHgsnat (reporter) | GGAACCCCCTTCTTCTATCC | GGAAAGGACAGTGGGAGTGG |
| mBlos (reporter) | CAAGGAGCTGCAGGAGAAGA | GGAAAGGACAGTGGGAGTGG |
| GFP | TAATACGACTCACTATAGGGAGA | TGCTCAGGTAGTGGTTGTC |
| mGapdh | TGAACGGGAAGCTCACTGG | GGTCCTCAGTGTAGCCCAAG |
| mBip | TCAGCATCAAGCAAGGATTG | AAGCCGTGGAGAAGATCTGA |
| mSdc4 | AACTGAGGTCTTGGCAGCTC | TCCCCAATAAGTCCAAGCAG |
| mCamI | GCGAGAAGAAGGTGAAGACG | TAAGTTCCTCGGGTTTCAGG |
| mTmem183a | CTCTTTGACTGGTGGCATCC | TTCAACCTTTCCACCTCCTG |
| mGalnt11 | ATGGCTCCTCCTCTCAACAG | CAGCAGCTCGGAAGTAAACC |
| mGorasp2 | CGAGAAGCCTGTGTCTGATG | CAGCCTCTTGCGTAGTTTCC |
| mSTT3 | CATCGTCCCCAAACAGAAGT | TGTACCCTTGGTGCTGTGAA |
| mCog2 | GGAGACGGTCAAGCAGAAAC | TATTGGTCCTGCGGTAAAGC |
| mYif1a | CCAAGGGAAGGACATAGTGC | TAGAGGTCAGGGGCATTGAG |
| mPlscr3 | GGCATCCCTTCCTTCCTAAG | CAAAGTCATCGGCATCTGTG |
| mSlc20a2 | GTTGCATCTTCCCATTGCTT | GACACCGAGTGGGACTTGAT |
| mBlos1 (endogenous plus reporter) | CCAGGCCTACATGAACCAGA | TAGACGTATTCCAGCGCAGT |
Supplementary Material
Acknowledgments
We thank C. Hagedorn, J. Rutter, and A. Maricq for gifts of cell lines, C. Sidrauski and P. Walter for ISRIB, and J. Lee, W. Morrison, R. Palu, and M. LaBella for discussions and critical reading of the manuscript.
Abbreviations used:
- ER
endoplasmic reticulum
- Ire1
inositol-requiring enzyme 1
- Perk
PKR-like endoplasmic reticulum kinase
- qPCR
quantitative real-time PCR
- RIDD
regulated Ire1-dependent decay
- SL
stem loop
- UPR
unfolded protein response
- Xbp1
X-box binding protein 1.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-02-0074) on June 24, 2015.
REFERENCES
- Bobrovnikova-Marjon E, Pytel D, Riese MJ, Vaites LP, Singh N, Koretzky GA, Witze ES, Diehl JA. PERK utilizes intrinsic lipid kinase activity to generate phosphatidic acid, mediate Akt activation, and promote adipocyte differentiation. Mol Cell Biol. 2012;32:2268–2278. doi: 10.1128/MCB.00063-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright MD, Itzhak DN, Wardell CP, Morgan GJ, Davies FE. Cleavage of BLOC1S1 mRNA by IRE1 is sequence specific, temporally separate from XBP1 splicing, and dispensable for cell viability under acute endoplasmic reticulum stress. Mol Cell Biol. 2015;35:2186–2202. doi: 10.1128/MCB.00013-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Cho JA, Lee AH, Platzer B, Cross BC, Gardner BM, De Luca H, Luong P, Harding HP, Glimcher LH, Walter P, et al. The unfolded protein response element IRE1alpha senses bacterial proteins invading the ER to activate RIG-I and innate immune signaling. Cell Host Microbe. 2013;13:558–569. doi: 10.1016/j.chom.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Clarke TF, Clark PL. Rare codons cluster. PLoS One. 2008;3:e3412. doi: 10.1371/journal.pone.0003412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho DS, Cairrão F, Zeng X, Pires E, Coelho AV, Ron D, Ryoo HD, Domingos PM. Xbp1-independent Ire1 signaling is required for photoreceptor differentiation and rhabdomere morphogenesis in Drosophila. Cell Rep. 2013;5:791–801. doi: 10.1016/j.celrep.2013.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drogat B, Auguste P, Nguyen DT, Bouchecareilh M, Pineau R, Nalbantoglu J, Kaufman RJ, Chevet E, Bikfalvi A, Moenner M. IRE1 signaling is essential for ischemia-induced vascular endothelial growth factor-A expression and contributes to angiogenesis and tumor growth in vivo. Cancer Res. 2007;67:6700–6707. doi: 10.1158/0008-5472.CAN-06-3235. [DOI] [PubMed] [Google Scholar]
- Gaddam D, Stevens N, Hollien J. Comparison of mRNA localization and regulation during endoplasmic reticulum stress in Drosophila cells. Mol Biol Cell. 2013;24:14–20. doi: 10.1091/mbc.E12-06-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- Hur KY, So JS, Ruda V, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Iwawaki T, Glimcher LH, Lee AH. IRE1alpha activation protects mice against acetaminophen-induced hepatotoxicity. J Exp Med. 2012;209:307–318. doi: 10.1084/jem.20111298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama H, Amagai Y, Ando H, Yamamoto S. Establishment of a clonal osteogenic cell line from newborn mouse calvaria. Jpn J Oral Biol. 1981:899–901. [Google Scholar]
- Kontos H, Napthine S, Brierley I. Ribosomal pausing at a frameshifter RNA pseudoknot is sensitive to reading phase but shows little correlation with frameshift efficiency. Mol Cell Biol. 2001;21:8657–8670. doi: 10.1128/MCB.21.24.8657-8670.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Korennykh AV, Behrman SL, Walter P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc Natl Acad Sci USA. 2010;107:16113–16118. doi: 10.1073/pnas.1010580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem. 2003;278:34864–34873. doi: 10.1074/jbc.M301107200. [DOI] [PubMed] [Google Scholar]
- Ma Y, Shimizu Y, Mann MJ, Jin Y, Hendershot LM. Plasma cell differentiation initiates a limited ER stress response by specifically suppressing the PERK-dependent branch of the unfolded protein response. Cell Stress Chaperones. 2010;15:281–293. doi: 10.1007/s12192-009-0142-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder M, Huang C, Snider MD, Komar AA, Tanaka J, Kaufman RJ, Krokowski D, Hatzoglou M. A novel feedback loop regulates the response to endoplasmic reticulum stress via the cooperation of cytoplasmic splicing and mRNA translation. Mol Cell Biol. 2012;32:992–1003. doi: 10.1128/MCB.06665-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KA, Hollien J. The unfolded protein response in secretory cell function. Annu Rev Genet. 2012;46:165–183. doi: 10.1146/annurev-genet-110711-155644. [DOI] [PubMed] [Google Scholar]
- Moore KA, Plant JJ, Gaddam D, Craft J, Hollien J. Regulation of sumo mRNA during endoplasmic reticulum stress. PLoS One. 2013;8:e75723. doi: 10.1371/journal.pone.0075723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi N, Pustylnik S, Harrison RE. Rab GTPase mediated procollagen trafficking in ascorbic acid stimulated osteoblasts. PLoS One. 2012;7:e46265. doi: 10.1371/journal.pone.0046265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B, Jacobson A. The intimate relationships of mRNA decay and translation. Trends Genet. 2013;29:691–699. doi: 10.1016/j.tig.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife. 2013;2:e00498. doi: 10.7554/eLife.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So JS, Hur KY, Tarrio M, Ruda V, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Lichtman AH, Iwawaki T, Glimcher LH, Lee AH. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012;16:487–499. doi: 10.1016/j.cmet.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens SB, Dodd RD, Lerner RS, Pyhtila BM, Nicchitta CV. Analysis of mRNA partitioning between the cytosol and endoplasmic reticulum compartments of mammalian cells. Methods Mol Biol. 2008;419:197–214. doi: 10.1007/978-1-59745-033-1_14. [DOI] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Walters R, Parker R. Quality control: is there quality control of localized mRNAs. J Cell Biol. 2014;204:863–868. doi: 10.1083/jcb.201401059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.