

The CREB3 transcription factors are a specialized set of regulatory proteins that are normally anchored in the endoplasmic reticulum and relay perturbation of function back to the nucleus. Phosphorylation and degradation modulate the response of one such factor (CREB-H), which is involved in secretion and metabolic homeostasis.
Abstract
CREB‑H, an endoplasmic reticulum–anchored transcription factor, plays a key role in regulating secretion and in metabolic and inflammatory pathways, but how its activity is modulated remains unclear. We examined processing of the nuclear active form and identified a motif around S87–S90 with homology to DSG-type phosphodegrons. We show that this region is subject to multiple phosphorylations, which regulate CREB-H stability by targeting it to the SCFFbw1a E3 ubiquitin ligase. Data from phosphatase treatment, use of phosophospecific antibody, and substitution of serine residues demonstrate phosphorylation of candidate serines in the region, with the core S87/S90 motif representing a critical determinant promoting proteasome-mediated degradation. Candidate kinases CKII and GSK-3b phosphorylate CREB-H in vitro with specificities for different serines. Prior phosphorylation with GSK-3 at one or more of the adjacent serines substantially increases S87/S90-dependent phosphorylation by CKII. In vivo expression of a dominant-negative Cul1 enhances steady-state levels of CREB‑H, an effect augmented by Fbw1a. CREB-H directly interacts with Fbw1a in a phosphorylation-dependent manner. Finally, mutations within the phosphodegron, when incorporated into the full-length protein, result in increased levels of constitutively cleaved nuclear protein and increased transcription and secretion of a key endogenous target gene, apolipoprotein A IV.
INTRODUCTION
The endoplasmic reticulum (ER) is a major cellular organelle system that regulates diverse aspects of cell metabolism and homeostasis, including protein synthesis and quality control, lipid and fatty acid synthesis, membrane incorporation, and constitutive and regulated secretion (McClellan et al., 2005; Thomas et al., 2010; Brodsky and Skach, 2011; Guerriero and Brodsky, 2012; Cnop et al., 2012). To maintain function and capacity, the ER possesses many integrated feedback systems that monitor diverse processes at the ER and regulate adaptive responses to alterations in the extracellular and intracellular environments. One such system involves intricate ER–nuclear signaling by which demand or perturbation is sensed at the ER via dedicated transmembrane transcription factors and relayed to the nucleus via processes including regulated intramembrane proteolysis (RIP). Examples include the sterol regulatory element-binding proteins (SREBPs), which control fatty acid and sterol homeostasis (Goldstein et al., 2006), and activating transcription factor 6 (ATF6), one of the components that regulate the unfolded protein response (Schroder and Kaufman, 2005; Ron and Walter, 2007). In both cases, these factors are retained in the ER by sensors of the particular pathways and, in response to fluctuation in those pathways, released from the ER, transported to the Golgi, and cleaved by resident enzymes S1P and S2P (Nohturfft et al., 1999; Ye et al., 2000; Yang et al., 2002; Shen et al., 2005). This releases the N-terminal products, which traffic to the nucleus to effect appropriate transcriptional responses.
The identification of the cAMP response element–binding protein 3 (CREB3) family of ER-anchored basic leucine zipper (bZip) transmembrane factors now extends the class of factors regulated by RIP and the spectrum of integrated systems involved in maintaining ER function (reviewed in Bailey and O'Hare, 2007). CREB3 factors are an ancient group, closely related to but distinct from ATF6 (Barbosa et al., 2013). Humans possess five related members: CREB3/Luman, CREB3L1/OASIS, CREB3L2/BBF2H7, CREB3L3/CREB‑H, and CREB3L4/CREB4/AIbZIP/ (Lu et al., 1997; Honma et al., 1999; Omori et al., 2001; Cao et al., 2002; Qi et al., 2002; Storlazzi et al., 2003; for ease of reference to previous work, the familiar names are used here rather than the systematic CREB3 nomenclature). Recent data from knockout animal models show that CREB3 proteins play critical roles in metabolic homeostasis, cell differentiation, and inflammation (Zhang et al., 2006, 2012; Murakami et al., 2009; Saito et al., 2009, 2012; Lee et al., 2010; Asada et al., 2012). They are expressed in a tissue-specific manner and control overlapping but distinct physiological responses involving the ER. For example, old astrocyte specifically induced substance (OASIS) is preferentially expressed in osteoblast and astrocytes, and OASIS-deficient mice exhibit a delay in osteoblast maturation and severe osteopenia (Murakami et al., 2009). OASIS plays additional roles, including in pancreatic beta cell secretion (Vellanki et al., 2010) and goblet cell differentiation in the large intestine (Asada et al., 2012). BBF2H7 is expressed in many cell types and plays a critical role in chondrocytes during chondrogenesis, inducing expression of targets, including Sec23a, a secretory coat protein involved in transport from the ER to the Golgi (Saito et al., 2009).
CREB-H is found mainly in cells of the liver and small intestine and has been shown to be a key factor in metabolic homeostasis, involved in maintaining normal levels of lipids and triglycerides (Lee et al., 2011; Lee, 2012; Zhang et al., 2012) and in hepatic glucose metabolism (Lee et al., 2010; Chanda et al., 2011). CREB-H target genes strongly overlap with those of OASIS and BBF2H7, including Sec23, Sec24, and extracellular matrix components, but also encompass distinct specific physiological targets in de novo lipogenesis, triglyceride and cholesterol biosynthesis, fatty acid elongation and oxidation, lipolysis, and lipid transport (Lee et al., 2011; Zhang et al., 2012; Barbosa et al., 2013; Xu et al., 2014). CREB-H promotes increased expression and secretion of specific cargoes, including apolipoproteins A I and A IV and phospholipase A 2. Lack of CREB-H expression in the liver results in elevated levels of fatty acids and triglycerides due to the failure to secrete these apolipoproteins and enzyme complexes, which otherwise help suppress plasma levels of these metabolites (Luebke-Wheeler et al., 2008; Lee et al., 2011; Lee, 2012; Zhang et al., 2012). Stimuli including saturated fatty acids, insulin, and an atherogenic high-fat diet promote CREB-H activation, leading to increased target gene transcription (Danno et al., 2010; Gentile et al., 2010; Lee et al., 2010, 2011; Zhang et al., 2012). Moreover, multiple nonsynonomous mutations in CREB3-H have been identified in humans with hypertriglyceridemia (Lee et al., 2011). Nevertheless, despite the recent demonstration of key physiological roles for CREB3 proteins, we have little mechanistic understanding of the signaling pathways involved or how their activities are controlled.
Protein stability represents a major determinant of transcription factor abundance and the magnitude and longevity of a signaling response. Here we examine features of the nuclear stability of one of the CREB3 members, CREB-H. Many transcription factors are short-lived proteins and are stabilized in response to specific signals (reviewed by Muratani and Tansey, 2003; Dhananjayan et al., 2005; Westermarck, 2010). For example, SREBP1 is regulated by multiple positive and negative phosphorylation events in response to metabolic cues. Phosphorylation by AKT promotes the release of SREBP–SCAP to the Golgi (Du et al., 2006; Yellaturu et al., 2009) and also promotes stabilization of the nuclear cleaved form, antagonizing GSK-3, which constitutively phosphorylates nuclear SREBP1, promoting its ubiquitination by a member of the Skp1/cullin/F-box proteins (SCF) class of E3 ubiquitin ligases and proteasomal degradation (Hirano et al., 2001, 2003; Sundqvist et al., 2005; Bengoechea-Alonso and Ericsson, 2009). Generally, phosphorylation-dependent recognition of substrate proteins by ubiquitin- conjugating complexes such as the SCF E3 ligase complexes and antagonism of that phosphorylation by further modification of either the substrate or the various kinases play a fundamental role in modulating the abundance and/or localization of transcription factors and thus the kinetics and duration of their activity (Muratani and Tansey, 2003; Dhananjayan et al., 2005). F-box proteins of the SCF complexes frequently bind their substrates at motifs termed phosphodegrons. The prototypical F-box factor, Fbw1a, binds the consensus degron Asp-Ser-Gly-Xaa-Xaa-Ser (in which Xaa is any amino acid and both Ser residues are phosphorylated; Pickart and Eddins, 2004; Frescas and Pagano, 2008; Skaar et al., 2013). Phosphodegrons can be phosphorylated by a single kinase in multiple residues or alternatively by multiple kinases that phosphorylate different residues or by use of priming kinases, which adds an increased complexity to substrate recognition by F-box proteins.
In this study, we examine degradation of nuclear CREB‑H and its interactions with the ubiquitin-proteasome system, providing insights into the regulatory mechanisms that modulate its transcriptional response in the nucleus. We identify a conserved motif, DSGIS, in the N-terminus of CREB‑H and propose that it acts as a phosphodegron, mediating SCF complex binding by the specific F-box factor βTrCP (Fbw1a) and promoting proteasome degradation of CREB‑H. We demonstrate that serine-to-alanine substitutions (DSGIS to DAGIA) within this motif significantly increase stability of the nuclear CREB‑H product and that expression of a dominant-negative mutant of Cul1 (the scaffold component of the SCF ubiquitin E3 ligase), particularly with coexpression of the βTrCP component, increases steady-state levels of CREB‑H. Sequential phosphorylation in vitro by GSK-3b and CKII kinases results in efficient DSG-dependent phosphorylation of CREB-H. Moreover, in vivo the addition of the drug CHIR99021, a potent inhibitor of GSK-3, substantially increases levels of nuclear CREB-H. Serine-to-alanine substitutions within this motif not only retained but had a pronounced stimulatory effect on CREB‑H functional activity in a transcription reporter assay, consistent with enhanced stabilization. Finally, we place these same serine substitutions in the context of full-length CREB-H and demonstrate increased transcription and secretion of a main endogenous physiological target, the apolipoprotein A IV (ApoA IV). These results are discussed in the overall context of CREB-H regulation and other family members in the coordination of signaling events during ER homeostatic responses.
RESULTS
Phosphorylation of CREB-H and identification of a conserved serine-rich motif
The cleaved form of CREB-H, termed CREB‑HΔTMC (see schematic in Figure 1a) represents the mature S2P-cleaved N-terminal product and is virtually exclusively located in the nucleus (Bailey et al., 2007). It migrates on SDS–PAGE gels as a series of bands that comigrate precisely with the major forms produced after brefeldin A–induced cleavage of the full-length ER-associated form (Figure 1b, lanes 1–3). The fastest-migrating form of CREB‑HΔTMC is labeled N throughout this work, and the main upper species combined are labeled N1. Additional minor, slower-migrating bands could also be discerned (Figure 1b, lane 1, small arrows), although their lower abundance and the resolving power of the gels limit their detection. We previously showed that the N species comigrates exactly with the primary in vitro–translated CREB‑HΔTMC product, whereas longer in vitro translation times result in the progressive appearance of N1. Consistent with previous results, we show that phosphatase treatment results in reduction of the N1 species and minor upper bands with concomitant increases in the N species, an effect prevented by the presence of phosphatase inhibitors (Figure 1c, lanes 1–9). We were able to observe stepwise reduction in N1 with increased amounts of species of intermediate mobility. These results indicate that N represents unmodified CREB‑HΔTMC, whereas the upper bands represent phosphorylation events, with the most abundant species N1, likely due to multiple phosphorylation events.
FIGURE 1:
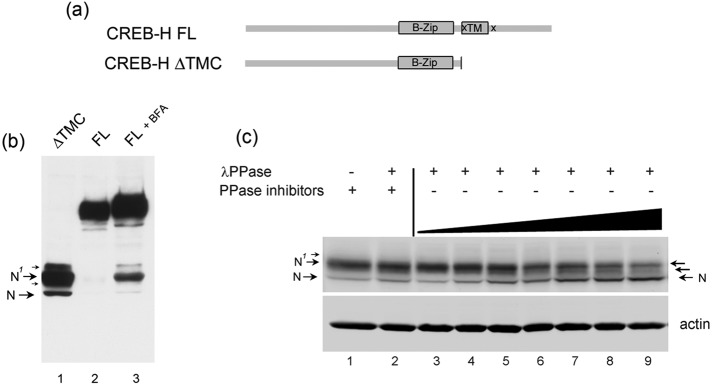
Multiple species of the N-terminal product of CREB-H due to phosphorylation. (a) Schematic indicating the conserved bZip and transmembrane (TM) domains, the S1P and S2P cleavage sites (x), and the mature cleaved product, termed CREB-HΔTMC. (b) Western blot showing CREB-HΔTMC after transfection in COS cells, migrating as multiple species (lane 1), which comigrate with those produced by brefeldin A (BFA)–induced cleavage of the full-length (FL) precursor form (lane 3), as discussed in the text. (c) Soluble extracts of cells expressing CREB-HΔTMC were treated with λ phosphatase for increasing times at 37°C (0, 2, 5, 7, 10, 15, 30 min; lanes 3–9) or as controls, without phosphatase (lane 1) or with phosphatase in the presence of phosphatase inhibitors (10 mM sodium orthovanadate and 20 mM NaF; lane 2). Phosphatase treatment converts the upper form, N1, to the N form, with species likely representing multiple phosphorylated forms (small arrows) closely migrating between N and N1.
Examination of the CREB-H sequence reveals >70 serine and threonine residues in the protein, although these are not well conserved in other CREB-H homologues and many have no similarity to consensus phosphorylation sites. This renders examination of potential phosphorylation sites difficult and analysis of every serine or threonine beyond the scope of this work. However, from comparative analysis, we identified a very highly conserved serine-rich motif in all CREB‑H homologues, consisting of a core 87-[DSGIS]-90 amino acid sequence with good conservation also in serine-rich flanking residues (Figure 2a, black bar indicates relative position; expanded sequence, Figure 2b). For CREB-H, the central DSG motif is highly conserved across diverse species (Figure 2b) and, apart from the bZip domain itself, is the most conserved feature within the N-terminus of the protein. Of note, in terms of phosphorylation and protein stability, this region fits the consensus (DSGX(1-5)S) for one of several types of phosphodegron that target substrates for proteasome-mediated degradation and indeed is very similar to the known phosphodegron in cdc25A (Figure 2c; Fuchs et al., 2004; Skaar et al., 2013).
FIGURE 2:

A conserved putative phosphodegron motif within the N-terminal domain. (a) Schematic of CREB-H with a short, solid bar indicating the relative position of the candidate motif, with sequence expanded below. (b) Sequence alignment of the putative phosphodegron motif in CREB‑H homologues from various species. White text on a black background indicates complete identity, allowing for S/T equivalence. White text on gray and black text on gray indicate progressively decreasing conservation. (c) Conservation of the putative DSG phosphodegron motif with that in the known phosphodegron of cdc25a. Asterisks indicate S/Ts that have been demonstrated to be involved in cdc25a phosphorylation and SCFFbw1a-mediated degradation. (d) Cells transfected with wt or the DSG mutant were treated −/+ cycloheximide 100 μg/ml (+Cyclo) with or without MG132 (10 μM) and samples harvested at the intervals indicated (minutes). Samples were analyzed as described in Materials and Methods. (e) Summary of the S → A substitutions made within the DSG region (DSG), the flanking serine-rich region (3S), or the two regions combined. For clarity, the term CREB-HΔTMC has been omitted, and the parental protein is termed wt. (f) Equivalent amounts (1 μg) of isogenic expression plasmids for each variant were transfected and total extracts analyzed by SDS–PAGE and Western blotting. Arrows indicate putative phosphorylated (N1) and unmodified (N) species as discussed in the text. (g) Relative steady-state levels of each of the mutant proteins calculated from quantitative analysis after Western blotting and detection by fluorochrome-coupled secondary antibodies using the LI-COR Odyssey system as described in Materials and Methods with wt levels set to 1. Actin used as a loading control (not shown) demonstrates that equal cell equivalents were analyzed. (h) Immunofluorescence analysis showing virtually exclusive nuclear localization of the wt and mutant variants with increasing number of cells or intensity per cell of the mutant species, consistent with the more quantitative conclusion from Western blotting.
Serines in the DSG motif promote proteasome-mediated degradation
We previously showed that CREB-HΔTMC was rapidly lost from cells when new protein synthesis was blocked by cycloheximide (Bailey et al., 2007). (For clarity and simplicity in nomenclature when comparing different mutants, the parental CREB-HΔTMC is termed wt throughout this work.) Here we show that this turnover is substantially prevented by treatment with the proteasome inhibitor MG132 (Figure 2d, compare wt, +cycloheximide −/+ MG132). The motif of the type described in the foregoing serves in many proteins as a phosphorylation-dependent targeting motif (DSGX(1-5)S, where S denotes phosphoserine) for interaction with E3 ubiquitin ligases of the SCF type, thus regulating proteasome-mediated degradation (Wu et al., 2003; Cardozo and Pagano, 2004; Fuchs et al., 2004; Skaar et al., 2013).
To examine whether serines within the CREB-H motif were in any way involved in promoting turnover, we simultaneously mutated both serines 87 and 90 to alanine in the mutant CREB-HΔTMC.DSG (termed DSG here for simplicity; Figure 2e). The result was clear, with a significant increase in the steady-state levels of the DSG mutant compared with the wt (Figure 2f, lanes 1 and 3; see also later discussions of Figures 3–9). This was accompanied by a slight upward shift of the N1 species and increased amounts of minor, more slowly migrating species, although these latter effects were more difficult to discern. Moreover, compared with the wt, the DSG mutant exhibited a significantly extended half-life in the presence of cycloheximide and a correspondingly reduced response to proteasome inhibition, since it was already comparatively stable (Figure 2d, compare wt vs. DSG +cycloheximide; also Figure 3).
FIGURE 3:

Stabilization of CREB-H after mutation of the phosphodegron region. (a) Cells transfected with wt or mutants were treated −/+ cycloheximide 100 μg/ml (+Cyclo), samples were harvested at the intervals indicated (minutes) and then analyzed by Western blotting, and detection used fluorochrome-coupled secondary antibodies as described in Materials and Methods. (b) Relative levels of each of the mutant proteins were calculated. For each mutant, the abundance at the beginning of treatment is normalized to 100. Quantitative estimates of half-lives were obtained using GraphPad Prism software with curve fitting and one-phase decay parameters.
FIGURE 4:

Phosphorylation of CREB-HΔTMC on serines 73 and 81. (a) Amino acid sequence of peptides used to generate phosphospecific antibody to CREB-HΔTMC species that are phosphorylated at serine 73 or 81. (b) Western blotting of wt protein without (lane 2) and with (lane 3) phosphatase treatment (λ phosphatase 400 U, 1 h). Phosphatase treatment in the presence of phosphatase inhibitors (10 mM sodium orthovanadate) is shown in lane 4. Untransfected samples are in lane 1. Detection was performed simultaneously on the same blot. Each channel was separated out for detailed analysis and merged in the lower panel to allow relative ratios to be visualized. (c) Western blotting of cells expressing each of the mutants as indicated using anti-SV5 epitope antibody to detect the total levels of the proteins and anti-pS73 to detect the phosphorylated forms. An asterisk indicates background bands detected with the anti-pS73 antibody. Whereas the anti-pS73 antibody detected multiple species of the wt protein, these showed a shift in relative levels from the species detected by SV5. Of greater importance, the anti-pS73 antibody completely failed to detect the mutant with a single S → A change at position 73, despite similar levels of the protein. Other conclusions are as discussed in the text. (d) Identical to c, but now using anti-pS81 antibody and anti-SV5.
FIGURE 5:

CREB-HΔTMC phosphorylation by CKII and GSK-3 on distinct motifs within and adjacent to the DSG region, respectively. (a) Equal amounts of purified GST, GST-wt, or GST-DSG protein bound to glutathione-agarose beads were incubated with purified GSK-3b or CKII and analyzed by SDS–PAGE and autoradiography. The amount of proteins in the reaction is shown by total protein staining (CBB), and the phosphorylated species is shown for each kinase. The GST control migrates at a lower position but has been transposed for ease of direct comparison. (b) Replicate equivalent samples of the GST-wt protein were incubated in vitro without or with a primary kinase as indicated (1st) and the beads washed to remove the kinase and exchange buffers and then incubated without or with a second kinase (2nd). The different combinations and order are indicated. Samples were then analyzed by SDS–PAGE and autoradiography. (c) Equal amounts of the GST-wt (lanes 1, 3, 5, and 7) and GST-DSG mutant (lanes 2, 4, 6, and 8), as in a, were incubated with the various combinations of primary and secondary kinases as indicated. (d) Equal amounts of purified GST, GST-3Sd, GST-DSG, or GST-3Sd+DSG protein were incubated with purified GSK-3 or CKII and subsequently analyzed by SDS–PAGE and autoradiography. The equalized amount of the proteins in the reaction is shown by total protein staining (CBB), and the phosphorylated species is shown for each kinase and mutant as indicated.
FIGURE 6:
CREB‑H is stabilized by dominant-negative Cul1. (a) CREB‑HΔTMC wt was expressed alone (lane 1) or coexpressed with Cul1 DN (Flag-tagged dominant-negative Cul1; lane 2), Fbw1a (Flag-tagged Fbw1a; lane 3), or a combination of the two (lane 4). Lysates were harvested and analyzed with anti-SV5 antibody to detect CREB‑H (top) or anti-Flag antibody to detect Cul1 DN and Fbw1a (bottom). (b) As for a, but now comparing the effects of Cul1 DN and Fbw1a on wt (lanes 1–3) vs. the DSG mutant (lanes 4–6).
FIGURE 7:
Direct phosphorylation-dependent interaction between CREB-H and Fbw1a. Cells were transfected with Flag-tagged Fbw1a and soluble extracts made. Equal amounts of purified GST, GST-wt, or GST-DSG protein bound to glutathione-agarose beads were incubated without (–) or with (+) purified GSK-3 and CKII for 30 min before the addition of the Fbw1a- transfected soluble extracts. After further incubation, the beads were isolated, washed extensively, and then analyzed by SDS–PAGE and Western blotting for Fbw1a and for GST fusion protein in the pull-down (Fbw1a and GST panels). The input sample (Inp) represents 1/50 of the total input, and 1/5 of the pull-down material was analyzed in each case. Equal amounts of total GST-fusion proteins were incubated in each case and present in the pull-down as detected by anti-GST antibody (bottom GST panel). Specific interaction with wt was observed but only after phosphorylation by GSK-3 and CKII. In parallel, no significant interaction was observed for the GST control or for GST-DSG.
FIGURE 8:

The GSK-3 inhibitor CHIR99021 increases the abundance of CREB-HΔTMC. (a) Cells transfected with the vector for CREB-HΔTMC under the control of the TK promoter were untreated or treated for 1 h (lanes 1–3) or 2 h (lanes 4 and 5) with 20 mM of NaCl or LiCl. (b) Cells were transfected with the vector for CREB-HΔTMC under the control of the CMV promoter and treated with either NaCl or LiCl for 1 h. Equal cell equivalents were analyzed for levels of CREB-HΔTMC using anti-SV5 or anti-pS73 antibodies. (c) Transfected cells were incubated in reduced serum (5% NBCS) 16 h before treatment with CHIR99021 (10 μM), LY294002 (10 μM), SB216763 (20 μM), TBCA (10 μM), or rapamycin (1 μM) for 24 h. Equal cell equivalents were analyzed for expression levels of CREB-HΔTMC using anti-SV5 antibody. (d) Transfected cells were incubated and treated as before with CHIR99021 (5, 10 μM) and LY294002 (5, 10 μM). Extracts were analyzed for total levels of CREB-HΔTMC using the anti-SV5 or for the pS73-phosphorylated form as before.
FIGURE 9:

CREB‑H phosphodegron mutants display enhanced functional activity on the apolipoprotein A IV target promoter. (a) Cells were cotransfected in triplicate with the apolipoprotein A IV–Luc target vector without or with 10 ng of the expression vectors for each of the CREB-HΔTMC protein as indicated. Samples were subsequently processed for luciferase activity. Results are plotted as absolute activity compared with reporter alone control, which was transfected with equal amount of pcDNA3 DNA. (b) Parallel samples from cells transfected with 100 ng of the corresponding vectors were analyzed for expression levels of each of the CREB-H variants. (c) Each of the mutants was incorporated into full-length proteins in isogenic expression vectors, which were transfected into HepG2 cells. The cells were solubilized and the cleaved nuclear product analyzed by Western blotting using fluorochrome-coupled secondary antibodies. Anti-actin was probed in parallel as a loading control. The N and N1 species detected were entirely consistent with the levels observed from the independent cleaved product. (d) Cells transfected with the wild type or mutants as indicated were harvested and RNA prepared and analyzed for control marker RNAs (TBP and actin), CREB-H RNA, and apolipoprotein A IV RNA. Absolute levels of apolipoprotein A IV RNA were normalized for the endogenous marker TBP and for CREB-H transcript levels. (e) In further analyses, the medium from similarly transfected cells was harvested, precipitated, and analyzed for total levels of secreted apolipoprotein A IV as described in Materials and Methods.
Other serines flanking the DSG motif also exhibited significant conservation in CREB-H homologues, notably serines 73, 77, and 81 (Figure 2b). To examine these also, we initially constructed individual substitutions at each serine but with only modest effects, although with some effect on mobility (see, e.g., later discussion of Figure 4c). We therefore constructed mutants in which all three serines were simultaneously substituted to alanine either alone (3S) or in combination with the DSG mutations (3S+DSG). A similar mutant in the 3S region was constructed inserting two alanines in place of the sequence for S73–S81 (termed 3Sd). Identical results were obtained for 3S and 3Sd, which was used for certain experiments in localization studies and in vitro phosphorylation (see later discussion). A summary of these changes and mutant names is given in Figure 2e.
There was a clear downward shift in migration of the 3S mutant, with the major population now migrating in a way equivalent to the N species, although still with a population of slower-migrating species (Figure 2f, lanes 1 and 4). There was also a consistent but relatively minor increase in the total abundance of the 3S mutant, although considerably less than that for DSG. For the 3S+DSG mutant, again there was a significant shift in migration, with this mutant migrating similarly to the 3S alone but now with even more increased abundance (Figure 2f, lanes 1, 2, and 4). A summary of the total steady-state levels observed in experiments throughout the course of this work is given in Figure 2g, showing increases of >10-fold for 3S+DSG, significant increases of five- to sixfold for DSG and a modest 1- to twofold for 3S. Furthermore, to estimate relative stability, we analyzed each of the wt and mutant proteins for abundance after cycloheximide treatment by quantitative Western blotting using fluorescent secondary antibodies and laser scanning densitometry (Figure 3, a and b). The results are consistent with the steady-state levels, showing an increase in half-life of the 3S mutant (83 min) over the wt (47 min) and more significant increases for the DSG (102 min) and 3S+DSG mutants (167 min). In addition, in localization studies, we found virtually exclusively nuclear accumulation for the wt protein and increased cell numbers and/or intensity for each of the mutants, with the greatest increase being for the combined mutant 3Sd+DSG (Figure 2h). The results were consistent with the more quantitative assessment obtained from the Western blot analyses.
The shift in migration of the 3S mutant (and the 3S+DSG mutant) to yield a more abundant species comigrating with the N species (i.e., equivalent to dephosphorylated wt) is consistent with phosphorylation at one or more of the mutated serines. Indeed S73, when individually mutated to alanine, showed a mobility shift, although not as great as the combined 3S mutant (Figure 4c, lanes 2 and 3). The 3S mutant had only a minor effect on overall abundance but did so both in the wt context and when combined with the DSG mutant. However, the DSG mutant by itself clearly had a very pronounced stabilizing effect in the context of both wt protein (Figure 2f, lanes 1 and 3) and the 3S mutant (lanes 2 and 4). Because the majority of 3S+DGS mutant species migrated above the unmodified N species, other phosphorylation sites apart from the five serines mutated in 3S+DGS are likely to occur. Consistent with this, phosphatase treatment of 3S+DSG further increased its migration to a form more completely comigrating with the N species (unpublished data).
To support the proposal for phosphorylation within this region, we synthesized a series of peptides containing phosphoserine in place of serine at the appropriate position in the attempt to generate antibodies specific for pS73, pS81, and pS87 (summarized in Figure 4a). In the event, the peptide for pS87 failed to generate any antibody that detected CREB-H, nor did other peptides designed for the S87–S90 region. However, the antibodies for both peptides at S73 and S81 were reactive with results that strongly supported the proposal for phosphorylation at these residues, as described later.
Anti-SV5 is a mouse monoclonal antibody, and the anti-peptide antibodies are rabbit, allowing simultaneous detection of the various species on the same blots using fluorochrome-labeled secondary antibodies. This facilitated conclusions about phosphorylation of the mutant species that would be difficult to obtain from standard chemiluminescence assays. CREB-HΔTMC was expressed and analyzed untreated or after treatment with λ phosphatase on a single membrane using anti-SV5 antibody to detect the total population (green channel) and in parallel with anti-pS73 antibody (red channel; Figure 4b). As described earlier, we detected the lower species N and upper doublet N1 and closely migrating bands (lanes 2, SV5, and merged). The anti-pS73 antibody selectively detected the upper species N1 with virtually no detection of the N species (lane 2, pS73). Furthermore upon phosphatase treatment, we saw the loss of the N1 species and a corresponding increase in the N species when detected by the SV5 antibody (lane 3, SV5) but almost complete loss of all species and virtually no detection of the increased N species when using the anti-pS73 antibody (lane 3, pS73; quantitation in Supplemental Figure S1a). The conversion from N1 to N was prevented by inclusion of phosphatase inhibitors (lane 4, SV5). Of interest, we noticed a slight shift in ratio of detection in the untreated sample (lanes 2) within the upper N1 species by the anti-pS73 antibody, which reacted slightly preferentially against the lower part of this doublet compared with SV5 antibody, better observed in the merged image showing yellow and green in this region (lane 2). Although this shift in ratio of the upper species is not key to the conclusions here, the observation adds weight to our interpretations (see later description and Discussion). What is clear from these data is that the anti-pS73 antibody detects the upper modified form of the protein, and this detection is completely lost upon phosphatase treatment. Combined with the mobility shift detected by SV5 upon phosphatase treatment and the mobility shift upon mutation, the data provide strong evidence for phosphorylation at S73. We repeated these phosphatase experiments for the anti-pS81 antibody with similar results (unpublished data; see also Figure 4, c and d).
To supply additional evidence that the anti-pS73 and anti-pS81 antibodies showed strong preference for the phosphorylated form of the protein, we expressed wt CREB-HΔTMC or single-substitution mutants S73A or S81A and used the anti-epitope tag antibody (SV5) or the anti-pS73 or anti-pS81 antibodies to detect phosphorylated species. Mutation of the single S73A increased mobility detectably (Figure 4c, SV5 lanes 2 and 3), although with no reproducible effect on abundance. In this blot, the anti-pS73 antibody detection of multiple upper species for the wt protein could be more easily seen, with a slightly different ratio of the closely migrating N1 species for the two antibodies (lane 2, single and merged channels). The single substitution of S73A substantially reduced (<10% remaining) detection by anti-pS73 (lanes 2 and 3) while having no or little effect on the S81A mutant (lane 4). Quantitative analysis of the relative detection of the wt and mutants is shown in Supplemental Figure S1b. Our results indicate that the major reactivity of the anti-pS73 antibody is against CREB-H species phosphorylated on S73.
We performed a similar analysis for the anti-pS81 antibody (Figure 4d). The S81A mutation had little effect on migration rate or overall abundance (SV5, lanes 2 and 4). The anti-pS81 antibody again detected the wt protein, while there was highly reduced detection of the S81A mutant (anti-pS81, lanes 2 and 4). There was residual detection by the anti-pS81 antibody for the S81A mutant, indicating a minor cross-reactivity to nonphosphorylated species. Nonetheless the results are again consistent with the anti-pS81 detecting mainly phosphorylated species. Noted that this antibody also failed to detect the S73A single mutant (lanes 2 and 3). One possible interpretation of this result is that phosphorylation on S81 is itself dependent on S73, possibly as a priming phosphorylated site. This proposal is consistent with results of the following sections examining candidate kinases that phosphorylate CREB-H.
Although we could not confirm phosphorylation at S87 and S90 by this route, results in the following section are also consistent with the proposal that S87/S90 is a site of phosphorylation, targeting CREB-H for degradation.
Phosphorylation at S87/90 by CKII and enhancement by GSK-3
For many substrates with DSG-type motifs, phosphorylation has been shown to be mediated by GSK-3. Phosphosite prediction algorithms indicated that the DSG site, in addition to possible GSK-3 phosphorylation, conformed to the consensus for CKII phosphorylation but did not have a good fit for any other kinase. To identify possible kinases, we first examined whether the DSG could act as a site for either GSK-3 or CKII phosphorylation in vitro. Fusion proteins consisting of the wt or DSG mutant linked to glutathione S-transferase (GST) were expressed and purified as described in Materials and Methods and subjected to in vitro phosphorylation with either GSK-3 or CKII and 32P-labeled γ-ATP (Figure 5). Equal amounts of the two fusion proteins or GST control were incubated (Figure 5a, CBB, total protein stain) and the products subjected to autoradiography (Figure 5a, GSK-3). Whereas the wt protein was phosphorylated by GSK-3, contrary to our expectation, we detected no difference in relative efficiency with the DSG mutant (GSK-3, lanes 1 and 2). This result indicated that although there was GSK-mediated phosphorylation within CREB-H, it resided at some different site. However, in parallel with CKII, whereas the wt protein was again phosphorylated, we observed a significant reduction when S87/90 were mutated, although with residual phosphorylation still occurring (CKII, lanes 1 and 2). Consistent with the fit to the consensus, the results are indicative of S87 and/or S90 being a direct CKII target site.
Because many of the phosphorylated sites that target substrates for degradation are themselves subject to priming at additional sites (Doble and Woodgett, 2003; Cardozo and Pagano, 2004; Skaar et al., 2013), we next examined whether prior phosphorylation with one of these two kinases would stimulate phosphorylation by the other. The GST-wt fusion protein was incubated with one kinase (or in controls, without the primary kinase), and after washing to remove the first kinase and exchange the buffer, the second kinase was added (or again as control, the second kinase omitted). The results demonstrated a very pronounced increase in overall phosphorylation when the wt protein was phosphorylated by GSK-3 before incubation with CKII compared with either kinase alone (Figure 5b, lanes 1, 2, and 4). This pronounced increase in wt phosphorylation was not observed when phosphorylation order was reversed, that is, CKII before phosphorylation by GSK-3, this combination being not significantly different from CKII alone (lanes 4 and 5). In further controls, no increase was observed with a different kinase, CKI, as the secondary kinase after GSK-3, with no difference observed compared with phosphorylation by CKI alone (lanes 1–3 and 7). Together these results indicate that the DSG motif is not itself likely to be a direct substrate for GSK-3, it can be phosphorylated by CKII, and prior GSK-3 phosphorylation elsewhere in the protein substantially increases phosphorylation by CKII, presumably at the DSG motif itself.
To test this proposal further, we performed sequential phosphorylation, comparing wt to the DSG mutant (Figure 5c). As shown earlier, the DSG mutant was phosphorylated with similar efficiency to wt by GSK-3 alone (lanes 7 and 8). In contrast, when GSK-3 phosphorylation was followed by CKII phosphorylation, in contrast to wt (lanes 3 and 7), the DSG mutant no longer showed any significant increase in phosphorylation (lanes 4 and 8). The difference in phosphorylation of the DSG mutant compared the wt by the combined kinase regime was now very pronounced (lanes 3 and 4). These results indicate that the enhanced phosphorylation seen when GSK-3 primes before CKII is due to subsequent CKII phosphorylation at the DSG site.
We next tested whether the flanking serines within the 3S region could be relevant sites of phosphorylation by GSK-3. In this case, we used the mutant 3Sd, which behaves identically to 3S but substitutes two alanines in place of S73–S81. The 3Sd mutant was fused to GST either alone or again in combination with the DSG mutation and tested for GSK-3 and CKII phosphorylation. Using equalized amounts of substrate protein (Figure 5d, CBB, total protein stain) we observed that the 3Sd mutant exhibited substantially reduced phosphorylation by GSK-3 (lanes 1 and 2; also for the combined mutant, lanes 1 and 4), whereas no change was observed for CKII phosphorylation (lanes 5 and 6). In contrast in parallel assays, the DSG mutant showed no difference compared with wt when phosphorylated by GSK-3 (lanes 1 and 3) while showing again a significant reduction when phosphorylated by CKII (lanes 5, 7, and 8). These results provide evidence for specificity in phosphorylation by each kinase.
Involvement of the SCF ubiquitin ligase complex in CREB‑H steady-state levels
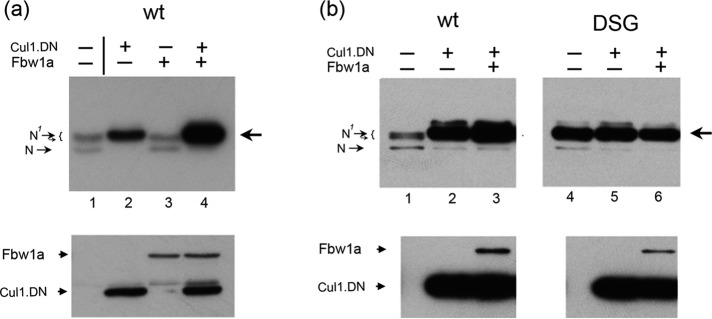
Considering that the phosphodegrons of the type identified generally target the substrate protein for SCF-mediated degradation, we next sought to address experimentally a role for the SCF complex in CREB-H turnover. Our initial attempts using small interfering RNA approaches to one of the components Cul1 (the main cullin adaptor of the SCF class of complexes) were not successful, and we achieved only modest reductions in Cul1 levels. However, another very useful strategy to confirm SCF complex involvement in degradation uses the expression of a Cul1 dominant-negative (Cul1 DN) construct that lacks the E2-binding domain and therefore affects half-life or steady-state levels when coexpressed with a respective target, either alone or in conjunction with the relevant F-box factor (Jin et al., 2005; Sundqvist et al., 2005). We therefore used the Cul1 DN strategy together with Fbw1a, the F-box shown mostly to target DSG-type motifs, including cdc25a, to examine involvement of the SCF E3 ubiquitin ligase complex in regulating the stability of CREB‑H (Figure 6).
In the absence of Cul1 DN, CREB‑HΔTMC wt was expressed at low levels, with a typical doublet pattern, as discussed earlier (Figure 6a, lane 1). On coexpression of the Cul1 DN, wt levels were significantly enhanced (lanes 1 and 2). Whereas Fbw1a alone had little effect, cotransfection of Cul1 DN with Fbw1a had an enhanced cumulative effect on steady-state levels of wt compared with Cul1 DN alone (lanes 1–4). Over the course of this work, when the independent Cul1 DN enhancement was pronounced, the additive effect of Fbw1a was generally less. The enhanced levels of wt protein were pronounced and specific, as Cul1 DN had little effect on the levels of cotransfected Fbw1a (Figure 6a, bottom; see also Figure 6b). Of interest, we also noted that the stabilized form of CREB-H had shifted predominantly to the upper N1 species, a result consistent with blocking degradation of the phosphorylated form (see Discussion). The fact that Fbw1a alone had little significant effect on the steady-state level of CREB‑H indicated that it is not a limiting factor for degradation of CREB‑HΔTMC wt, whereas the augmented effect with the Cul1 DN is consistent with previous results that its expression increases the effective abundance of the DN complex. Controls demonstrated that the levels of expression of the Cul1 DN and Fbw1a had no effect on one another (Figure 6a, bottom).
These results provide strong support for the proposal that CREB‑H is being degraded by the SCF E3 ligase complex. To further examine the specificity of the effect of dominant-negative Cul1 and whether the activity involved recognition of phosphorylated serine residues on the DSG motif, we next performed a parallel comparison of the effect of Cul1 DN either alone or in conjunction with Fbw1a on the wt and DSG mutant (Figure 6b). As described earlier, the wt protein levels were significantly elevated by the coexpression of the Cul1 DN mutant, with a modest but still enhanced cumulative effect by the additional presence of Fbw1a (Figure 6b, lanes 1–3). In parallel assays for the DSG mutant, enhanced steady-state levels of expression were already observed in the absence of any coexpression (Figure 6b, lanes 1 and 4), as described earlier. In contrast, the DSG mutant showed no significant increase in steady-state levels in the presence of Cul1 DN whether alone or in combination with Fbw1a (Figure 6b, lanes 4–6). These results demonstrate specificity in stabilization by Cul1 DN and that the effect of SCF/Cul1 in destabilizing the wt protein involves the DSG motif.
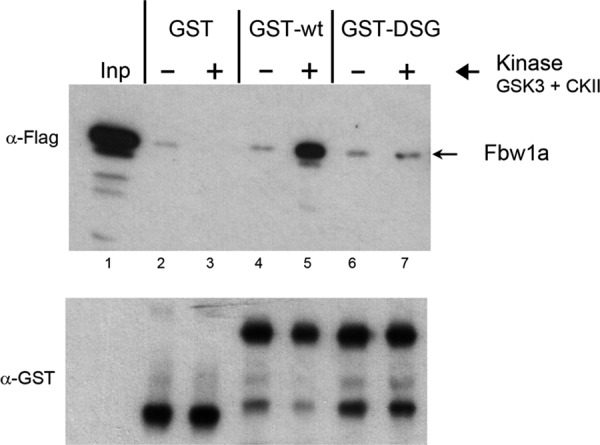
To expand on these data, we next examined possible direct interaction between CREB-H and Fbw1a. We incubated soluble extracts from cells expressing flag-tagged Fbw1a with equal amounts of the GST-wt or GST-DSG proteins or GST alone as a control. Furthermore, we analyzed interaction before or after in vitro phosphorylation by the combined action of GSK-3 and CKII (Figure 7, indicated by −/+ kinase). Specific interaction with wt was observed, but only after phosphorylation by GSK-3 and CKII (lanes 4 and 5). In parallel, no interaction was observed for the GST control, and, of importance, no significant interaction was observed with GST-DSG. Although it could formally be proposed that the interaction was indirect, via some adaptor protein binding specifically to GST-wt in a phosphorylation-dependent manner and this adaptor then binding Fbw1a, this seems unlikely and has not been tested further. In addition, although beyond the scope of the present work, it is formally possible that other F-box proteins could also be involved, for example, via distinct degrons (see Discussion). These data provide strong supporting evidence that CREB-H is processed through the ubiquitin-proteasome pathway, mediated through SCFFbw1a activity, with direct interaction of CREB-H by Fbw1a, dependent on the DSGIS motif in a phosphorylation-dependent manner involving GSK-3 and CKII.
Increased abundance of CREB-HΔTMC after inhibition of GSK-3
To provide further evidence that the abundance of CREB-HΔTMC is regulated by GSK-3, we tested the effect of various inhibitors on the overall abundance of protein (Figure 8), with quantitative analysis of various experiments shown in Supplemental Figure S2. We first tested the effects of the GSK inhibitor LiCl on the expression levels of CREB-H from the relatively weak thymidine kinase (TK) promoter (Figure 8a). Treatments for either 1 h (lanes 1–3) or 2 h (lanes 4–6) resulted in significant increases of approximately threefold in CREB-H over those seen in untreated cells or cells treated with the control (NaCl). It would be predicted that the relative levels of phosphorylated CREB-H would decrease accompanying the increase in protein abundance. However, in these experiments with relatively weak expression from the TK promoter, levels of detection with the phosphospecific antibodies were too low for easy detection. We repeated these experiments using the stronger cytomegalovirus (CMV) promoter (Figure 8b), and in this case, whereas the increase in total abundance was slightly lower, we could now analyze pS73 phosphorylation (lanes 3 and 4) in parallel. In this case, whereas S73 was still phosphorylated in the presence of the inhibitor, we observed a decline in relative ratio of the phosphorylated species to the total levels of protein (see also Supplemental Figure S2b). Moreover, LiCl treatment was accompanied by appearance of a more rapidly migrating species within the upper N1 species (SV5, arrow, lane 2), which was not detected by the phosphospecific antibody (pS73, lane 4). This result is consistent with the increased abundance but lower phosphorylation status of the N1 bands.
To confirm these results, we next compared a series of inhibitors, including the more recently developed and more specific GSK inhibitor CHIR99021 (Figure 8c). Of the series of drugs tested, we observed a very pronounced increase in abundance in the presence of CHIR99021 (Figure 8c, lanes 1 and 2). Consistent with the more potent and specific inhibition by CHIR99021, the increase in protein levels was considerably more pronounced than in our general observations with lithium chloride. Moreover, in the presence of the inhibitor within the slower-migrating N1 species, we again observed the appearance of a more rapidly migrating species (arrow, lane 2) indicating increased abundance but lower relative phosphorylation status of the N1 bands. A more modest effect was observed with an independent GSK-3 inhibitor SB16763 (SB, lane 4), but we consistently observed a very pronounced effect with CHIR99021 in numerous independent experiments. Over the course of this work, CHIR resulted in ∼2.5- to 6-fold increases in CREB-H abundance (see also Supplemental Figure S2, a, c, and d). Furthermore, we found a modest but detectable decrease in the CREB-HΔTMC protein steady-state levels when cells were incubated with the phosphatidylinositol-3 (PI3) kinase inhibitor LY29400 (LY, lane 3). This result is consistent with the known inhibitory effect of PI3 kinase on GSK-3b by phosphorylation of the Ser-9 residue (Cross et al., 1995). No significant effect was observed with rapamycin (lane 6), whereas a modest increase in the steady-state protein levels was seen with tetrabromocinnamic acid (TBCA), an inhibitor of CKII.
Considering the phosphospecific antibody to pS73, one of the candidate sites, we therefore next compared the effect of CHIR and LY (as a control) on the total abundance of the protein (assessed by the epitope tag) and on pS73 (assessed by the phosphospecific antibody), analyzed in identical samples on the same blot simultaneously. The results (Figure 8d) confirmed the pronounced increase in abundance of CREB-HΔTMC in the presence of CHIR and a modest decrease with LY (SV5, lanes 1–5). In parallel, we found that although there was also an increase in pS73-positive protein abundance in the presence of CHIR, this increase was reproducibly less than that in total protein. This effect was modest, however, with less than twofold difference in the absence and presence of CHIR (Supplemental Figure S2d). A possible explanation for these data is expanded upon in the Discussion, where we propose that although the effects of GSK-3 inhibition were clear, it is also possible that additional kinases and phosphorylation sites were involved in CREB-H modification.
Serine 87/90 substitution mutants display enhanced transcriptional activity
We sought to address the formal possibility that stabilization due to S87/90A substitutions in the DSG motif was not due to an unrelated effect—for example, by disrupting the functional activity of the protein after mutating a conserved region. This seemed an unlikely explanation, since mutated proteins are rather more likely to be malfolded and degraded than stabilized and the protein also localized normally to the nucleus. Nevertheless we measured the functional activity of CREB‑HΔTMC wt versus DSG mutant in a transcriptional activation assay on the native promoter for one of the main physiological targets of CREB-H identified by us and others, namely apolipoprotein A IV (Lee et al., 2011; Barbosa et al., 2013; Xu et al., 2014). The results (Figure 9a) show that apolipoprotein A IV–Luc transcription was massively increased by the wt protein, with increases in absolute activity of greater than several thousandfold. Consistent with the increases in steady-state levels (Figure 9b), activation by each of the mutants was increased, with the greatest being for the 3S+DSG mutant. However, whereas the mutants were all clearly active, the increases in transcriptional activity of the target were modest and not proportional to the relative increases in protein levels. A reasonable explanation for this is that the nuclear wt product is already highly active on the apolipoprotein A IV promoter–Luc target and that some other limitation in the system or the phenomenon of squelching by overabundant activators (Gill and Ptashne, 1988; Prywes and Zhu, 1992) limits the dynamic range of the assay and the ability to measure further increases in activity.
To pursue this, we next asked whether the mutant products would be stabilized upon production from the full-length protein and furthermore whether this would result in increased levels of the endogenous target apolipoprotein A IV. We previously showed that apolipoprotein A IV is induced and secreted by CREB-H (Barbosa et al., 2013), and secreted apolipoprotein A IV is a sensitive assay for CREB-H activity, measuring an endogenous physiologically relevant readout. We transfected isogenic constructs expressing full-length versions of the wt and mutants and analyzed relative levels of the cleaved products (Figure 9c). Consistent with our previous results (Bailey et al., 2007; Bailey and O'Hare, 2007; Llarena et al., 2010) and those of other laboratories (Danno et al., 2010; Lee et al., 2010; Xu et al., 2014), the full-length wt protein produces some but minor amounts of the cleaved product, again almost all of which is in the N1 form (Figure 9c, lane 1). Each of the mutants 3S+DSG, DSG, and 3S showed elevated levels of the cleaved products, with the greatest being for 3S+DSG, followed by DSG and less but still significant increases for 3S, completely consistent with earlier results. Actin served as a loading control and was analyzed simultaneously on the same blots (Figure 9c). We performed quantitative analysis by reverse transcription-PCR of the levels of endogenous apolipoprotein A IV transcripts (Figure 9d). The results demonstrate a pronounced increase in apolipoprotein A IV transcript levels of 12- and 6-fold for the 3S+DSG and DSG mutants, respectively, compared with wt and a modest but detectable increase for the 3S mutant. Moreover, we also analyzed absolute levels of the native secreted apolipoprotein A IV protein in the medium from similarly transfected cells (Figure 9e). Consistent with the results from analysis of transcript levels, significant increases in apolipoprotein A IV levels were observed, with increases again showing a good correspondence with the relative levels of the cleaved proteins. Thus the elevated steady-state levels of the products from the full-length protein reflected properties we observed in the isolated cleaved protein. Of importance, the serine-rich region examined here is involved in modulating the stability and thus overall activity of CREB-H in a manner relevant to expression of a key physiological target.
DISCUSSION
CREB-H plays a key role in metabolic homeostasis, in particular in maintaining normal levels of lipids and triglycerides (Lee et al., 2011; Zhang et al., 2012) and in hepatic glucose metabolism (Lee et al., 2010). Although it is activated by stimuli including fatty acids, fasting, and insulin, there is limited mechanistic understanding of the determinants or pathways involved. We propose that the region in and around 86.DSGISE.91 functions as a phosphodegron signal to mediate phosphorylation-dependent destruction and attenuation of the active nuclear form. Our evidence is as follows: 1) the presence and conservation of the motif; 2) the pronounced effect of DSG mutation conferring increased CREB-HΔTMC stability; 3) phosphatase treatment of the wt protein resulting in increased mobility; 4) increased mobility upon mutation of the 3S region; 5) phosphospecific antibody demonstrating phosphorylation at least at S73 and S81 in vivo; 6) in vitro phosphorylation showing reciprocal specificities by candidate kinases GSK-3b and CKII within the motif; 7) enhanced levels of CREB-HΔTMC upon expression of Cul1 DN, specifically dependent on the DSG motif; 8) specific interaction of Fbw1a with CREB-HΔTMC requiring both the DSG-motif and phosphorylation; 9) pronounced increase in steady-state levels of CREB-HΔTMC in the presence of CHIR99021, an inhibitor of GSK-3; and 10) elevated steady-state levels of the cleaved protein after DSG mutation in the context of the full-length protein, leading to increased expression of a key physiological target, apolipoprotein A IV. These data supply strong support for the main proposition of this work and give mechanistic insight into CREB-H regulation that is consistent with previous data. Conclusions and speculations are summarized in Figure 10.
FIGURE 10:
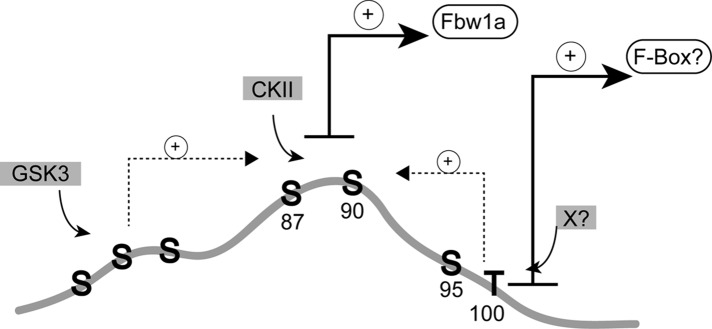
Model for CREB‑H phosphorylation at the S87/90 phosphodegron. The model illustrates the serine-rich area centering around the highly conserved residues S87–S90 in the N-terminus of CREB-HΔTMC, which we show is degraded in a proteasome-dependent manner and stabilized by substitution of S87A/S90A or inhibition of GSK-3 kinase or by interference with SCF activity by a dominant-negative strategy. Residues S87/90 are illustrated to be a key site for interaction with Fbw1a and thus for degradation by the SCFFbw1a complex in a manner dependent on phosphorylation by CKII but with augmented phosphorylation by GSK-3 at other sites, including the adjacent serine residues around S73–S81. Because GSK inhibition has a much more profound effect on CREB-H stabilization than mutation of the S73–S81 region, we propose that other sites within this region in downstream serines (e.g., 95–100) or indeed elsewhere in the protein may be involved, either as independent sites dictating interaction with SCF or in promoting the recognition of the key S87/90 site. As with other regulatory system, other kinases (x) are likely also to play a role, conferring flexibility upon CREB-H regulation and the ability to respond and integrate multiple metabolic or other stimuli.
The CREB-H phosphodegron
The DSGISE motif in CREB‑H fits the expanded consensus sequence for Fbw1a and is extremely similar to those in the erythropoietin receptor (Meyer et al., 2007) and the Yes-associated protein (Zhao et al., 2010) and identical to that found in Nrf2 (DSGIS; Rada et al., 2011), all of which are known phosphodegrons regulated by phosphorylation and SCFFbw1a. Nrf2 is particularly apposite regarding the possibility of multiple modes of regulation of CREB-H. Nrf2, a redox-sensitive transcription factor, is regulated by at least two independent mechanisms, one involving a redox-dependent (but phosphorylation-independent) interaction with an E3 ligase adaptor (Keap1) and a second, Keap1-independent pathway involving GSK phosphorylation at the DSG phosphodegron and SCFFbw1a-mediated degradation (Rada et al., 2011). The possibility of multiple modes of CREB-H regulation is discussed later.
Our results do not, however, directly demonstrate phosphorylation of the DSG itself. Despite the increase in abundance of the DSG mutant, we did not observe increased electrophoretic mobility, nor were we able to generate phosphospecific antibody against the DSG region. However, the increased mobility of the 3S mutant and detection of the wt by phosphospecific antibodies supply convincing evidence that at the least S73 and S81 are phosphorylated in vivo. Moreover, GSK-3b in vitro preferentially phosphorylated residues within S73 to S81. We note that S73, S77, and S81 could all be targets of GSK-3 since they each match the consensus sequence, S/TxxxS/TP (Doble and Woodgett, 2003; Cohen and Goedert, 2004). Conversely, in vitro studies suggest that serines S87 and/or S90 from the DSG motif are likely targets of the CKII kinase, with phosphorylation significantly more efficient after primary phosphorylation by GSK-3, likely in serines S73, S77, and S81. These results are consistent with the in vivo increase in CREB-H levels upon treatment with kinase inhibitors in particular for the GSK-3.
One puzzling feature of our analysis, however, is the modest effect of the 3S mutation on overall steady-state levels of the protein in vivo. If GSK-3 were a critical priming kinase, then we would expect the 3S mutation to yield as pronounced an increase in protein levels as the DSG mutation itself. However, in other examples, it is not unusual to have complex systems involving independent kinases with overlapping specificities interdigitated within an extended region or in distinct regions of a substrate (Skaar et al., 2013). Thus, even with substitution of the 3Ss, the DSG motif might still be able to bind the SCFFbw1a E3 ligase, perhaps because it would still be phosphorylated using unidentified priming site(s). It is also possible that the DSG motif binds and targets CREB-H for destruction even without phosphorylation, as reported for certain substrates (Skaar et al., 2013). However, this would not explain the significant stabilization observed by in vivo inhibition of GSK activity, which suggests that this kinase has an important role regulating levels of nuclear CREB-H. Moreover, 3S mutation did have an effect, and the 3S+DSG mutant was more abundant than the DSG mutant and the most abundant mutant protein detected. Therefore we favor an explanation seen with many examples, such as Nrf2a, cdc25a, and SREBP, for which regulatory motifs encompass multiple overlapping determinants that can be phosphorylated by a single kinase or several kinases, with the potential for phosphorylation without priming, overlapping with independent sites involving priming, sometimes in a complex, interrelated manner. Thus, as summarized in the model in Figure 8, the DSG motif at 87/90 is a key site (if not the only site) for dictating interaction with the SCFFbw1a complex, and it can be phosphorylated mainly by CKII. Other kinases, notably GSK-3, help promote phosphorylation at 87/90 by potentially redundant or overlapping sites not limited to the 3S site at 73–81. For example, there are additional serines and a conserved threonine downstream of 87/90. These could contribute by promoting phosphorylation of the 87/90 site or by promoting interaction with alternative SCF complexes as indicated. In any case, these data suggest that CREB‑H has parallels to other systems that use sequential or multisite phosphorylation to regulate the rate of degradation (Jin et al., 2003; Jia et al., 2005; Tempe et al., 2006; Hao et al., 2007). Such complex overlapping phosphorylation controls allow responses to multiple types of signals with fine “gain” control and integration with (or independence from) distinct stimuli.
The parallels between CREB-H and SREBPs are particularly intriguing. Both are involved in ER–Golgi transport and metabolic regulation. For SREBP, the cleaved nuclear form is highly unstable (Wang et al., 1994) and subject to proteasome-mediated degradation by an SCF E3 ligase using in this case the F-box Fbw7, regulated by GSK-3 phosphorylation (Sundqvist et al., 2005). SREBP1 is also regulated by multiple positive and negative phosphorylation events in response to other metabolic cues, including, for example, insulin, which activates AKT, thus suppressing GSK-3 and promoting stabilization of the nuclear form (Sundqvist et al., 2005). It has been demonstrated that insulin induces the accumulation of the nuclear form of CREB-H (Zhang et al., 2012). Considering that GSK-3 is constitutively active and suppressed by insulin signaling, our results are now consistent with and could provide a mechanistic explanation for these observations. However, levels of the nuclear form of CREB-H are also stimulated under fasting conditions in vivo in the absence of any change in the full-length form (Lee et al., 2011) and suppressed by refeeding (Danno et al., 2010). Thus, as for SREBP, broad, integrated metabolic signals are likely to combine to modulate the levels and activity of CREB-H. Despite the complexities, our results provide the first mechanistic insight into CREB-H phosphorylation, SCF interaction, and regulated stability. Future work will dissect the motifs and other potential kinases and pathways and also investigate the influence of phosphorylation on the intricate ER–Golgi transport of CREB-H, which is likely to be coordinated with the modulation of the nuclear form.
MATERIALS AND METHODS
Cells
HepG2 cells were grown on collagen-coated plates in MEM containing 0.1 mM nonessential amino acids (NEAA), 10% fetal calf serum, 2 mM l-glutamine, 1 mM sodium pyruvate, and penicillin and streptomycin at 100 U/ml and 100 μg/ml, respectively (Pen/Strep). (All tissue materials were purchased from Life Technologies, Paisley, UK, unless otherwise stated.) COS-1 cells were grown in DMEM supplemented with 10% newborn calf serum (NBCS) and Pen/Strep. Routine transfection assays were performed in COS cells cultured at 37°C in a 5% CO2 environment under standard conditions unless otherwise stated.
Plasmids
The plasmid for expression of the SV5 epitope-tagged truncated nuclear form of CREB-H, termed CREB‑HΔTMC (pDJB125), under the control of the low-level TK promoter has been described previously (Bailey et al., 2007). A series of mutants was constructed by site-directed mutagenesis or PCR-based mutagenesis in which individual or combined serines were substituted by alanines. For most constructs, a two-stage PCR nested reaction was used in which primers 1 and 2 generated the 5′ fragment with a mutation at the 3′ end, and primers 3 and 4 generated the 3′ fragment with a corresponding mutation at the 5′ end. After denaturing and annealing of the resulting fragments, the final fragment was produced by PCR using the external primers 1 and 4. The external primers contained restriction sites for natural unique sites within the CREB-H gene, NheI and XbaI or ClaI. In a modified version of this strategy for efficient mutagenesis (Ko and Ma, 2005), primers 2 and 3 contained, in addition to the appropriate mutation, a sequence for digestion by the enzyme EarI. Digestion by EarI and ligation of the two fragments results in removal of the intervening region and production of a single fragment with only the desired mutation, cloned then by conventional means directly or via an intermediate as before. For certain vectors, the amplified fragments were cloned into the intermediate vector pCRBlunt II-TOPO, from which they were then cloned into pDJB125. For combinations of mutations, the same strategy was used, except that the mutant fragments were cloned into plasmids that already contained mutations, resulting in combined mutations. Alternatively, mutants were combined by digestion with unique enzymes NheI and ClaI (which flank the S73–S91 region under investigation) and swapping into the appropriate recipient plasmid. For the 3SA mutant and the 3S deletion mutant (see later discussion and main text), a single combined mutant was made by the same PCR strategy as described. All mutant plasmids were confirmed by sequencing.
We constructed fusion proteins in which CREB-HΔTMC (residues 1–323) or the DSG mutant were fused in frame to that of bacterial GST for the purposes of purification and subsequent analysis of in vitro phosphorylation. CREB-HΔTMC was amplified from the full-length vector pSM20 (Bailey et al., 2007) using a 5′ primer for insertion in-frame to GST and a 3′ primer that introduced a termination codon at the S2P site together with an XbaI site for cloning into the commercial vector pGex-6P-3 (GE Life Sciences, Little Chalfont, UK). This vector is termed pGST-wt (pDJB126). A mutant version containing S87A and S91A was constructed by insertion of the BamHI-XbaI fragment from the corresponding eukaryotic expression vector constructed (pDJB136) into pDJB126.
The luciferase reporter vector for one of the key CREB-H targets, apolipoprotein A IV, contained the promoter region from −2399 to +85 and was created by amplification from human cell genomic DNA using the primers 5′-GCTCGAGATCTATGTCACCTCATAGTCACTAG-3′ and 5′-TGCCAAGCTTCCTGAGCTGCTTGCTGGGCT-3′ and insertion into the pGL3-basic vector (Promega) at the BglII and HindIII restriction sites. This vector was termed pGL3-hApoA-IV-LUC.
The pcDNA3.1 Flag expression vector for the F-box factor Fbw1a/βTrCP (Busino et al., 2003) was obtained from Michele Pagano (Department of Pathology, NYU School of Medicine, New York, NY). The dominant-negative Flag-tagged Cul1, pcDNA-HCUL1-N452-FLAG (Jin et al., 2005), was obtained from Wade Harper (Harvard Medical School, Boston, MA).
Transfections
Transfections were performed using Genejammer (Agilent Technologies, Cheshire, UK) or FuGENE (Promega, Southampton, UK) transfection reagents according to the manufacturer's instructions or the calcium phosphate precipitation procedure modified by the use of {N,N-bis(2-hydroxyetyhl)-2-aminoethanesulfonic acid}-buffered saline (pH 7.06) as previously described (Batchelor and O'Hare, 1992). Routinely, 0.5–1 μg of the appropriate expression vector was transfected with amounts of DNA normalized using pUC19 carrier DNA.
Expression of recombinant proteins
GST-wt and GST-ΔDSG were expressed in Escherichia coli BL21 after induction of cells with isopropyl-β-d-thiogalactoside (1 mM) for 4 h at 30°C. Bacteria were harvested, resuspended in lysis buffer (phosphate-buffered saline [PBS], 1% Triton, 1 mM dithiothreitol, and protease inhibitors [1× Complete Protease; Roche, Sigma-Aldrich, Gillingham, UK]), and sonicated. The lysates were incubated for 1 h with glutathione Sepharose 4B (GE Life Sciences), and the beads then washed extensively with lysis buffer and stored at 4°C for assay. In each case, samples were equalized by either optical density reading of purified protein or SDS–PAGE analysis and quantitation of the stained bands. Equal amounts of substrates were then used for the in vitro kinase assays as described next.
In vitro kinase assays
Bacterially expressed proteins that were bound to glutathione Sepharose 4B were washed with the appropriate reaction buffer for either CKII or GSK-3β. The CKII reaction buffer contained 20 mM Tris-HCl, 50 mM KCl, 10 mM MgCl2, 200 μM ATP, and 500 μCi/μmol γ-32P-ATP, pH 7.5. The GSK-3 reaction buffer contained 25 mM Tris-HCl, 12 mM MgCl2, 2 mM dithiothreitol, 5 mM β-glycerophosphate, 100 μM sodium orthovanadate, 200 μM ATP, and 500 μCi/μmol γ-32P-ATP, pH 7.5. Samples were incubated with 100 U of purified CKII (New England Biolabs, Hitchin, UK) in 40 μl of reaction buffer or in certain cases with 100 μl of cellular lysate (and 200 μM ATP, 500 μCi/μmol γ-32P-ATP) for 45 min at 30°C. For sequential phosphorylation, samples were first incubated with one kinase in the appropriate reaction buffer. The GST-fusion protein substrates were then purified on the glutathione-agarose beads, washed extensively, and then incubated with the second kinase in the appropriate buffer. Reactions were stopped by washing in reaction buffer and boiling in SDS sample buffer. Proteins were resolved by SDS–PAGE, the gels were stained with Coomassie brilliant blue (CBB) for total protein assessment, and dried gels were exposed to autoradiography.
Generation of phosphospecific antibodies
Nonoverlapping peptides were selected for the generation of antibodies to phosphorylated forms of S73, S81, and S87. The peptides were coupled at their N-terminal end to keyhole limpet hemocyanin with the incorporation of a cysteine residue for coupling purposes and with phosphoserine in place of serine as follows: for pS81, (C)LGSGD[pS]LPS; for pS81, (C)PLW[pS]PEGSD; and for pS87, (C)PEGSD[pS]GI. Rabbits were immunized by standard protocols (Thermo Scientific) and specific antibody to phosphorylated species isolated by positive-negative selection. Phosphospecific antibody was first positively selected by binding to columns with immobilized phosphorylated form of the immunizing peptide. After washing to remove nonspecific and unreactive material, peptide-specific antibodies were then eluted. From this eluate, phosphospecific antibodies were then enriched by negative selection on columns with immobilized form of the native unmodified peptide, where in this case the flow through unbound material contains the phosphospecific antibody. Confirmation of reactivity to the intact protein and the phosphospecific nature of the detection were then confirmed both by site-directed mutagenesis of the specific serine residue and the sensitivity of reactivity to phosphatase treatment as described in Results.
Phosphatase treatment
Cells transfected with expression vectors for wt and mutant CREB-HΔTMC were washed with ice-cold PBS, scraped, and pelleted. After suspension in PBS, samples were lysed in buffer for λ phosphatase containing 0.4% NP-40, 0.5% Triton X-100, protease inhibitors (1× Complete Protease; Sigma-Aldrich), and 1 mM phenylmethylsulfonyl fluoride. As controls, two aliquots of the sample were treated with orthovanadate (10 mM) and sodium fluoride (20 mM) and incubated at 37°C either with or without added phosphatase for 30 min to reflect the starting material before incubation with phosphatase alone. Parallel samples were then incubated with 400 U of λ phosphatase and incubated at 37°C for different times up to 30 min. The reactions were terminated by adding SDS sample buffer to 1× and boiling for 5 min. Samples were analyzed by SDS–PAGE and Western blotting.
Western blot analysis
Proteins were separated by electrophoresis in 10 or 12% SDS–PAGE gels and transferred to nitrocellulose membranes, which were blocked with PBS/0.05% Tween 20 (PBST) containing 5% nonfat dried milk or 5% bovine serum albumin (BSA). After blocking, membranes were incubated in PBST/5% dried milk or 5% BSA overnight at 4°C with primary antibodies anti-V5, 1:10,000 (Life Technologies); anti–actin 20-36, 1:2500; anti–actin AC-40, 1:2500; anti–CREB-H pS73 and pS81, 1:100 (this work); anti–apolipoprotein A IV 1D6B6 (1:2000, Cell Signaling, Leiden, The Netherlands); or anti-Flag, 1:1000. Antibodies were from Sigma-Aldrich unless otherwise stated, Membranes were washed three times in PBS/0.05% Tween and incubated for a further 1 h in PBST/5% dried milk or 5% BSA containing the appropriate horseradish peroxidase–conjugated secondary antibodies (for detection by conventional chemiluminescence detection) or antibodies conjugated with Dylight680 or Dylight800 (Fisher Scientific, Lutterworth, UK) for detection by laser scanning using the LI-COR Odyssey Image system. This latter method was routinely used for CREB-H detection using the phosphospecific antibodies since it allowed simultaneous analysis on one blot of total CREB-H species with the mouse antibody to the epitope tag in one channel and the phosphorylated forms with antibody to the rabbit antibody to the phosphorylated peptide in a second channel. For quantitative estimates of protein half-lives (Figure 3), blots were scanned in the appropriate channel, background subtracted, and normalized for loading using actin abundance in the separate channel. Data were analyzed in the GraphPad software package using nonlinear regression analyses for one-phase decay.
Inhibitor treatments
Brefeldin A (Sigma-Aldrich) was prepared as 10 mg/ml stocks in methanol and added to cells at a final concentration from 1 μg/ml for 30 min. Cycloheximide (Sigma-Aldrich) was prepared as 100 mg/ml stock in ethanol and used at a final concentration of 100 μg/ml for times indicated in the text. MG132 was dissolved in dimethyl sulfoxide (DMSO) and used at a final concentration of 10 μM as stated in the text and added together with cycloheximide. SB216763 (Tocris Biosciences, Bristol, UK) was prepared at 20 mM in DMSO and added at 20 μM. CHIR99021 (Tocris Biosciences), LY29400 (Calbiochem, Nottingham, UK), and TBCA (Calbiochem) were prepared at 10 mM in DMSO and added at 10 or 5 μM as indicated. Rapamycin (Calbiochem) was prepared in 10 mM DMSO and added at 1 μM in appropriate media.
Transcription activation assays
COS-1 cells were seeded into 24-well plates and transfected in triplicate with expression vectors for CREB‑H.ΔTMC or mutants together with the target luciferase reporter vector. The luciferase reporter vector (pGL3-hApoA-IV-LUC) for the principal CREB-H target, ApoA IV, was created as described. A series of preliminary dose–response experiments was carried out to optimize parameters of target and expression vector concentration and time. In each case, total DNA was equalized with appropriate amount of pUC19 plasmid DNA. Cell lysates were prepared 48 h after transfection by addition of Glo Lysis buffer (Promega). Firefly luciferase activity was assayed using the Bright-Glo assay system as described by the manufacturer and activity measured using a Victor luminescence reader. Assays were routinely performed in triplicate and readings calculated as fold increase relative to control alone.
Analysis of apolipoprotein A IV transcription
For quantitative PCR, total RNA was prepared using RNeasy isolation kit according to the manufacturer's instructions including DNase treatment (Qiagen, Hilden, Germany). Quality control (absorbance ratio 280/260 nm) was ∼2.0 for all samples analyzed. cDNA was synthesized from 2 μg of total RNA using anchored oligo(dT)12-18 primers and SuperScript III Reverse Transcriptase (Life Technologies). For quantitative real-time reverse transcription PCR (qPCR), primers were designed using qPrimerDepot software (primerdepot.nci.nih.gov/), and specificity against genomic DNA was validated by conventional PCR with Thermo 2x Reddymix PCR master mix (Thermo Scientific, Lutterworth, UK). qPCR was performed with SYBR select mastermix for CFX (Life Technologies) using the Bio-Rad CFX96 Detection System instrument and software (Bio-Rad Laboratories, Hemel Hempstead, United Kingdom). In all, 0.5 μl of cDNA was used per 20 μl of reaction with the following cycling conditions: an initial activation at 95°C for 10 min, followed by 50 cycles of denaturation at 95°C for 15 s and 60°C for 1 min. A melt curve analysis was included as the final step, which consisted of temperature increments of 0.5°C from 55 to 95°C. Each sample was analyzed in triplicate and normalized to TATA box–binding protein housekeeping gene. Primer sequences were as follows, with forward primer indicated first and reverse primer indicated second:
ApoA IV: CCGTGGAACATCTCCAGAAA, AAAGGGCACCAGCTTCTTCT
CREBH: TGCAATCTCACCGTGAAAGA, GAGCACCAGCTCCTGACAGT
TBP: TATAATCCCAAGCGGTTTGC, GCTGGAAAACCCAACTTCTG
Analysis of apolipoprotein A IV secretion
Apolipoprotein A IV levels were analyzed as described previously (Barbosa et al., 2013). Medium was harvested from the test cells and proteins precipitated with 15% trichloroacetic acid. Precipitates were centrifuged at 15,000 × g for 15 min and pellets washed twice with 100% ice-cold acetone or ethanol. Pellets were air-dried and subsequently resuspended in 2× SDS sample buffer. Apolipoprotein A IV levels were analyzed by SDS–PAGE and Western blotting as described.
Immunofluorescence analysis
Cells (1 × 105 cells/35-mm well) were plated on glass coverslips placed in plastic tissue culture vessels and processed ∼40 h after transfection. For routine immunofluorescence analysis, cells were washed in PBS, fixed with ice-cold methanol, and blocked in PBS/10% NBCS for 45 min. Primary antibodies were diluted in PBS/10% NBCS and applied for 45 min. Primary antibodies used were anti-SV5 (1:1000; Life Technologies); fluorochrome (Alexa 488)–conjugated secondary antibodies of appropriate specificity (Fisher Scientific, Loughborough, UK) were diluted 1:200 in PBS/10% NBCS and added for 45 min. After washing, cells were mounted in Mowiol and visualized using a Zeiss LSM 410 confocal microscope imaging system and a Zeiss Plan-Apochromat (63×, 1.4 numerical aperture) lens. Images for each channel were captured sequentially with eightfold averaging at an image size of 512 × 512 pixels. Composite illustrations were prepared using Adobe software. Example images shown are representative of numerous images gathered for each test construct and condition.
Supplementary Material
Acknowledgments
We are grateful to Rick Randall for supplying monoclonal antibodies for the detection of the SV5-epitope tag. We thank J. W. Harper (Cul1 DN construct) and M. Pagano (F-box constructs) for vectors supplied to aid this analysis. This work was supported by the Marie Curie Cancer Care and the Medical Research Council, MR/K017926/1.
Abbreviations used:
- AKT
protein kinase B
- ATF6
activating transcription factor 6
- BFA
brefeldin A
- bZIP
basic and leucine zipper domain
- CBB
Coomassie brilliant blue
- CHX
cycloheximide
- CKII
casein kinase II
- COS-1
SV40 transformed African green monkey kidney cells
- CREB-H
cyclic AMP response element binding factor-H
- Cul1
Cullin 1
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- GSK3
glycogen synthase kinase 3
- LiCl
lithium chloride
- MG132
carbobenzoxy-l-leucyl-l-leucyl-l-leucinal
- NBCS
newborn calf serum
- RIP
regulated intramembrane proteolysis
- S1P
site-1 protease
- S2P
site-2 protease
- SCF
Skp, Cullin, F-box containing complex
- SREBP
sterol regulatory element–binding protein
- UPR
unfolded protein response
- wt
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-04-0247) on June 24, 2015.
*These authors contributed equally to this work.
†Present address: Institute of Cancer Research, London SM2 5NG, United Kingdom.
REFERENCES
- Asada R, Saito A, Kawasaki N, Kanemoto S, Iwamoto H, Oki M, Miyagi H, Izumi S, Imaizumi K. The endoplasmic reticulum stress transducer OASIS is involved in the terminal differentiation of goblet cells in the large intestine. J Biol Chem. 2012;287:8144–8153. doi: 10.1074/jbc.M111.332593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey D, Barreca C, O'Hare P. Trafficking of the bZIP transmembrane transcription factor CREB-H into alternate pathways of ERAD and stress-regulated intramembrane proteolysis. Traffic. 2007;8:1796–1814. doi: 10.1111/j.1600-0854.2007.00654.x. [DOI] [PubMed] [Google Scholar]
- Bailey D, O'Hare P. Transmembrane bZIP transcription factors in ER stress signaling and the unfolded protein response. Antioxid Redox Signal. 2007;9:2305–2321. doi: 10.1089/ars.2007.1796. [DOI] [PubMed] [Google Scholar]
- Barbosa S, Fasanella I, LLarena M, Fox RM, Andrew DJ, O'Hare P. An orchestrated programme regulating secretory pathway genes and cargos by the transmembrane transcription factor CREB-H. Traffic. 2013;14:382–398. doi: 10.1111/tra.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor AH, O'Hare P. Localization of cis-acting sequence requirements in the promoter of the latency-associated transcript of herpes simplex virus type 1 required for cell-type-specific activity. J Virol. 1992;66:3573–3582. doi: 10.1128/jvi.66.6.3573-3582.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengoechea-Alonso MT, Ericsson J. A phosphorylation cascade controls the degradation of active SREBP1. J Biol Chem. 2009;284:5885–5895. doi: 10.1074/jbc.M807906200. [DOI] [PubMed] [Google Scholar]
- Brodsky JL, Skach WR. Protein folding and quality control in the endoplasmic reticulum: Recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol. 2011;23:464–475. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, Draetta GF. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature. 2003;426:87–91. doi: 10.1038/nature02082. [DOI] [PubMed] [Google Scholar]
- Cao G, Ni X, Jiang M, Ma Y, Cheng H, Guo L, Ji C, Gu S, Xie Y, Mao Y. Molecular cloning and characterization of a novel human cAMP response element-binding (CREB) gene (CREB4) J Hum Genet. 2002;47:373–376. doi: 10.1007/s100380200053. [DOI] [PubMed] [Google Scholar]
- Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- Chanda D, Kim DK, Li T, Kim YH, Koo SH, Lee CH, Chiang JY, Choi HS. Cannabinoid receptor type 1 (CB1R) signaling regulates hepatic gluconeogenesis via induction of endoplasmic reticulum-bound transcription factor cAMP-responsive element-binding protein H (CREBH) in primary hepatocytes. J Biol Chem. 2011;286:27971–27979. doi: 10.1074/jbc.M111.224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. 2012;18:59–68. doi: 10.1016/j.molmed.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Danno H, Ishii KA, Nakagawa Y, Mikami M, Yamamoto T, Yabe S, Furusawa M, Kumadaki S, Watanabe K, Shimizu H, et al. The liver-enriched transcription factor CREBH is nutritionally regulated and activated by fatty acids and PPARalpha. Biochem Biophys Res Commun. 2010;391:1222–1227. doi: 10.1016/j.bbrc.2009.12.046. [DOI] [PubMed] [Google Scholar]
- Dhananjayan SC, Ismail A, Nawaz Z. Ubiquitin and control of transcription. Essays Biochem. 2005;41:69–80. doi: 10.1042/EB0410069. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Kristiana I, Wong J, Brown AJ. Involvement of Akt in ER-to-Golgi transport of SCAP/SREBP: a link between a key cell proliferative pathway and membrane synthesis. Mol Biol Cell. 2006;17:2735–2745. doi: 10.1091/mbc.E05-11-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs SY, Spiegelman VS, Kumar KG. The many faces of beta-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene. 2004;23:2028–2036. doi: 10.1038/sj.onc.1207389. [DOI] [PubMed] [Google Scholar]
- Gentile CL, Wang D, Pfaffenbach KT, Cox R, Wei Y, Pagliassotti MJ. Fatty acids regulate CREBh via transcriptional mechanisms that are dependent on proteasome activity and insulin. Mol Cell Biochem. 2010;344:99–107. doi: 10.1007/s11010-010-0533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill G, Ptashne M. Negative effect of the transcriptional activator GAL4. Nature. 1988;334:721–724. doi: 10.1038/334721a0. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell. 2007;26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Murata S, Tanaka K, Shimizu M, Sato R. Sterol regulatory element-binding proteins are negatively regulated through SUMO-1 modification independent of the ubiquitin/26 S proteasome pathway. J Biol Chem. 2003;278:16809–16819. doi: 10.1074/jbc.M212448200. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Yoshida M, Shimizu M, Sato R. Direct demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathway. J Biol Chem. 2001;276:36431–36437. doi: 10.1074/jbc.M105200200. [DOI] [PubMed] [Google Scholar]
- Honma Y, Kanazawa K, Mori T, Tanno Y, Tojo M, Kiyosawa H, Takeda J, Nikaido T, Tsukamoto T, Yokoya S, Wanaka A. Identification of a novel gene, OASIS, which encodes for a putative CREB/ATF family transcription factor in the long-term cultured astrocytes and gliotic tissue. Brain Res Mol Brain Res. 1999;69:93–103. doi: 10.1016/s0169-328x(99)00102-3. [DOI] [PubMed] [Google Scholar]
- Jia J, Zhang L, Zhang Q, Tong C, Wang B, Hou F, Amanai K, Jiang J. Phosphorylation by double-time/CKIepsilon and CKIalpha targets cubitus interruptus for Slimb/beta-TRCP-mediated proteolytic processing. Dev Cell. 2005;9:819–830. doi: 10.1016/j.devcel.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Jin J, Ang XL, Shirogane T, Wade Harper J. Identification of substrates for F-box proteins. Methods Enzymol. 2005;399:287–309. doi: 10.1016/S0076-6879(05)99020-4. [DOI] [PubMed] [Google Scholar]
- Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, Harper JW. SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17:3062–3074. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JK, Ma J. A rapid and efficient PCR-based mutagenesis method applicable to cell physiology study. Am J Physiol Cell Physiol. 2005;288:C1273–C1278. doi: 10.1152/ajpcell.00517.2004. [DOI] [PubMed] [Google Scholar]
- Lee AH. The role of CREB-H transcription factor in triglyceride metabolism. Curr Opin Lipidol. 2012;23:141–146. doi: 10.1097/MOL.0b013e3283508fed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Giannikopoulos P, Duncan SA, Wang J, Johansen CT, Brown JD, Plutzky J, Hegele RA, Glimcher LH, Lee AH. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat Med. 2011;17:812–815. doi: 10.1038/nm.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MW, Chanda D, Yang J, Oh H, Kim SS, Yoon YS, Hong S, Park KG, Lee IK, Choi CS, et al. Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab. 2010;11:331–339. doi: 10.1016/j.cmet.2010.02.016. [DOI] [PubMed] [Google Scholar]
- Llarena M, Bailey D, Curtis H, O'Hare P. Different mechanisms of recognition and ER retention by transmembrane transcription factors CREB-H and ATF6. Traffic. 2010;11:48–69. doi: 10.1111/j.1600-0854.2009.00997.x. [DOI] [PubMed] [Google Scholar]
- Lu R, Yang P, O'Hare P, Misra V. Luman, a new member of the CREB/ATF family, binds to herpes simplex virus VP16-associated host cellular factor. Mol Cell Biol. 1997;17:5117–5126. doi: 10.1128/mcb.17.9.5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebke-Wheeler J, Zhang K, Battle M, Si-Tayeb K, Garrison W, Chhinder S, Li J, Kaufman RJ, Duncan SA. Hepatocyte nuclear factor 4alpha is implicated in endoplasmic reticulum stress-induced acute phase response by regulating expression of cyclic adenosine monophosphate responsive element binding protein H. Hepatology. 2008;48:1242–1250. doi: 10.1002/hep.22439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan AJ, Tam S, Kaganovich D, Frydman J. Protein quality control: chaperones culling corrupt conformations. Nat Cell Biol. 2005;7:736–741. doi: 10.1038/ncb0805-736. [DOI] [PubMed] [Google Scholar]
- Meyer L, Deau B, Forejtnikova H, Dumenil D, Margottin-Goguet F, Lacombe C, Mayeux P, Verdier F. beta-Trcp mediates ubiquitination and degradation of the erythropoietin receptor and controls cell proliferation. Blood. 2007;109:5215–5222. doi: 10.1182/blood-2006-10-055350. [DOI] [PubMed] [Google Scholar]
- Murakami T, Saito A, Hino S, Kondo S, Kanemoto S, Chihara K, Sekiya H, Tsumagari K, Ochiai K, Yoshinaga K, et al. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat Cell Biol. 2009;11:1205–1211. doi: 10.1038/ncb1963. [DOI] [PubMed] [Google Scholar]
- Muratani M, Tansey WP. How the ubiquitin-proteasome system controls transcription. Nat Rev Mol Cell Biol. 2003;4:192–201. doi: 10.1038/nrm1049. [DOI] [PubMed] [Google Scholar]
- Nohturfft A, DeBose-Boyd RA, Scheek S, Goldstein JL, Brown MS. Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proc Natl Acad Sci USA. 1999;96:11235–11240. doi: 10.1073/pnas.96.20.11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori Y, Imai J, Watanabe M, Komatsu T, Suzuki Y, Kataoka K, Watanabe S, Tanigami A, Sugano S. CREB-H: a novel mammalian transcription factor belonging to the CREB/ATF family and functioning via the box-B element with a liver- specific expression. Nucleic Acids Res. 2001;29:2154–2162. doi: 10.1093/nar/29.10.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Prywes R, Zhu H. In vitro squelching of activated transcription by serum response factor: evidence for a common coactivator used by multiple trancriptional activators. Nucleic Acids Res. 1992;20:513–520. doi: 10.1093/nar/20.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Fillion C, Labrie Y, Grenier J, Fournier A, Berger L, El-Alfy M, Labrie C. AIbZIP, a novel bZIP gene located on chromosome 1q21.3 that is highly expressed in prostate tumors and of which the expression is up- regulated by androgens in LNCaP human prostate cancer cells. Cancer Res. 2002;62:721–733. [PubMed] [Google Scholar]
- Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31:1121–1133. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Saito A, Hino S, Murakami T, Kanemoto S, Kondo S, Saitoh M, Nishimura R, Yoneda T, Furuichi T, Ikegawa S, et al. Regulation of endoplasmic reticulum stress response by a BBF2H7-mediated Sec23a pathway is essential for chondrogenesis. Nat Cell Biol. 2009;11:1197–1204. doi: 10.1038/ncb1962. [DOI] [PubMed] [Google Scholar]
- Saito A, Kanemoto S, Kawasaki N, Asada R, Iwamoto H, Oki M, Miyagi H, Izumi S, Sanosaka T, Nakashima K, Imaizumi K. Unfolded protein response, activated by OASIS family transcription factors, promotes astrocyte differentiation. Nat Commun. 2012;3:967. doi: 10.1038/ncomms1971. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman R. The mammalian unfolded protein response. Ann Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Shen J, Snapp EL, Lippincott-Schwartz J, Prywes R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol Cell Biol. 2005;25:921–932. doi: 10.1128/MCB.25.3.921-932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14:369–381. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storlazzi CT, Mertens F, Nascimento A, Isaksson M, Wejde J, Brosjo O, Mandahl N, Panagopoulos I. Fusion of the FUS and BBF2H7 genes in low grade fibromyxoid sarcoma. Hum Mol Genet. 2003;12:2349–2358. doi: 10.1093/hmg/ddg237. [DOI] [PubMed] [Google Scholar]
- Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7) Cell Metab. 2005;1:379–391. doi: 10.1016/j.cmet.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Tempe D, Casas M, Karaz S, Blanchet-Tournier MF, Concordet JP. Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol Cell Biol. 2006;26:4316–4326. doi: 10.1128/MCB.02183-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SE, Dalton LE, Daly ML, Malzer E, Marciniak SJ. Diabetes as a disease of endoplasmic reticulum stress. Diabetes/Metab Res Rev. 2010;26:611–621. doi: 10.1002/dmrr.1132. [DOI] [PubMed] [Google Scholar]
- Vellanki RN, Zhang L, Guney MA, Rocheleau JV, Gannon M, Volchuk A. OASIS/CREB3L1 induces expression of genes involved in extracellular matrix production but not classical endoplasmic reticulum stress response genes in pancreatic beta-cells. Endocrinology. 2010;151:4146–4157. doi: 10.1210/en.2010-0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Sato R, Brown MS, Hua X, Goldstein JL. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell. 1994;77:53–62. doi: 10.1016/0092-8674(94)90234-8. [DOI] [PubMed] [Google Scholar]
- Westermarck J. Regulation of transcription factor function by targeted protein degradation: an overview focusing on p53, c-Myc, and c-Jun. Methods Mol Biol. 2010;647:31–36. doi: 10.1007/978-1-60761-738-9_2. [DOI] [PubMed] [Google Scholar]
- Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell. 2003;11:1445–1456. doi: 10.1016/s1097-2765(03)00234-x. [DOI] [PubMed] [Google Scholar]
- Xu X, Park JG, So JS, Hur KY, Lee AH. Transcriptional regulation of apolipoprotein A-IV by the transcription factor CREBH. J Lipid Res. 2014;55:850–859. doi: 10.1194/jlr.M045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Yellaturu CR, Deng X, Park EA, Raghow R, Elam MB. Insulin enhances the biogenesis of nuclear sterol regulatory element-binding protein (SREBP)-1c by posttranscriptional down-regulation of Insig-2A and its dissociation from SREBP cleavage-activating protein (SCAP). SREBP-1c complex. J Biol Chem. 2009;284:31726–31734. doi: 10.1074/jbc.M109.050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wang G, Zheng Z, Maddipati KR, Zhang X, Dyson G, Williams P, Duncan SA, Kaufman RJ, Zhang K. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology. 2012;55:1070–1082. doi: 10.1002/hep.24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP) Genes Dev. 2010;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.