

Phosphorylation on well-recognized, regulatory connexin43 amino acid residues by a series of kinases serves as an early molecular signal that triggers not only inhibition of gap junction (GJ)–mediated cell-to-cell communication but also GJ internalization. The findings contribute to the newly evolving dynamic picture of GJs.
Abstract
Gap junctions (GJs) exhibit a complex modus of assembly and degradation to maintain balanced intercellular communication (GJIC). Several growth factors, including vascular endothelial growth factor (VEGF), have been reported to disrupt cell–cell junctions and abolish GJIC. VEGF directly stimulates VEGF-receptor tyrosine kinases on endothelial cell surfaces. Exposing primary porcine pulmonary artery endothelial cells (PAECs) to VEGF for 15 min resulted in a rapid and almost complete loss of connexin43 (Cx43) GJs at cell–cell appositions and a concomitant increase in cytoplasmic, vesicular Cx43. After prolonged incubation periods (60 min), Cx43 GJs reformed and intracellular Cx43 were restored to levels observed before treatment. GJ internalization correlated with efficient inhibition of GJIC, up to 2.8-fold increased phosphorylation of Cx43 serine residues 255, 262, 279/282, and 368, and appeared to be clathrin driven. Phosphorylation of serines 255, 262, and 279/282 was mediated by MAPK, whereas serine 368 phosphorylation was mediated by PKC. Pharmacological inhibition of both signaling pathways significantly reduced Cx43 phosphorylation and GJ internalization. Together, our results indicate that growth factors such as VEGF activate a hierarchical kinase program—including PKC and MAPK—that induces GJ internalization via phosphorylation of well-known regulatory amino acid residues located in the Cx43 C-terminal tail.
INTRODUCTION
Gap junctions (GJs) are specialized domains in the plasma membrane that consist of clusters of channels that allow direct intercellular communication between neighboring cells. These channels permit ions and small molecules to be transferred from cell to cell (Goodenough et al., 1996; Kumar and Gilula, 1996). Gap junction–mediated intercellular communication (GJIC) is crucial for normal cell function, including coordination of development (Guthrie and Gilula, 1989), tissue homeostasis, growth, and differentiation (Mehta et al., 1986; Loewenstein and Rose, 1992). Aberrant function of GJs and reduction of cell–cell coupling via GJs are associated with many pathological conditions, including cancer (Simon, 1999; Sulkowski et al., 1999; Chang et al., 2003; Leithe et al., 2006; Cronier et al., 2009). Microscopic observations performed in living cells expressing fluorescent-tagged connexin43 (Cx43), the most ubiquitously expressed and an essential GJ protein, have shown that GJs are highly dynamic structures. To maintain normal cell-to-cell communication, recruitment of newly synthesized GJ channels along the outer edge of GJ plaques and simultaneous removal of older channels from plaque centers are precisely regulated (Gaietta et al., 2002; Lauf et al., 2002; Falk et al., 2009; Rhett et al., 2011; Dunn and Lampe, 2014). Dynamic assembly and degradation of GJs correlates with the short half-life of Cx proteins of 1–5 h determined in situ as well as in cultured cells (Fallon and Goodenough, 1981; Beardslee et al., 1998; Berthoud et al., 2004; Falk et al., 2009). A characteristic feature of GJs is that docked double-membrane-spanning GJ channels cannot be separated into individual, single-membrane-spanning hemichannels (connexons) under physiological conditions (Goodenough and Gilula, 1974; Ghoshroy et al., 1995). However, cells can constitutively turn over GJs by either internalizing small double-membrane domains from central areas of plaques (Jordan et al., 2001; Gaietta et al., 2002; Falk et al., 2009; Cone et al., 2014) or internalizing entire plaques or large portions of plaques (Piehl et al., 2007). Moreover, cells can acutely internalize GJs in response to inflammatory agents or growth factors (Leithe and Rivedal, 2004; Thuringer, 2004; Baker et al., 2008; Fong et al., 2014). Earlier studies showed that GJ turnover uses the clathrin-mediated endocytosis (CME) machinery (Larsen et al., 1979; Naus et al., 1993; Huang et al., 1996; Piehl et al., 2007; Gumpert et al., 2008; Nickel et al., 2008, 2013; Gilleron et al., 2011), and internalization results in the formation of cytoplasmic double-membrane GJ vesicles, termed annular gap junctions (AGJs) or connexosomes (reviewed in Thévenin et al., 2013; Falk et al., 2014; Su and Lau, 2014).
Cx43 is a phosphoprotein that is known to be phosphorylated in situ on at least 15 different serine and tyrosine residues located in its C-terminal domain that are known to regulate all steps of GJ assembly and turnover (Solan and Lampe, 2007, 2014; Thévenin et al., 2013). However, the cellular signals and molecular mechanisms that regulate these events are poorly understood.
Vascular endothelial growth factor (VEGF) is an angiogenic growth factor that is highly specific for endothelial cells. The VEGF-specific tyrosine kinase receptors VEGFR-1 (Flt-1), VEGFR-2 (Flk-1/KDR), and VEGFR-3 (Flt-4, in lymphangiogenesis) activate several intracellular signaling pathways upon VEGF binding (de Vries et al., 1992). Proliferation and migration of endothelial cells are primarily mediated by VEGFR-2 (Waltenberger et al., 1994). Activation of VEGF receptors promotes phosphorylation of a number of downstream proteins in endothelial cells, including phospholipase Cγ (Takahashi et al., 2001), phosphatidylinositol 3-kinase (PI3-kinase; Qi and Claesson-Welsh, 2001), and guanine 5′-triphosphate and GTPase-activating protein (Suzuma et al., 2000). Furthermore, VEGF induces protein kinase C (PKC), which results in an increase in intracellular calcium ions and stimulates inositol-1,4,5-triphosphate accumulation (Brock et al., 1991). VEGF has also been reported to transiently disrupt GJIC in endothelial cells via the activation of VEGFR-2, which activates mitogen-activated protein kinase (MAPK) and c-Src activity (Kevil et al., 1998; Suarez and Ballmer-Hofer, 2001). In addition, it was reported that the VEGF-induced disruption of GJIC correlated with the rapid internalization of Cx43 and its tyrosine phosphorylation in rat coronary capillary endothelium (Thuringer, 2004). Although VEGF-mediated endothelial response has been investigated previously, inconsistent results related to timing of recovery and kinases involved have been reported (Kevil et al., 1998; Suarez and Ballmer-Hofer, 2001; Thuringer, 2004), hinting at endothelial cell type–specific responses and possibly technical issues.
Furthermore, potential mechanisms, including posttranslational modifications of Cx43 such as phosphorylation or ubiquitination, that may be important for VEGF-induced inhibition of GJIC and whether VEGF-mediated inhibition of GJIC involves GJ internalization have not been investigated. Elucidating the mechanisms that down-regulate GJIC in response to VEGF is important for understanding, for example, the biology of cancer development, including changes in cell cycle progression, angiogenesis, and tumor cell metastasis.
To test the hypothesis that phosphorylation of known regulatory amino acid residues in the Cx43 C-terminus might serve as an early signal to initiate channel closure and GJ internalization, we investigated whether treating cells with VEGF would induce phosphorylation of specific C-terminal Cx43 serine residues. We used primary porcine pulmonary artery endothelial cells (PAECs) as a model, as these cells express abundant levels of Cx43 and VEGF receptors. We found that in these cells, recombinant human VEGF-165 (VEGF-A) significantly inhibits GJIC and rapidly induces Cx43 GJ internalization via CME. Our results demonstrate that VEGF treatment induces the phosphorylation of Cx43 at serines 255, 262, 279/282, and 368 by MAPK and PKC that correlates with GJIC inhibition and Cx43-based GJ internalization. Pharmacological inhibition of both kinases significantly reduced Cx43 phosphorylation and GJ internalization, indicating that phosphorylation of Cx43 by both kinases on at least two different sites is required to initiate GJ internalization.
RESULTS
VEGF induces efficient internalization of Cx43 GJs
Previous studies showed that treatment with VEGF leads to a transient disruption of GJIC that coincides with a disorganization of junctional proteins (Kevil et al., 1998; Suarez and Ballmer-Hofer, 2001); however, whether VEGF treatment induces GJ internalization has not been known. Thus, we first investigated whether VEGF induces endocytosis of Cx43 GJs in PAECs. Cells were grown on coverslips, incubated with VEGF for 5–60 min as indicated, and subjected to immunofluorescence to determine the subcellular distribution of Cx43 (Figure 1). In untreated cells, the majority of the Cx43 signal was detected at cell–cell contacts (visualized by counterstaining for zonula occludens-1 [ZO-1]), with the typical appearance of GJs assembled in these cells endogenously expressing Cx43 (Baker et al., 2008). In addition, a faint punctate distribution of Cx43 was detected in the cytoplasm, especially in areas surrounding the cell nucleus (including the Golgi apparatus), as is typical for secretory Cx43 in transit to the plasma membrane (Musil and Goodenough, 1993; Jordan et al., 1999; Lauf et al., 2002; Falk et al., 2009; Figure 1A, a–c). VEGF treatment for only 5 min induced a rapid disruption of Cx43 GJs at cell–cell contacts and at the same time a significant increase of vesicular Cx43 in the cytoplasm (Figure 1, A, d–f, and B). At 15 min, almost all GJs at cell–cell contacts had disappeared and vesicular cytoplasmic Cx43 staining peaked (Figure 1, A, g–i, and B). However, after 30 min of VEGF treatment, the Cx43 signal in the cytoplasm had decreased, and GJs began to reappear at cell–cell contacts (Figure 1, A, j–l, and B). At 60 min after VEGF treatment, most cells showed an almost complete restoration of GJs at cell–cell contacts, and the vesicular Cx43 signal in the cytoplasm had returned to levels comparable to what was observed before treatment (Figure 1, A, m–o, and B). In contrast, the staining pattern for ZO-1 at cell–cell contact areas remained largely unchanged over the entire course of VEGF treatment (Figure 1A, b, e, h, k, and n). Quantitative intracellular Cx43 fluorescence intensity analyses performed on >300 cells of three independent experiments revealed a significant average increase of (2.4 ± 0.17)- and (3.4 ± 0.86)-fold at 5 and 15 min of treatment, which returned to (1.5 ± 0.25)- and (1.4 ± 0.34)-fold after 30–60 min of treatment, respectively (Figure 1B). Taken together, these analyses suggest a rapid but transient internalization of Cx43 GJs in response to VEGF receptor activation that was restored within 1–2 h.
FIGURE 1:

VEGF induces rapid, transient Cx43-GJ internalization in PAECs. (A) Double staining of Cx43 (green) and ZO-1 (red) in untreated cells (a–c), or in cells treated with 100 ng/ml VEGF for 5 (d–f), 15 (g–i), 30 (j-l), or 60 (m–o) min. Nuclei were stained with Hoechst 33342 (blue). Representative images acquired with identical camera settings are shown. Note the acute transient loss of plasma membrane–localized GJs and a simultaneous increase in punctate, intracellular Cx43 fluorescence. (B) Normalized quantitative analyses of intracellular fluorescence intensity of Cx43 (an indirect measure of reduced plasma membrane fluorescence intensity) in untreated and VEGF-treated cells at indicated time points of three independent experiments (a total of >300 cells). Significant differences are depicted with asterisks.
Ultrastructural analyses reveal a rapid reorganization of lateral junctional membranes in response to VEGF treatment that is followed by GJ internalization and AGJ formation
The loss in membrane fluorescence upon VEGF treatment, the concomitant increase in cytoplasmic staining, and the spherical appearance of the newly formed cytoplasmic structures appearing larger and brighter than secretory Cx43-containing transport vesicles (Lauf et al., 2002; Falk et al., 2009) suggest that GJs were internalized as complete plaques to form AGJs, as observed previously and determined to be typical for GJs (Jordan et al., 2001; Piehl et al., 2007; Baker et al., 2008; Nickel et al., 2008, 2013; Fong et al., 2014). To further support this assumption, we fixed untreated cells at 5 and 15 min after VEGF treatment and performed ultrastructural analyses by transmission electron microscopy (TEM). A number of GJs were detected in untreated cells (Figure 2, A–C, arrows) and exhibited the characteristic pentalaminar staining pattern and small size that is typical for these endogenously Cx43-expressing cells (Baker et al., 2008). In cells treated for 5 min with VEGF, plasma membranes and GJs were largely rearranged from a straight orientation to an undulating, highly folded orientation suggestive of GJs in the process of endocytosis (Figure 2D, arrows). Some AGJ vesicles showing the characteristic pentalaminar staining pattern typical for double-membrane-spanning GJs were also detected in the cytoplasm of these cells, and their number increased in the longer-treated cells (Figure 2, E and F, arrowheads). Of interest, patches of organized protein coats indicative of clathrin coats were detected on some AGJ vesicles, as observed earlier in HeLa and rabbit granulosa cells (Larsen et al., 1979; Piehl et al., 2007; Figure 2F, open arrowhead; enlarged in inset on the right). Taken together, these results suggest that GJs after VEGF treatment internalized as whole plaques, resulting in the formation of AGJs in the cytoplasm.
FIGURE 2:

VEGF induces a rapid reorganization of lateral junctional membranes and internalization of GJs into annular GJs. PAECs were processed untreated or after treatment with VEGF for 5 and 15 min and examined by thin-section electron microscopy. (A–C) GJs with distinctive pentalaminar-striped staining pattern (labeled with arrows), in general small in size, as characteristic for these endogenously Cx43-expressing cells, were detectable in untreated cells. (D) Cells in the 5-min VEGF-treated samples largely exhibited undulating lateral membranes suggestive of GJs in the process of internalization (arrows). (E, F) Cytoplasmic AGJs with vesicular, double-membrane GJ morphology (arrowheads) were also detected, especially in the VEGF-treated cells. On some AGJs, a patchy protein coat similar in appearance to clathrin coats was detected (open arrowhead in F, enlarged on the right). M, mitochondria; N, cell nucleus; PMs, lateral plasma membranes.
Activation of MAPK and PKC signaling pathways mediates VEGF-induced GJ internalization
Previous studies reporting inhibition of GJIC in response to VEGF treatment implied that MAPK and other kinases were involved in this process (Suarez and Ballmer-Hofer, 2001; Thuringer, 2004). To address whether GJ internalization in PAECs in response to VEGF is dependent on MAPK and PKC signaling, we examined the effects of inhibiting MEK (an upstream kinase of the MAPK signaling cascade; see later discussion of Figure 8) and PKC on Cx43 GJ internalization. PAECs grown on coverslips were left untreated or were VEGF treated after addition of MEK inhibitor PD98059 and PKC inhibitor GF109203X before immunofluorescence analyses to detect Cx43. As observed and shown in Figure 1, untreated cells exhibited robust Cx43 GJ staining at cell–cell appositions (Figure 3, Aa and B), whereas cells exposed to VEGF for 15 min had internalized their GJs (Figure 3, Ab and B). In contrast, GJ internalization was significantly inhibited in cells that were preincubated with the MEK inhibitor PD98059 or the PKC inhibitor GF109203X before VEGF treatment. This indicates that both kinase pathway inhibitors counteracted the effect of VEGF in inducing Cx43 GJ internalization (Figure 3, A, c and d, and B). Quantitative intracellular Cx43 fluorescence intensity analyses, again performed on >300 cells of three independent experiments, showed that the fluorescence intensity of Cx43 in the cytosol was increased to (2.6 ± 0.10)-fold after VEGF treatment and that the VEGF-induced internalization of Cx43 was reduced to (1.7 ± 0.11)- and (1.6 ± 0.10)-fold in the presence of PD98059 and GF109203X, respectively (Figure 3B). These results demonstrate that in response to VEGF-receptor activation, MAPK and PKC signaling cascades are activated in PAECs, triggering Cx43 GJ internalization.
FIGURE 8:

Schematic representation illustrating VEGF-induced, MAPK/PKC-mediated GJ internalization in PAECs. VEGF binds to, phosphorylates, and activates VEGF receptors, receptor tyrosine kinases located in the plasma membranes of endothelial cells. Activated VEGF-receptor actuates MAPK and PKC signaling cascades to phosphorylate Cx43 at Ser-255, Ser-262, Ser-279/282, and Ser-368. Phosphorylated Cx43 can then interact with and bind clathrin adaptors and clathrin to mediate GJ endocytosis.
FIGURE 3:

Inhibition of MEK (MAPK signaling pathway) and PKC prevents VEGF-induced Cx43 GJ internalization. (A) PAECs were (a) left untreated, (b) VEGF treated for 15 min, or preincubated with (c) MEK inhibitor PD98059 or (d) PKC inhibitor GF109203X 1 h before VEGF treatment. Cells were then fixed and stained for Cx43. Representative images acquired with identical camera settings. (B) Normalized quantitative analyses of intracellular fluorescence intensity of Cx43 in untreated, VEGF-treated, and VEGF-treated cells preincubated with PD98059 or GF109203X. Data represent analyses from three independent experiments (a total of >300 cells). Note the highly significant reduction in GJ internalization in cells in which MAPK and PKC signaling pathways were blocked.
VEGF-induced GJ internalization correlates with GJIC inhibition
The aforementioned data strongly suggest that GJs are internalized in response to VEGF receptor stimulation. To examine whether GJ internalization correlates with inhibition of GJIC, we performed scrape-loading dye transfer assays using the established GJ permeable dye Lucifer yellow (LY; Figure 4A). Within 5 min after scraping, LY robustly spread to many rows of adjacent cells in untreated PAECs, consistent with the high level of Cx43 GJs in these cells (Figure 4, Aa and B; cut depicted with a red dotted line; fluorescence intensity measured along the white line is depicted graphically on the right). In contrast, LY dye transfer was significantly inhibited in cells that were treated for 15 min with VEGF (Figure 4, Ab and B). In cells treated either with VEGF receptor inhibitor or with MAPK (PD98059) or PKC (GF109203X) inhibitor before VEGF addition, LY dye transfer was comparably high to that for untreated cells (Figure 4, A, c–e, and B). Quantitative fluorescence analyses performed along lines placed perpendicular to the cuts in three independent experiments revealed that GJIC in VEGF-treated cells was significantly reduced to 34.5 ± 8.46% (n = 3) compared with the untreated group (100%, n = 3), whereas dye transfer was 88 ± 2.18% (n = 3) in VEGF receptor inhibitor, 83.9 ± 9.74% (n = 3) in PD98059, and 78.9 ± 1.82 (n = 3) in GF109203X-treated cells, respectively (Figure 4B). Dye transfer was restored within 1–2 h after VEGF treatment to levels almost as high as observed in untreated cells (72.5 ± 9.3% after 1 h and 76.8 ± 4.0% after 2 h; n = 5; Supplemental Figure S1). These data correlate with the reappearance of GJs in the plasma membranes as described earlier and previously published observations in VEGF-treated human umbilical vein cells and Ea.hy926 endothelial cells (Suarez and Ballmer-Hofer, 2001).
FIGURE 4:

VEGF inhibits GJIC in PAECs. (A) PAECs were (a) left untreated or treated with (b) VEGF for 15 min without inhibitor, (c) VEGF receptor inhibitor, (d) PD98059, or (e) GF109203X, administered 1 h before VEGF treatment, followed by Lucifer yellow (LY) scrape loading dye transfer assays. Representative LY fluorescence images for each group from three independent experiments acquired with identical camera settings are shown in A, and quantitative analyses of relative dye transfers (representative of GJIC efficiencies) are shown in B. Scalpel cuts through the cell monolayers leading to cell wounding and dye uptake are depicted by dashed lines. Fluorescence intensity profiles along lines (solid) placed in representative areas perpendicular to the cuts are shown beside the images. Note efficient dye transfer, similar to untreated cells, when VEGF receptors or MAPK or PKC signaling pathways were blocked.
VEGF treatment induces phosphorylation of Cx43 on serines 255, 262, 279/282, and 368
To explore whether VEGF-induced internalization of Cx43 GJs might be triggered by phosphorylation of Cx43 and whether it involves MAPK and/or PKC signaling pathways, we examined the level of phosphorylation of known regulatory serine residues located in the Cx43 C-terminal domain. We again exposed PAECs to VEGF for 5, 15, 30, and 60 min before lysing the cells and comparing Cx43 phosphorylation levels to untreated cells by Western blot analyses using Cx43 phosphospecific antibodies. Representative Western blots are shown in Figure 5A, and quantitative analyses of three independent experiments are shown in Figure 5B. We found that VEGF induced significant phosphorylation of Cx43 serines 255, 262, 279/282, and 368 within 5 min after VEGF exposure. Phosphorylation levels peaked at 15 min after VEGF treatment and then decreased after 30–60 min, reaching levels comparable to those of untreated cells (Ser-279/282 and especially Ser-368 phosphorylation levels appeared to be delayed in reverting back to untreated levels, remaining at [1.40 ± 0.03]- and [1.87 ± 0.08]-fold after 1 h) and correlating with the general time frame observed for GJ internalization and restoration (Figure 5, A and B). In this set of experiments (n = 3), Cx43 phosphorylation reached a maximum of (2.21 ± 0.15)-fold for Ser-255, (2.73 ± 0.14)-fold for Ser-262, (2.51 ± 0.001)-fold for Ser-279/282, and (2.55 ± 0.03)-fold for Ser-368 at 15 min after VEGF treatment (Figure 5B). Levels of total Cx43 protein detected at all time points remained largely unchanged (Figure 5A, top). Levels of α-tubulin were analyzed for equal loading (Figure 5A, bottom).
FIGURE 5:

VEGF treatment induces efficient phosphorylation of Cx43 serine residues 255, 262, 279/282, and 368. (A) PAECs were left untreated or treated with VEGF for indicated times before cell lyses and Western blot analyses. Phosphorylation of Cx43 was detected using phosphospecific antibodies directed against Ser-255, Ser-262, Ser-279/282, and Ser-368. Phosphorylation (activation) of ERK1/2 was also detected using phosphospecific anti-ERK1/2 antibodies (pERK1/2). Representative Western blots. (B) Normalized quantitative analyses of phosphorylated Ser-255, Ser-262, Ser-279/282, and Ser-368 of Cx43 of three independent experiments. Note the significant increase in Cx43 serine phosphorylation, which peaked 15–30 min after VEGF treatment. (C) Quantitative kinetic analyses of VEGF-induced ERK1/2 phosphorylation/activation in PAECs over the course of a 1 h treatment as indicated by Western blot (shown in A) and immunofluorescence analyses (representative images acquired with identical camera settings are shown in C) using pERK1/2-specific antibodies. Note that ERK1/2 activation correlates with Cx43 phosphorylation.
VEGF-induced Cx43 phosphorylation is mediated by MAPK and PKC
Next we examined the potential signaling cascades that up-regulate phosphorylation of respective Cx43 serine residues upon VEGF treatment. We directly monitored activation of MAPK signaling by analyzing activation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2; downstream kinases of the MAPK signaling cascade [see later discussion of Figure 8] that are phosphorylated when active) by Western blot and immunofluorescence techniques using ERK1/2 phosphospecific antibodies (pERK1/2; Figure 5, A and C). We found that in three independent experiments, increased ERK1/2 phosphorylation was detectable 5 min after VEGF treatment, which peaked at (4.3 ± 0.002)-fold at 15 min and then dropped to (2.6 ± 0.01)-fold and levels comparable to those detected in untreated cells after 30 and 60 min, respectively (Figure 5C).
We further verified that MAPK and PKC are indeed the kinases that mediate phosphorylation of respective Cx43 serine residues by analyzing Cx43 phosphorylation patterns in cells in which VEGF receptors and MAPK and PKC signaling cascades were inhibited using the previously applied pharmacological inhibitors. Figure 6 shows that cells in this set of experiments (n = 3) treated in control with VEGF alone for 15 min exhibited significant increases in pSer-255, pSer-262, pSer-279/282, and p-Ser368 to ∼1.84 ± 0.02, 2.81 ± 0.10, 2.14 ± 0.08, and 2.35 ± 0.06, respectively, comparable to levels detected before and described earlier (Figure 5, A and B). The VEGF receptor inhibitor significantly inhibited VEGF-induced phosphorylation of ERK1/2 (Figure 6A, next to bottom) and all examined serine residues (Figure 6). The MEK inhibitor (MAPK pathway) PD98059 significantly inhibited VEGF-induced phosphorylation of ERK1/2, Ser-255, Ser-262, and Ser-279/282 but not that of Ser-368 (Figure 6). The PKC inhibitor GF109203X significantly inhibited VEGF-induced phosphorylation of Ser-368, from (∼2.35 ± 0.06)-fold (without GF109203X) to (∼1.0 ± 0.03)-fold (with GF109203X), which was similar to the basal level of Ser-368 phosphorylation observed in untreated cells (Figure 5, A and B). Of interest, the PKC inhibitor GF109203X also significantly decreased VEGF-induced phosphorylation of Ser-279/282 and Ser-255 (Figure 6), suggesting that PKC-mediated phosphorylation of Ser-368 might occur before the phosphorylation of serines 255, 262, and 279/282 by MAPK. Taken together, these results indicate that VEGF receptor activation causes a significant up-regulation of known Cx43 regulatory serine residues that correlates with and induces GJ internalization.
FIGURE 6:
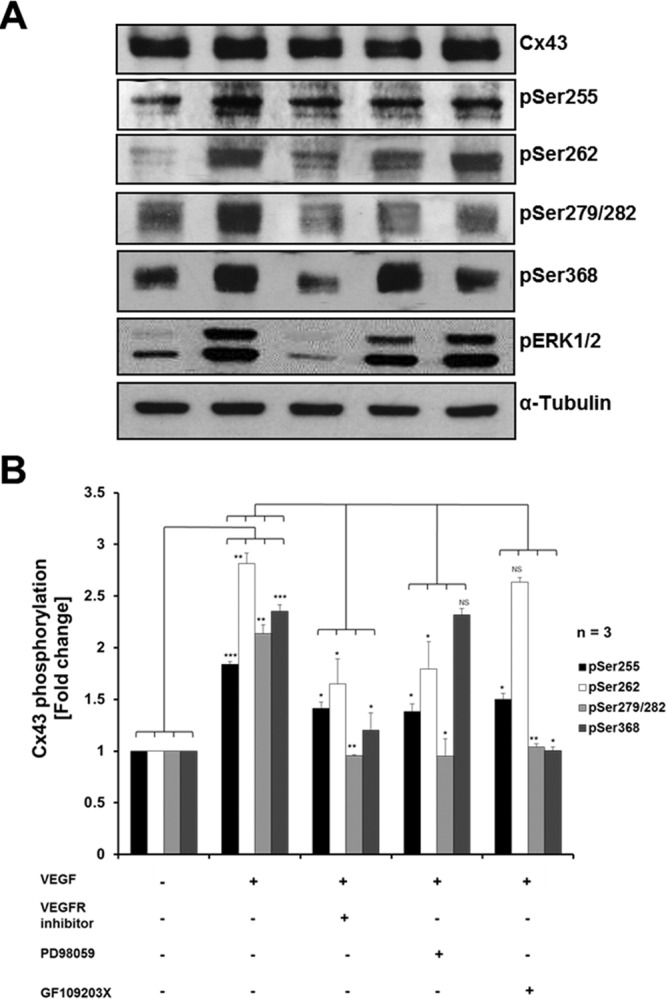
MAPK and PKC mediate VEGF-induced Cx43 phosphorylation. (A) PAECs were left untreated, treated with VEGF for 15 min, or incubated with VEGF receptor inhibitor PD98059 or GF109203X 1 h before VEGF treatment prior to cell lysis and Western blot analyses. Phosphorylation of Cx43 was detected using phosphospecific antibodies directed against Ser-255, Ser-262, Ser-279/282, and Ser-368. Phosphorylation of ERK1/2 was also detected using anti–phospho ERK1/2–specific antibodies. Representative Western blots. (B) Normalized quantitative analyses of phosphorylated Ser-255, Ser-262, Ser-279/282, and Ser-368 of Cx43 of three independent experiments. Note that VEGF-mediated Cx43 phosphorylation is reduced significantly when VEGF receptors or MAPK or PKC signaling pathways were inhibited.
VEGF-induced GJ internalization is clathrin mediated
Previous studies in our and other laboratories have revealed that Cx43 GJs are internalized utilizing the CME machinery (Piehl et al., 2007; Baker et al., 2008; Gumpert et al., 2008; Gilleron et al., 2011; Fong et al., 2013, 2014; Nickel et al., 2013). We therefore reasoned that VEGF-induced Cx43 GJ internalization is also clathrin mediated. It has been established that hypertonic conditions can efficiently prevent the budding of clathrin-coated vesicles from the plasma membrane and that this eventually will significantly inhibit CME (Heuser and Anderson, 1989; Hansen et al., 1993; Wu et al., 2001). We therefore inhibited CME by exposing PAECs to hypertonic culture medium (0.45 M sucrose) before and during VEGF exposure. As shown in Figure 7Ac and quantified in Figure 7B, VEGF-induced Cx43 GJ internalization was almost completely inhibited under these hypertonic culture conditions as compared with normal conditions (Figure 7, A, a–c, and B). Because transferrin receptor endocytosis is known to be dependent on CME (Motley et al., 2003), we used Alexa 568–conjugated transferrin uptake as a control to verify that our conditions successfully blocked CME. Significant inhibition of Alexa 568–transferrin uptake was observed under these conditions as well (Supplemental Figure S2). We also found that phosphorylation of ERK1/2 occurred after 15 min of VEGF exposure under these hypertonic conditions (unpublished data), suggesting that MAPK and PKC signaling pathways were not affected by the treatment. Similar results were obtained when VEGF-treated cells in addition were incubated with other known CME inhibitors such as dynasore, a dynamin-specific small-molecule inhibitor (Marcia et al., 2006), Pitstop 2, a small-molecule inhibitor that inhibits both clathrin-dependent and -independent endocytic processes (Dutta et al., 2012), or ikarugamycin, a macrocyclic antibiotic known to block CME (Luo et al., 2001), also resulting in significant inhibition of GJ internalization (Figure 7A, d–f). Moreover, coimmunofluorescence analyses using anti-Cx43 (green) and anti–clathrin heavy chain antibodies (red) indicated that, after 15 min of VEGF exposure, intracellular Cx43 vesicles localized close to cell–cell contacts and colocalized with clathrin, as indicated by their yellow appearance in merged images (Figure 7C, arrows). Together with the ultrastructural analyses (Figure 2F), these results suggest that VEGF-induced Cx43 GJ internalization most likely also represents a CME-mediated process.
FIGURE 7:

VEGF induces Cx43 GJ internalization via CME. (A) PAECs were (a) untreated or treated with VEGF for 15 min at 37°C in HEPES-buffered, serum-free DMEM without (b) or with (c) 0.45 M sucrose (hypertonic medium, a potent inhibitor of endocytosis) or with inhibitors of CME (d) 80 μM dynasore, (e) 30 μM Pitstop 2, or (f) 4 μM ikarugamycin before fixation and immunostaining cells for Cx43. Representative images acquired with identical camera settings are shown. (B) Quantitative analyses of plasma membrane–localized Cx43 GJ fluorescence of seven independent measurements. Note significant inhibition of Cx43 GJ internalization in cells in which CME was blocked. (C) Coimmunolocalization of Cx43 (green) and clathrin (red) in PAECs treated for 15 min with VEGF. Several colocalizing cytoplasmic vesicles (yellow) are depicted with arrows.
DISCUSSION
Integrity of blood vessel walls must be maintained precisely to prevent the leakage of plasma components and blood cells into surrounding tissues while simultaneously permitting extravasation of leukocytes and other immune cells at sites of inflammation. The GJ is one type of dynamic intercellular junction located at cell–cell contacts that regulates the selective permeability of endothelial cells. VEGF is a potent endothelial cell mitogen, with a significant role in regulating angiogenesis (Ferrara, 2004). VEGF has been reported to disrupt GJIC in endothelial cells (Kevil et al., 1998; Suarez and Ballmer-Hofer, 2001; Thuringer, 2004); however, underlying molecular mechanisms were not characterized. We investigated cellular and molecular effects of VEGF-induced GJ reorganization and characterized relevant signaling pathways in PAECs, which express high levels of endogenous Cx43 and VEGF receptors. We found that exposure to 100 ng/ml VEGF induced a rapid (within 5–15 min), almost complete but transient loss of Cx43-based GJ plaques from cell–cell appositions that correlated with a significant increase of vesicular Cx43 in the cytoplasm of the treated cells and a significant loss of GJIC (Figures 1 and 4), consistent with a study by Suarez and Ballmer-Hofer (2001). Within 30–60 min after VEGF treatment, both plasma membrane GJs and cytoplasmic fluorescence were restored, possibly based on VEGF-receptor desensitization. Ultrastructural analyses indicated a rapid reorganization of lateral junctional membranes, which included the formation of deep invaginations and a number of AGJs in the cytoplasm of the treated cells (Figure 2). This suggests that GJs internalized after VEGF treatment and formed cytoplasmic AGJs, as is typical for the removal of GJs from the plasma membrane (Jordan et al., 2001; Piehl et al., 2007; Nickel et al., 2008; Falk et al., 2009; Gilleron et al., 2009; Cone et al., 2014). Hypertonic conditions and incubation of cells in dynasore, Pitstop 2, or ikarugamycin, known potent inhibitors of CME (Heuser and Anderson, 1989; Hansen et al., 1993; Luo et al., 2001; Wu et al., 2001; Marcia et al., 2006; Dutta et al., 2012), significantly inhibited internalization (Figure 7 and Supplemental Figure S2). This suggests that VEGF-mediated GJ internalization is also clathrin driven, as has been characterized for GJ internalization under various other conditions (Piehl et al., 2007; Baker et al., 2008; Gumpert et al., 2008; Gilleron et al., 2009; Fong et al., 2013; Nickel et al., 2013; Cone et al., 2014). No obvious change in the cellular distribution of the tight junction protein ZO-1 was observed. This suggests that VEGF may preferentially affect GJ internalization.
At least seven different kinases (Akt, CK1, PKA, PKC, MAPK, CDC2, and Src) of different signal transduction pathways have been characterized that phosphorylate numerous regulatory C-terminal serine and tyrosine residues of Cx43 at various stages of its life cycle. Of these, PKC, MAPK, CDC2 (during mitosis), and Src have been reported to induce GJ channel closure and inhibition of GJIC (Kanemitsu et al., 1998; Lampe et al., 2000; Petrich et al., 2002; Polontchouk et al., 2002; Leykauf et al., 2003; Solan and Lampe, 2007, 2014; Solan et al., 2007; Sirnes et al., 2009; Thévenin et al., 2013). We therefore wondered whether in addition to the described GJIC inhibition, the GJ internalization that we observed to also occur in response to VEGF treatment might be triggered by the phosphorylation of Cx43 in GJs. Using phosphospecific Cx43 antibodies, we detected a significant, up to 2.8-fold increase in the phosphorylation of Cx43 serine residues 255, 262, 279/282, and 368 soon after VEGF exposure, which correlated with GJIC inhibition and GJ internalization. Increased phosphorylation was transient and returned within 1–2 h upon GJ restoration to levels comparable to what was detected before VEGF exposure (77%; Figures 5 and 6 and Supplemental Figure S1). Kinetics of Cx43 serine phosphorylation correlated with phosphorylation of ERK1/2, and pretreating PAECs with VEGF-receptor inhibitor abolished the VEGF-induced phosphorylation of these Cx43 serine residues. We further found that pharmacological inhibition of MEK-1 (an upstream kinase of the MAPK signaling cascade) and PKC significantly inhibited VEGF-mediated Cx43 GJ internalization (Figure 6). In addition, we detected direct activation (i.e., phosphorylation) of ERK1/2 (downstream kinases of the MAPK signaling cascade; Figure 5), indicating that MAPK- and PKC-mediated Cx43 phosphorylation events together trigger VEGF-mediated Cx43 GJ internalization. Our observations are in accordance with previous findings showing that VEGF transiently reduces GJIC via the activation of VEGF receptor-2 (VEGFR-2), eventually inducing a tyrosine kinase of the Src family to activate ERK1/2 (Suarez and Ballmer-Hofer, 2001). In addition, VEGF-mediated activation of the MAPK signaling pathway, which we characterized in pulmonary artery endothelial cells, is in agreement with a study that instead used aortic endothelial cells (Kroll and Waltenberger, 1997).
We did not analyze Cx43 tyrosine phosphorylation or activation of c-Src in our study; however, we concur that c-Src mediated phosphorylation likely occurs in response to VEGF treatment (see later discussion), as observed previously (Suarez and Ballmer-Hofer, 2001; Thuringer, 2004). Surprisingly, in one of the earlier studies, a series of specific inhibitory drugs that blocked PI3-kinase, eNOS, or PKCs had no effect on VEGF-induced GJ disruption (Suarez and Ballmer-Hofer, 2001), an observation that differs from the significantly increased Ser-368 phosphorylation and the role of PKC in response to VEGF treatment that we observed. In that study, the identity of the drug and data supporting that statement were not given, making a potential explanation for this discrepant observation impossible. In addition, the same authors report that in their hands, VEGF apparently did not change morphology and number of plasma membrane–localized GJs (Suarez and Ballmer-Hofer, 2001), an observation that does not conform to the pronounced transient loss of GJs we observed in VEGF-treated PAECs. However, the authors mention that the anti-Cx43 antibodies they used in Ea.hy926 endothelial cells may have complicated detection of transient GJ internalization, giving high cytoplasmic fluorescence and detecting only a very small number of puncta suggestive of GJs. Finally, Thuringer (2004) investigated VEGF-mediated disruption of GJs in capillary endothelial cells and observed reestablishment of GJIC when cells were treated with bradykinin (another hyperpermeabilizing agent) but not VEGF. Although these, in some instances inconsistent observations require further analyses, all studies (Kevil et al., 1998; Suarez and Ballmer-Hofer, 2001; Thuringer, 2004), including our own, report a rapid inhibition of GJIC in response to VEGF that correlates with activation of several kinases, including MAPK.
Pretreatment of PAECs with PD98059 (ERK1/2 inhibitor) before VEGF exposure significantly reduced activation/phosphorylation of ERK1/2, Ser-255, Ser-262, and Ser-279/282 but not Ser-368, suggesting that VEGF induces phosphorylation of these residues via the activation of ERK1/2 (Figure 6). This is consistent with previous observations reporting that Ser-255, Ser-262, and Ser-279/282 of Cx43 are MAPK phosphorylation sites that cause channel closure and decrease GJIC when phosphorylated (Warn-Cramer et al., 1996; Leykauf et al., 2003; Sirnes et al., 2009). Serine 368 is a well-known PKC target site, and, consistent with this, phosphorylation of this residue was abolished when cells were exposed to VEGF in the presence of the known PKC inhibitor GF109203X. Of interest, GF109203X also significantly inhibited phosphorylation of Ser-279/282 and Ser-255. Thus, it is likely that in response to VEGF, PKC is activated first, causing the phosphorylation of Ser-368 to initiate GJ channel closure (Cone et al., 2014). Then members of the MAPK pathway are activated, which leads to subsequent phosphorylation of Ser-255, Ser-262, and Ser-279/282 to trigger GJ internalization. Taking these results together, this is the first study that reports that Ser-255, Ser-262, Ser-279/282, and Ser-368, well-known Cx43 MAPK and PKC target sites, are specifically phosphorylated in response to VEGF stimulation and that phosphorylation on all or a subset of these sites induces CME-mediated GJ internalization (Figure 8). A recent related study in our laboratory showed that treating mouse embryonic stem cell colonies (which express endogenous Cx43) with epidermal growth factor (EGF) also resulted in a significant inhibition of intercellular communication (GJIC) and activation of MAPK and PKC signaling cascades to phosphorylate serines 262, 279/282, and 368 of Cx43, leading to Cx43-based GJ endocytosis (Fong et al., 2014). This study and additional studies in phorbol ester–treated (PKC-activated) Cos7 cells (Cone et al., 2014) and GJ assembly–impaired BxPC3 and Capan-1 pancreatic cancer cells (Johnson et al., 2013) also describe the serine phosphorylation–mediated internalization of Cx43 GJs. Thus, phosphorylation on well-recognized C-terminal regulatory Cx43 amino acid residues by a series of different kinases emerges as an early molecular signal that triggers GJ internalization. PKC-mediated phosphorylation on S368 may serve as the lead phosphorylation event that then triggers subsequent MAPK-mediated phosphorylation on serines 255, 262, and 279/282 to allow AP-2 and clathrin to access juxtaposed Cx43-binding sites (Fong et al., 2013; Thévenin et al., 2013), resulting in GJ internalization. Precisely which of the MAPK sites (255, 262, 279/282) are phosphorylated may be stimulant or cell-type specific. In addition, Src kinase–mediated phosphorylation, which phosphorylates Cx43 on Tyr-247 and Tyr-265, is known to inhibit GJIC (Swenson et al., 1990; Lin et al., 2001), making it likely that this kinase too is a member of the “kinase program” (Solan and Lampe, 2014) that is activated in cells to trigger GJ internalization.
Stimulation with growth factors such as VEGF, EGF, and platelet-derived growth factor, inflammatory mediators such as thrombin and endothelin, and phorbol esters (12-O-tetradecanoylphorbol-13-acetate [TPA] and derivatives, analogues of the second messenger molecule diacylglycerol that activate PKC) or ischemia, wounding, or oncogene activation efficiently down-regulates GJIC (Kevil et al., 1998; Postma et al., 1998; Suarez and Ballmer-Hofer, 2001; Thuringer, 2004; Warn-Cramer and Lau, 2004; Pahujaa et al., 2007; Sirnes et al., 2009; Solan and Lampe, 2014). For some stimulants (EGF, VEGF, thrombin, endothelin, and phorbol-12,13-dibutyrate, a TPA derivative), concomitant GJ internalization has been shown (Leithe and Rivedal, 2004; Baker et al., 2008; Cone et al., 2014; Fong et al., 2014; this study). On the basis of these observations, we postulate that inhibition of GJIC in general is accompanied by GJ internalization that is initiated by specific Cx43 C-terminal phosphorylation events as described here. Elucidating in detail the molecular signals that trigger GJ internalization and turnover will be critical for understanding the many essential and dynamic roles of GJs in physiology/pathology.
MATERIALS AND METHODS
Materials
Recombinant human vascular endothelial growth factor-165 (VEGF-165, VEGF-A) was purchased from Millipore (Billerica, MA; GF315). VEGF tyrosine kinase receptor inhibitor (N-(4-chlorophenyl)-2-[(pyridin-4-ylmethyl) amino] benzamide) was from Calbiochem (La Jolla, CA; 676481). MEK inhibitor (PD98059) was from Sigma-Aldrich (St. Louis, MO; P215). PKC inhibitor (GF109203X) was from Enzo Life Sciences (Farmingdale, NY; BML-EI246). Dynasore was from Tocris Bioscience (Ellisville, MO; 2897), Pitstop 2 was from Abcam (Cambridge, MA; AB120687), and ikarugamycin was from Biomol (Plymouth Meeting, PA; El313). Lucifer yellow was from Invitrogen (Grand Island, NY; L682). Rabbit polyclonal anti-Cx43 antibody (3512), phosphospecific rabbit anti–Cx43 Ser-368 antibody (3511), and phosphospecific rabbit anti–p44/42 MAPK (ERK1/2) monoclonal antibody (4370) were from Cell Signaling Technology (Danvers, MA). Mouse monoclonal anti–ZO-1 antibody was from Zymed Laboratories (San Francisco, CA; A11008). Rabbit polyclonal anti–Cx43 Ser-255 antibody (sc-12899-R), rabbit polyclonal anti–Cx43 Ser-262 antibody (Sc-17219-R), and rabbit polyclonal anti–Cx43 Ser-279/282 antibody (12900-R) were from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse anti–α-tubulin monoclonal antibody was from Sigma-Aldrich. Mouse anti–clathrin heavy chain monoclonal antibody was from BD (Franklin Lakes, NJ; 610499). Hoechst 33342, 4′,6-diamidino-2-phenylindole (DAPI), and Alexa 568–conjugated transferrin (T23365) were from Invitrogen.
Cell culture
PAECs were previously isolated in our laboratory as described (Baker et al., 2008) or purchased from Cell Applications (San Diego, CA; Cat. No. P302). PAECs were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 1.0% l-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin in humidified atmosphere at 37˚C, 5% CO2. PAECs were passaged and cultured in 6-cm-diameter dishes for no more than six to eight passages. Confluent cells were incubated in serum-free medium for 3 h before exposure to VEGF or kinase inhibitors. Cells were incubated with kinase inhibitors 1 h before VEGF treatment.
Immunofluorescence
PAECs were grown on glass coverslips coated with poly-l-lysine and then were left untreated or treated with 100 ng/ml VEGF (with or without MEK or PKC inhibitors). Cells were fixed and permeabilized with −20°C cold methanol for 10 min, blocked with 10% FBS/phosphate-buffered saline (PBS) for 30 min, and incubated with 1:200 of an anti-Cx43 polyclonal antibody and 1:200 of an anti–ZO-1 monoclonal antibody at 4°C overnight. After three PBS washes, cells were incubated with 1:500 secondary antibodies (Alexa 488–conjugated goat anti-rabbit and Alexa 568–conjugated goat anti-mouse) for 1 h at room temperature. Cell nuclei were counterstained with 1 μg/ml Hoechst 33342 or 1 μg/ml DAPI. Cells were mounted using Fluoromount-G (0100-01; SouthernBiotech, Birmingham, AL). Observations were performed on a Nikon EclipseTE 2000-U inverted fluorescence microscope equipped with 40× and 60× Plan-Apochromat, numerical aperture 1.4 oil-immersion objectives (Nikon, Tokyo, Japan), and micrographs were captured with a SPOT RT KE camera (Scientific Instrument Co., Campbell, CA). Quantitative image analyses were performed using ImageJ software (National Institutes of Health, Bethesda, MD).
Dye transfer assays
Confluent PAECs in 6-cm-diameter dishes were serum starved as described and treated with 100 ng/ml of VEGF, with or without the presence of inhibitors, for 15 min at room temperature. Incisions through monolayers were performed in the presence of 0.05% Lucifer yellow using a fresh razor blade, and cells were incubated for 5 min at room temperature. After three quick washes with PBS, cells were fixed with 4% paraformaldehyde for 15 min at room temperature. Then cells were washed three times with PBS containing Ca2+ and Mg2+. Observations were performed using a Nikon Eclipse TE 2000-U fluorescence microscope equipped with a 10× objective, and micrographs were captured with a SPOT RT KE camera.
Endocytosis inhibition assays
PAECs grown to confluency on poly-l-lysine–coated cover glasses were treated with 100 ng/ml VEGF for 15 min at 37°C in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)–buffered and serum-free DMEM without or with the presence of 0.45 M sucrose. To inhibit CME, cells were treated with 100 ng/ml VEGF for 15 min at 37°C in serum-free DMEM containing 30 μM Pitstop 2, 80 μM dynasore, or 4 μM ikarugamycin. Cells were fixed and immunostained with anti-Cx43 antibodies and visualized using fluorescence microscopy. Transferrin was used as a positive control to determine efficiency of CME inhibition. Cells were incubated with 50 μg/ml Alexa 568–conjugated transferrin in the absence or presence of 0.45 M sucrose (for 45 min) or pharmacological CME inhibitors (for 15 min) at 37°C. Cells were then fixed and mounted. Immunofluorescence images were captured as described.
Electron microscopic analyses
PAECs cultured in 3.5-cm-diameter dishes were left untreated or treated with VEGF as described and then fixed with 2.5% glutaraldehyde (G7651; Sigma-Aldrich) in 0.1 M sodium cacodylate buffer at room temperature for 2 h. Cells were washed, treated with tannic acid, dehydrated, uranyl acetate-stained and flat-embedded in the dishes as described (Falk, 2000; Fong et al., 2014). Embedded cells were mounted, trimmed, thin-sectioned, poststained, and examined with a Phillips CM100 electron microscope.
Western blot analyses
PAEC cell lysates were prepared by adding 1× reducing Laemmli buffer into the sample dishes. Samples were collected and heated at 98°C for 10 min and then separated in a 10% gel by SDS–PAGE and electroblotted onto nitrocellulose. Membranes were blocked with 5% nonfat dry milk in TBS-T (0.02 mol/l Tris-HCl, pH 7.6, 0.137 mol/l NaCl, and 0.1% [wt/vol] Tween 20) at room temperature for 1 h. Membranes were then incubated with primary antibodies at 4°C overnight. Primary antibodies included 1:3000 of a rabbit anti-Cx43 antibody, 1:500 of a phosphospecific rabbit anti–Cx43 Ser-255 antibody, 1:500 of a phosphospecific rabbit anti–Cx43 Ser-262 antibody, 1:500 of a phosphospecific rabbit anti–Cx43 Ser-279/282 antibody, 1:2000 of a phosphospecific rabbit anti–Cx43 Ser-368 antibody, 1:3000 of anti–phospho-ERK1/2 antibody, and 1:5000 of an anti–α-tubulin antibody. After three washes with TBS-T, membranes were incubated with 1:5000 of horseradish peroxidase–conjugated secondary antibodies for 1 h at room temperature. Immune complexes were detected using enhanced chemiluminescence reagent. Intensity of immunoreactive bands was analyzed and quantified using ImageJ software.
Statistical analyses
Unpaired Student's t tests were performed to analyze intracellular and plasma membrane fluorescence intensities (Figures 1, 3, and 7) and distances of dye transfer (Figure 4 and Supplemental Figure S1), and analysis of variance was used to compare changes in Cx43 serine phosphorylation (Figures 5 and 6) using Excel's statistics package (Microsoft, Redmond, WA). Data are presented as mean ± SD. In all analyses, a p value (*p < 0.05, **p < 0.01, and ***p < 0.001) was considered statistically significant. NS indicates nonsignificant results.
Supplementary Material
Acknowledgments
We thank Paul Lampe (Fred Hutchinson Cancer Research Center, Seattle, WA) for generously providing the pSer279/282-specific antibodies that were used in initial experiments; Malcolm Wood (Microscopy Facility, Scripps Research Institute, La Jolla, CA) for electron microscopy analyses; and Falk lab members for constructive discussions and critical reading of the manuscript. This work was supported by National Institutes of Health/National Institute of General Medical Sciences Grant GM55725 to M.M.F. and funds from Lehigh University.
Abbreviations used:
- AGJ
annular gap junction
- CME
clathrin-mediated endocytosis
- Cx
connexin
- GJ
gap junction
- GJIC
gap junction intercellular communication
- LY
Lucifer yellow
- MAPK
mitogen-activated protein kinase
- PAEC
pulmonary artery endothelial cell
- PKC
protein kinase C
- VEGF
vascular endothelial growth factor
- ZO-1
zonula occludens-1.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-06-1105) on June 10, 2015.
*Present address: Department of Biology, Faculty of Science, Chiang Mai University, Muang Chiang Mai 50202, Thailand.
REFERENCES
- Baker SM, Kim N, Gumpert AM, Segretain D, Falk MM. Acute internalization of gap junctions in vascular endothelial cells in response to inflammatory mediator-induced G-protein coupled receptor activation. FEBS Lett. 2008;582:4039–4046. doi: 10.1016/j.febslet.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardslee MA, Laing JG, Beyer EC, Saffitz JE. Rapid turnover of connexin43 in the adult rat heart. Circ Res. 1998;83:629–635. doi: 10.1161/01.res.83.6.629. [DOI] [PubMed] [Google Scholar]
- Berthoud VM, Minogue PJ, Laing JG, Beyer EC. Pathways for degradation of connexins and gap junctions. Cardiovasc Res. 2004;62:256–267. doi: 10.1016/j.cardiores.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Brock TA, Dvorak HF, Senger DR. Tumor-secreted vascular permeability factor increases cytosolic Ca2+ and von Willebrand factor release in human endothelial cells. Am J Pathol. 1991;138:213–221. [PMC free article] [PubMed] [Google Scholar]
- Chang EH, Van Camp G, Smith RJ. The role of connexins in human disease. Ear Hear. 2003;24:314–323. doi: 10.1097/01.AUD.0000079801.55588.13. [DOI] [PubMed] [Google Scholar]
- Cone AC, Cavin G, Ambrosi C, Hakozaki H, Wu-Zhang AX, Kunkel MT, Newton AC, Sosinsky GE. Protein kinase cdelta-mediated phosphorylation of connexin43 gap junction channels causes movement within gap junctions followed by vesicle internalization and protein degradation. J Biol Chem. 2014;289:8781–8798. doi: 10.1074/jbc.M113.533265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronier L, Crespin S, Strale PO, Defamie N, Mesnil M. Gap junctions and cancer: new functions for an old story. Antioxid Redox Signal. 2009;11:323–338. doi: 10.1089/ars.2008.2153. [DOI] [PubMed] [Google Scholar]
- de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 1992;255:989–991. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- Dunn CA, Lampe PD. Injury-triggered Akt phosphorylation of Cx43: a ZO-1-driven molecular switch that regulates gap junction size. J Cell Sci. 2014;127:455–464. doi: 10.1242/jcs.142497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Williamson CD, Cole NB, Donaldson JG. Pitstop 2 is a potent inhibitor of clathrin-independent endocytosis. PLoS One. 2012;7:e45799. doi: 10.1371/journal.pone.0045799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MM. Connexin-specific distribution within gap junctions revealed in living cells. J Cell Sci. 2000;113:4109–4120. doi: 10.1242/jcs.113.22.4109. [DOI] [PubMed] [Google Scholar]
- Falk MM, Baker SM, Gumpert AM, Segretain D, Buckheit RW., 3rd Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol Biol Cell. 2009;20:3342–3352. doi: 10.1091/mbc.E09-04-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MM, Kells RM, Berthoud VM. Degradation of connexins and gap junctions. FEBS Lett. 2014;588:1221–1229. doi: 10.1016/j.febslet.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon RF, Goodenough DA. Five-hour half-life of mouse liver gap-junction protein. J Cell Biol. 1981;90:521–526. doi: 10.1083/jcb.90.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- Fong JT, Kells RM, Falk MM. Two tyrosine-based sorting signals in the Cx43 C-terminus cooperate to mediate gap junction endocytosis. Mol Biol Cell. 2013;24:2834–2848. doi: 10.1091/mbc.E13-02-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong JT, Nimlamool W, Falk MM. EGF induces efficient Cx43 gap junction endocytosis in mouse embryonic stem cell colonies via phosphorylation of Ser262, Ser279/282, and Ser368. FEBS Lett. 2014;588:836–844. doi: 10.1016/j.febslet.2014.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- Ghoshroy S, Goodenough DA, Sosinsky GE. Preparation, characterization, and structure of half gap junctional layers split with urea and EGTA. J Membr Biol. 1995;146:15–28. doi: 10.1007/BF00232677. [DOI] [PubMed] [Google Scholar]
- Gilleron J, Carette D, Fiorini C, Benkdane M, Segretain D, Pointis G. Connexin 43 gap junction plaque endocytosis implies molecular remodelling of ZO-1 and c-Src partners. Commun Integr Biol. 2009;2:104–106. doi: 10.4161/cib.7626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilleron J, Carette D, Fiorini C, Dompierre J, Macia E, Denizot JP, Segretain D, Pointis G. The large GTPase dynamin2: a new player in connexin 43 gap junction endocytosis, recycling and degradation. Int J Biochem Cell Biol. 2011;43:1208–1217. doi: 10.1016/j.biocel.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Goodenough DA, Gilula NB. The splitting of hepatocyte gap junctions and zonulae occludentes with hypertonic disaccharides. J Cell Biol. 1974;61:575–590. doi: 10.1083/jcb.61.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough DA, Goliger JA, Paul DL. Connexins, connexons, and intercellular communication. Annu Rev Biochem. 1996;65:475–502. doi: 10.1146/annurev.bi.65.070196.002355. [DOI] [PubMed] [Google Scholar]
- Gumpert AM, Varco JS, Baker SM, Piehl M, Falk MM. Double-membrane gap junction internalization requires the clathrin-mediated endocytic machinery. FEBS Lett. 2008;582:2887–2892. doi: 10.1016/j.febslet.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie SC, Gilula NB. Gap junctional communication and development. Trends Neurosci. 1989;12:12–16. doi: 10.1016/0166-2236(89)90150-1. [DOI] [PubMed] [Google Scholar]
- Hansen SH, Sandvig K, van Deurs B. Clathrin and HA2 adaptors: effects of potassium depletion, hypertonic medium, and cytosol acidification. J Cell Biol. 1993;121:61–72. doi: 10.1083/jcb.121.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Anderson RG. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol. 1989;108:389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XD, Horackova M, Pressler ML. Changes in the expression and distribution of connexin 43 in isolated cultured adult guinea pig cardiomyocytes. Exp Cell Res. 1996;228:254–261. doi: 10.1006/excr.1996.0324. [DOI] [PubMed] [Google Scholar]
- Johnson KE, Mitra S, Katoch P, Kelsey LS, Johnson KR, Mehta PP. Phosphorylation on Ser-279 and Ser-282 of connexin43 regulates endocytosis and gap junction assembly in pancreatic cancer cells. Mol Biol Cell. 2013;24:715–33. doi: 10.1091/mbc.E12-07-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan K, Chodock R, Hand AR, Laird DW. The origin of annular junctions: a mechanism of gap junction internalization. J Cell Sci. 2001;114:763–773. doi: 10.1242/jcs.114.4.763. [DOI] [PubMed] [Google Scholar]
- Jordan K, Solan JL, Dominguez M, Sia M, Hand A, Lampe PD, Laird DW. Trafficking, assembly, and function of a connexin43-green fluorescent protein chimera in live mammalian cells. Mol Biol Cell. 1999;10:2033–2050. doi: 10.1091/mbc.10.6.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemitsu MY, Jiang W, Eckhart W. Cdc2-mediated phosphorylation of the gap junction protein, connexin43, during mitosis. Cell Growth Differ. 1998;9:13–21. [PubMed] [Google Scholar]
- Kevil CG, Payne DK, Mire E, Alexander JS. Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. J Biol Chem. 1998;273:15099–15103. doi: 10.1074/jbc.273.24.15099. [DOI] [PubMed] [Google Scholar]
- Kroll J, Waltenberger J. The vascular endothelial growth factor receptor KDR activates multiple signal transduction pathways in porcine aortic endothelial cells. J Biol Chem. 1997;272:32521–32527. doi: 10.1074/jbc.272.51.32521. [DOI] [PubMed] [Google Scholar]
- Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381–388. doi: 10.1016/s0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;149:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen WJ, Tung HN, Murray SA, Swenson CA. Evidence for the participation of actin microfilaments and bristle coats in the internalization of gap junction membrane. J Cell Biol. 1979;83:576–587. doi: 10.1083/jcb.83.3.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauf U, Giepmans BN, Lopez P, Braconnot S, Chen SC, Falk MM. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci USA. 2002;99:10446–10451. doi: 10.1073/pnas.162055899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leithe E, Rivedal E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J Cell Sci. 2004;117:1211–1220. doi: 10.1242/jcs.00951. [DOI] [PubMed] [Google Scholar]
- Leithe E, Sirnes S, Omori Y, Rivedal E. Downregulation of gap junctions in cancer cells. Crit Rev Oncog. 2006;12:225–256. doi: 10.1615/critrevoncog.v12.i3-4.30. [DOI] [PubMed] [Google Scholar]
- Leykauf K, Durst M, Alonso A. Phosphorylation and subcellular distribution of connexin43 in normal and stressed cells. Cell Tissue Res. 2003;311:23–30. doi: 10.1007/s00441-002-0645-5. [DOI] [PubMed] [Google Scholar]
- Lin R, Warn-Cramer BJ, Kurata WE, Lau AF. v-Src phosphorylation of connexin 43 on Tyr247 and Tyr265 disrupts gap junctional communication. J Cell Biol. 2001;154:815–827. doi: 10.1083/jcb.200102027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewenstein WR, Rose B. The cell-cell channel in the control of growth. Semin Cell Biol. 1992;3:59–79. doi: 10.1016/s1043-4682(10)80008-x. [DOI] [PubMed] [Google Scholar]
- Luo T, Fredericksen BL, Hasumi K, Endo A, Garcia JV. Human immunodeficiency virus type 1 Nef-induced CD4 cell surface downregulation is inhibited by ikarugamycin. J Virol. 2001;75:2488–2492. doi: 10.1128/JVI.75.5.2488-2492.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Mehta PP, Bertram JS, Loewenstein WR. Growth inhibition of transformed cells correlates with their junctional communication with normal cells. Cell. 1986;44:187–196. doi: 10.1016/0092-8674(86)90497-6. [DOI] [PubMed] [Google Scholar]
- Motley A, Bright NA, Seaman MN, Robinson MS. Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol. 2003;162:909–918. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil LS, Goodenough DA. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 1993;74:1065–1077. doi: 10.1016/0092-8674(93)90728-9. [DOI] [PubMed] [Google Scholar]
- Naus CC, Hearn S, Zhu D, Nicholson BJ, Shivers RR. Ultrastructural analysis of gap junctions in C6 glioma cells transfected with connexin43 cDNA. Exp Cell Res. 1993;206:72–84. doi: 10.1006/excr.1993.1122. [DOI] [PubMed] [Google Scholar]
- Nickel B, Boller M, Schneider K, Shakespeare T, Gay V, Murray SA. Visualizing the effect of dynamin inhibition on annular gap vesicle formation and fission. J Cell Sci. 2013;126:2607–2616. doi: 10.1242/jcs.116269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel BM, DeFranco BH, Gay VL, Murray SA. Clathrin and Cx43 gap junction plaque endoexocytosis. Biochem Biophys Res Commun. 2008;374:679–682. doi: 10.1016/j.bbrc.2008.07.108. [DOI] [PubMed] [Google Scholar]
- Pahujaa M, Anikin M, Goldberg GS. Phosphorylation of connexin43 induced by Src: regulation of gap junctional communication between transformed cells. Exp Cell Res. 2007;313:4083–4090. doi: 10.1016/j.yexcr.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Petrich BG, Gong X, Lerner DL, Wang X, Brown JH, Saffitz JE, Wang Y. c-Jun N-terminal kinase activation mediates downregulation of connexin43 in cardiomyocytes. Circ Res. 2002;91:640–647. doi: 10.1161/01.res.0000035854.11082.01. [DOI] [PubMed] [Google Scholar]
- Piehl M, Lehmann C, Gumpert A, Denizot JP, Segretain D, Falk MM. Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell. 2007;18:337–347. doi: 10.1091/mbc.E06-06-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polontchouk L, Ebelt B, Jackels M, Dhein S. Chronic effects of endothelin 1 and angiotensin II on gap junctions and intercellular communication in cardiac cells. FASEB J. 2002;16:87–89. doi: 10.1096/fj.01-0381fje. [DOI] [PubMed] [Google Scholar]
- Postma FR, Hengeveld T, Alblas J, Giepmans BN, Zondag GC, Jalink K, Moolenaar WH. Acute loss of cell-cell communication caused by G protein-coupled receptors: a critical role for c-Src. J Cell Biol. 1998;140:1199–1209. doi: 10.1083/jcb.140.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi JH, Claesson-Welsh L. VEGF-induced activation of phosphoinositide 3-kinase is dependent on focal adhesion kinase. Exp Cell Res. 2001;263:173–182. doi: 10.1006/excr.2000.5102. [DOI] [PubMed] [Google Scholar]
- Rhett JM, Jourdan J, Gourdie RG. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell. 2011;22:1516–1528. doi: 10.1091/mbc.E10-06-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon AM. Gap junctions: more roles and new structural data. Trends Cell Biol. 1999;9:169–170. doi: 10.1016/s0962-8924(99)01547-0. [DOI] [PubMed] [Google Scholar]
- Sirnes S, Kjenseth A, Leithe E, Rivedal E. Interplay between PKC and the MAP kinase pathway in Connexin43 phosphorylation and inhibition of gap junction intercellular communication. Biochem Biophys Res Commun. 2009;382:41–45. doi: 10.1016/j.bbrc.2009.02.141. [DOI] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membr Biol. 2007;217:35–41. doi: 10.1007/s00232-007-9035-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014;588:1423–1429. doi: 10.1016/j.febslet.2014.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez S, Ballmer-Hofer K. VEGF transiently disrupts gap junctional communication in endothelial cells. J Cell Sci. 2001;114:1229–1235. doi: 10.1242/jcs.114.6.1229. [DOI] [PubMed] [Google Scholar]
- Su V, Lau AF. Connexins: mechanisms regulating protein levels and intercellular communication. FEBS Lett. 2014;588:1212–1220. doi: 10.1016/j.febslet.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski S, Sulkowska M, Skrzydlewska E. Gap junctional intercellular communication and carcinogenesis. Pol J Pathol. 1999;50:227–233. [PubMed] [Google Scholar]
- Suzuma K, Naruse K, Suzuma I, Takahara N, Ueki K, Aiello LP, King GL. Vascular endothelial growth factor induces expression of connective tissue growth factor via KDR, Flt1, and phosphatidylinositol 3-kinase-akt-dependent pathways in retinal vascular cells. J Biol Chem. 2000;275:40725–40731. doi: 10.1074/jbc.M006509200. [DOI] [PubMed] [Google Scholar]
- Swenson KI, Piwnica-Worms H, McNamee H, Paul DL. Tyrosine phosphorylation of the gap junction protein connexin43 is required for the pp60v-src-induced inhibition of communication. Cell Regul. 1990;1:989–1002. doi: 10.1091/mbc.1.13.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 2001;20:2768–2778. doi: 10.1093/emboj/20.11.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenin AF, Kowal TJ, Fong JT, Kells RM, Fisher CG, Falk MM. Proteins and mechanisms regulating gap junction assembly. internalization and degradation. Physiology. 2013;28:93–116. doi: 10.1152/physiol.00038.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuringer D. The vascular endothelial growth factor-induced disruption of gap junctions is relayed by an autocrine communication via ATP release in coronary capillary endothelium. Ann NY Acad Sci. 2004;1030:14–27. doi: 10.1196/annals.1329.003. [DOI] [PubMed] [Google Scholar]
- Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- Warn-Cramer BJ, Lampe PD, Kurata WE, Kanemitsu MY, Loo LW, Eckhart W, Lau AF. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J Biol Chem. 1996;271:3779–3786. doi: 10.1074/jbc.271.7.3779. [DOI] [PubMed] [Google Scholar]
- Warn-Cramer BJ, Lau AF. Regulation of gap junctions by tyrosine protein kinases. Biochim Biophys Acta. 2004;1662:81–95. doi: 10.1016/j.bbamem.2003.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zhao X, Baylor L, Kaushal S, Eisenberg E, Greene LE. Clathrin exchange during clathrin-mediated endocytosis. J Cell Biol. 2001;155:291–300. doi: 10.1083/jcb.200104085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.