

Abstract
Western blot analysis is a commonly employed technique for detecting and quantifying protein levels. However, for small tissue samples, this analysis method may not be sufficiently sensitive to detect a protein of interest. To overcome these difficulties, we examined protocols for obtaining protein from adult human cardiac valves and modified these protocols for the developing early embryonic mouse counterparts. In brief, the mouse embryonic aortic valve regions, including the aortic valve and surrounding aortic wall, are collected in the minimal possible volume of a Tris-based lysis buffer with protease inhibitors. If required based on the breeding strategy, embryos are genotyped prior to pooling four embryonic aortic valve regions for homogenization. After homogenization, an SDS-based sample buffer is used to denature the sample for running on an SDS-PAGE gel and subsequent western blot analysis. Although the protein concentration remains too low to quantify using spectrophotometric protein quantification assays and have sample remaining for subsequent analyses, this technique can be used to successfully detect and semi-quantify phosphorylated proteins via western blot from pooled samples of four embryonic day 13.5 mouse aortic valve regions, each of which yields approximately 1 μg of protein. This technique will be of benefit for studying cell signaling pathway activation and protein expression levels during early embryonic mouse valve development.
Keywords: Developmental Biology, Issue 91, heart, valve, embryonic, mouse, development, protein, western blot
Introduction
Being able to identify and quantify protein expression levels is a standard technique for animal- and cell-based experiments. However, despite a long-standing interest in early embryonic cardiac valve development, evaluating protein expression in this specific tissue during development is currently limited to immunohistochemistry in both the chick and mouse1,2. Part of the difficulty of quantifying protein expression in the developing valves of most model organisms (e.g., chick and mouse) is the small size of the valves, which limits the quantity of protein that can be obtained. Thus, for quantitative analyses, researchers typically rely on RNA extraction and amplification for subsequent quantitative PCR or microarray analysis2-5. However, RNA and protein expression levels are not wholly correlative6, so focusing on RNA expression cannot provide a rigorous account of the numerous changes that occur in any given signaling pathway at various times during developments. Based on this limit in the currently available methodology, the goal of this procedure was to develop a protocol for reliably obtaining sufficient amounts of protein from the developing embryonic mouse cardiac valve regions for quantitative analysis of changes that occur in various signaling pathways that are important in the maturation of this tissue.
Embryonic valves are already commonly dissected from mice for RNA isolation and subsequent gene expression analysis2-5. However, these studies have been limited to using gene expression as a read-out of signaling pathway activation, which does not allow the detection of detect post-translational protein modifications that may affect downstream signaling. Using the RNA isolation techniques as a starting point, we began with dissecting the regions of interest. Because our interest was detecting phosphorylated proteins that were indicative of signaling pathway activity during a specific period of aortic valve development (E13.5-14.5), we performed all dissections in phosphate-free Tris buffer and collected the valves in a Tris-based lysis buffer with phosphatase and protease inhibitors. In our specific case, only the aortic valve regions were collected, but the pulmonary valve region could easily be obtained at the same time. The valve regions were then homogenized and combined with a sample buffer that is currently used to study protein expression in adult cardiac valves7. By using small sample volumes (e.g., 2 μl) and pooling valve regions from embryos with the same genotype, we were able to detect phosphorylated and nonphosphorylated proteins at embryonic day 13.5 8. Because the valve regions can be frozen and stored in lysis buffer, embryos can be genotyped if needed before pooling.
This technique broadens the set of tools that are available for evaluating cell signaling pathways during development and provides a quantitative compliment to immunohistochemistry, specifically for the developing cardiac valves. This technique should be of benefit not only to developmental cardiologists but also to all developmental biologists who work with early stage embryos are interested in regions that contain limited tissue.
Protocol
NOTE: All experiments were approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill.
1. Excise the Aortic Valve
Using an approved euthanasia technique for the embryonic stage of interest, euthanize a pregnant mouse with pups at the desired embryonic day (E) of development.
Use 70% ethanol to sterilize the abdomen of the pregnant female. Lift the skin and muscle of the lower abdomen up and away from the internal organs, and dissect open the female’s lower abdominal cavity to allow access to the uterine horn.
To retrieve the uterine horn, hold the cervix with forceps and carefully cut caudal to the forceps. Lift the uterine horn, cutting any connective tissue that keeps the horn in place, and complete the removal by cutting the junction of the uterine horn with the oviduct.
Place the dissected uterine horn in a Petri dish containing cold 0.1 M Tris buffer (pH 7.6) and rinse as needed. Then, cut the uterine horn open length-wise to expose the embryonic sacs.
To expose the embryo, cut open an embryonic sac at the junction of the placenta and the embryo and cut the umbilical vessels to free the embryo. Place the dissected embryo in a second Petri dish with cold 0.1 M Tris buffer. Return the first Petri dish with the remaining undissected embryos to ice until ready for the next embryo.
To improve chest wall access within the embryo, decapitate the embryo. If genotyping is necessary, remove and save an extra tissue piece in an Eppendorf tube placed on ice.
Place the embryo on its back, and cut the chest wall vertically along the side of the rib cage, near a forelimb, and horizontally above the diaphragm to visualize the heart. Then, open the chest wall to expose the heart.
Using forceps to hold high along the great vessels, lift the heart using forceps and cut the vessels below. Then, cut above the forceps to free the heart. If the pulmonary vessels remain uncut, the lungs may be removed with the heart and should be removed before continuing.
Cut and remove the pulmonary artery above the level of the valve; at the embryonic stages described herein, the pulmonary artery is slightly opaque, which aids in its discrimination from the valve region. Cut just below the pulmonary valve, avoiding the trabeculated ventricular myocardium and collect as described in step 1.10 if this region is also of interest. Here and in the next step, use capillary action to draw excess Tris buffer away from the region of interest prior to collecting in lysis buffer.
Carefully remove the aorta distal to the aortic valve, just above the level of the valve; like the pulmonary artery, the aorta remains slightly opaque at these embryonic stages, aiding in its removal. Cut just below the aortic valve, again avoiding the trabeculated ventricular myocardium, and transfer the valve using forceps to an Eppendorf tube with 2 μl lysis buffer (described in the Materials table) containing protease and phosphatase inhibitors on ice.
Repeat steps 1.6-1.10 until the valves have been excised from all embryos, collecting each valve in a separate Eppendorf tube. If all embryos have the same genotype, all valves may be collected in a single Eppendorf tube.
If genotyping is to be performed to determine which valves to pool together, immediately place samples in lysis buffer at -80 °C until further use. Samples can then be pooled after genotyping (step 2.1.1).
2. Protein Extraction
- Thaw collected tissues on ice and centrifuge at 13,100 x g for 1 min to collect the tissues in lysis buffer at the bottom of the tube.
- If embryos were genotyped, use a pipette to gently collect and pool lysis buffer containing the valve regions from embryos with similar genotypes. To ensure a strong band, pooling 3-4 valve regions from E13.5 embryos per sample is recommended.
- If all embryos were of the same genotype and all valve regions were collected together in Step 1.10, lyse the entire sample, as described in Step 2.2.
Check the volume of the resulting pooled sample and add lysis buffer to a total volume of 40 μl; then, disrupt and homogenize samples using 5 mm stainless steel beads in Eppendorf tubes. Operate the lyser for 2-4 min at 50 Hz, as per the manufacturer’s instructions. Spin tubes briefly, (~13,100 x g for 1 min), and transfer samples to new tubes, leaving behind the insoluble debris.
Prepare and separate samples for SDS-PAGE and transfer for subsequent western blot analysis, following a protocol such as Eslami et al9. Blotting for a loading control protein is essential for protein quantification.
Representative Results
Using this preparation technique, we were able to detect phosphorylated Smad1,5,8 (pSmad) in single aortic valve regions from E13.5 embryos. As shown in Figure 1A, the protein isolated from even a single valve region is sufficient to detect a faint pSmad band. Signal intensity increases proportionately to the number of valve regions that are pooled. Importantly, the pSmad/β-actin ratio remains nearly constant across the different sample sizes (Figure 1C). Due to the low levels of protein available, the same blot cannot be stripped and reprobed for total Smad. However, a separate blot showing total Smad 1 and β-actin shows the same expression pattern (Figure 1B).
These valve regions are almost entirely devoid of contaminating ventricular myocardium. Using this preparation technique in combination with samples of the right ventricle, we were unable to detect ventricular myocardium marker ANP in aortic valve regions, whereas this marker was readily detected in the ventricle (Figure 2). Further, the ventricular myocardial marker myosin light chain 2V is also readily detected in the ventricular sample but is barely detectable in the aortic valve regions. These results support the preciseness of the dissection.
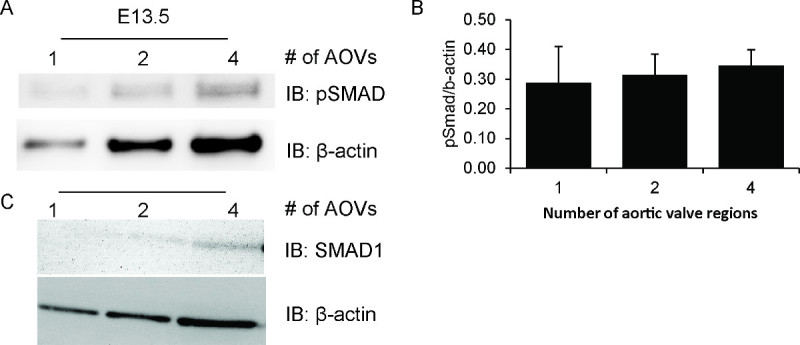 Figure 1. Smad phosphorylation in embryonic aortic valve regions. Samples were prepared with increasing numbers of pooled embryonic mouse aortic valve regions, ranging from 1 to 4 valve regions per sample. Each sample includes all proteins isolated from the entire valve region(s) in a total volume of 25-28 μl. Prepared tissues were then subjected to western blot analysis for phosphorylated Smad1,5,8 (pSmad) (A), total Smad1 (C), and β-actin (A, C). Protein was detected using Lumigen ECL Ultra and imaged with UVM VisionWorks software. The band intensity of pSmad and β-actin is proportional to the amount of starting material, as quantified in (B) using ImageJ software. The data presented are the mean and standard deviation of two separate blots.Please click here to view a larger version of this figure.
Figure 1. Smad phosphorylation in embryonic aortic valve regions. Samples were prepared with increasing numbers of pooled embryonic mouse aortic valve regions, ranging from 1 to 4 valve regions per sample. Each sample includes all proteins isolated from the entire valve region(s) in a total volume of 25-28 μl. Prepared tissues were then subjected to western blot analysis for phosphorylated Smad1,5,8 (pSmad) (A), total Smad1 (C), and β-actin (A, C). Protein was detected using Lumigen ECL Ultra and imaged with UVM VisionWorks software. The band intensity of pSmad and β-actin is proportional to the amount of starting material, as quantified in (B) using ImageJ software. The data presented are the mean and standard deviation of two separate blots.Please click here to view a larger version of this figure.
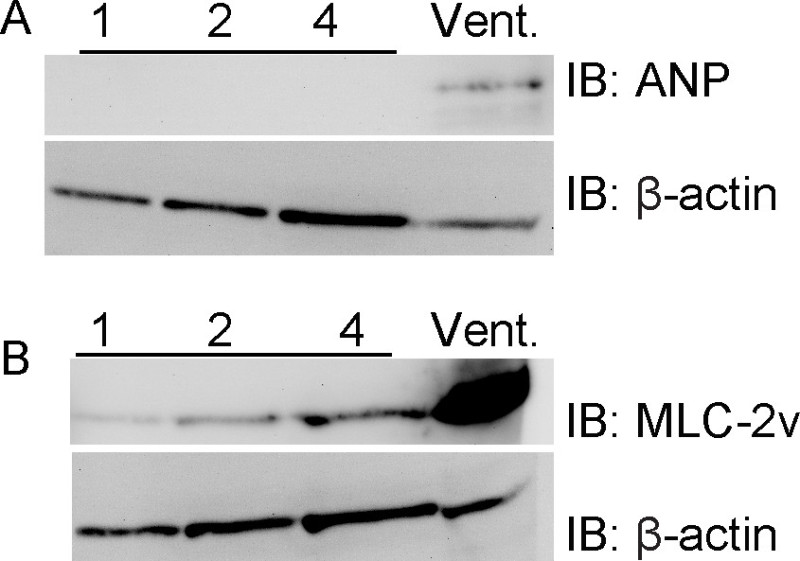 Figure 2. Aortic valve regions do not express ventricular myocardium markers. Samples were prepared with increasing numbers of pooled embryonic mouse aortic valve regions, ranging from 1 to 4 valve regions per sample, or with a sample of the right ventricle. Samples were processed as described in Figure 1 and blotted for the ventricular myocardium markers ANP (A) and myosin light chain (MLC)-2v (B). ANP is completely restricted to the ventricular sample, and MLC-2v is expressed at extremely low levels compared with the ventricular sample. β-actin is shown as a loading control.
Figure 2. Aortic valve regions do not express ventricular myocardium markers. Samples were prepared with increasing numbers of pooled embryonic mouse aortic valve regions, ranging from 1 to 4 valve regions per sample, or with a sample of the right ventricle. Samples were processed as described in Figure 1 and blotted for the ventricular myocardium markers ANP (A) and myosin light chain (MLC)-2v (B). ANP is completely restricted to the ventricular sample, and MLC-2v is expressed at extremely low levels compared with the ventricular sample. β-actin is shown as a loading control.
Discussion
The ability to quantify protein levels in early embryonic mouse and chick cardiac valve regions provides an additional tool for understanding the critical cellular signaling events for valve development. Our protocol described herein does not differ greatly from standard protein isolation procedures. However, by modifying some key steps, we have successfully obtained phosphorylated proteins from extremely small sample sizes. To achieve this outcome, the following steps are of particular importance. To ensure that quality protein is obtained, it is crucial to keep samples chilled, whether on ice or by using ice-cold buffers, in the presence of protease and, if necessary, phosphatase inhibitors. Further, homogenization with a device that is designed to process small sample volumes is essential for adequately disrupting cell membranes. Even with these precautions, the obtained yield of protein may be too low to detect via the Bradford assay with a spectrophotometer or a protein quantification kit and still have sample available for subsequent analyses. We were able to determine that each aortic valve region contains approximately 1 μg of protein by pooling 7 aortic valve regions and using the entire sample for quantification. Despite this limitation, sufficient protein is obtained to detect phosphorylated and non-phosphorylated proteins, though the lack of prior quantification makes a loading control essential. We specifically utilized β-actin because it is a high abundance protein, which allowed us to detect it even after stripping the membrane numerous times; its molecular weight differed from our proteins of interest; and it is ubiquitously expressed. A trial western blot with different numbers of pooled samples is recommended to determine how many tissues must be pooled for either embryonic cardiac valve regions at other stages of development or for other small tissues of interest. In addition, we recommend pooling at least three tissue samples for each protein sample to account for variability in the size of the dissected tissues as well as in the length of time each embryo sat on ice prior to dissection. If the protein of interest cannot be detected via western blot even after pooling samples, we recommend reducing sample volume and increasing the homogenization time to ensure that all potential protein is available for detection. Additionally, protease inhibitors may also be added to ice-cold Tris buffer during dissections for unstable or easily degraded proteins. If the protein of interest is expressed at low levels, additional troubleshooting during the western blot portion of the experiment, such as adjusting the primary antibody incubation or the substrate detection method, may help with visualization.
If the evidence (e.g., immunohistochemistry or in situ hybridization) suggests that a protein of interest is expressed both in the cardiac valves and the surrounding myocardium, the aortic or pulmonary artery myocardium can be teased apart gently with tungsten needles, even from valves from E12.5 embryos5. Because this additional step will lengthen the time of dissection and add significant technical difficulty, we suggest employing it as needed only. In our original study, we began with an immunohistochemical analysis that showed that Smad phosphorylation was only changed in the valve mesenchyme; thus, we were confident that any differences observed via western blot were due to changes in expression in the valve mesenchyme8. Further, because the developing outflow tract valves sit at the junction of the great arteries and the ventricles, performing a control blot for ventricular contamination using a ventricular myocardium-specific antibody such as ANP or MLC-2v is recommended.
Because the valves undergo dramatic reorganization during their development, we offer the following stage-specific recommendations for the cushions/valves and their surrounding tissue. For the early cushions (E9.5-11.5), both the outflow tract and atrioventricular cushions are easily visualized and dissected due to the translucency of the myocardium10; however, these embryos are small, and dissecting needles are recommended instead of forceps and microscissors. For mid-stage differentiation (E12.5-14.5)10, the semilunar valves (i.e., the aortic and pulmonary artery valves) will be more readily accessible than the atrioventricular valves (i.e., the mitral and tricuspid valves). However, the semilunar valves must be carefully dissected because the aorta and pulmonary artery are septating and rotating during this time frame11,12. Careful attention should be given to the rotation of the base of the aorta and pulmonary artery, and the midpoint between these arteries should be consistently identified. At later stages of valve maturation and condensation (E15.5 and later)10, the mitral and tricuspid valves can be teased away from inside the ventricles; however, the semilunar valves should be more easily accessible.
Western blot analysis is a long-standing technique for quantifying protein expression levels in adult cardiac valves13, where the biopsy size is large. In contrast, the complimentary early embryonic analyses have focused on either non-quantitative immunohistochemistry to detect protein expression or reverse transcription PCR, where the small starting amount of isolated mRNA can be amplified prior to analysis. These currently employed techniques provide some pertinent information regarding changes in signaling and gene expression but lack the ability to quantitatively assess protein levels. With this protocol, we can now add quantitative protein expression data to correlate with the quantitative mRNA expression data and immunohistochemical analyses.
This protocol compliments established techniques for isolating mRNA from the embryonic cardiac valves and imaging protein expression. As such, combining these techniques will allow for more complete analyses of the up- and down-regulation of signaling pathways, from mRNA to post-translational protein modification. Further, this technique allows for quantification of protein expression, which will enhance currently utilized immunohistochemistry techniques. Together, we believe that this technique will be of interest for any researcher interested in the cardiac valves or preparing other particularly small tissue explants for quantitative western blot analysis.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to thank Andrea Portbury and Davin Townley-Tilson for critical reading of the manuscript and the NIH (grant # R01HL061656) for funding support.
References
- Wirrig EE, Hinton RB, Yutzey KE. Differential expression of cartilage and bone-related proteins in pediatric and adult diseased aortic valves. J Mol Cell Cardiol. 2011;50:561–569. doi: 10.1016/j.yjmcc.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, et al. Twist1 promotes heart valve cell proliferation and extracellular matrix gene expression during development in vivo and is expressed in human diseased aortic valves. Dev Biol. 2010;347:167–179. doi: 10.1016/j.ydbio.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MP, Yutzey KE. Twist1 directly regulates genes that promote cell proliferation and migration in developing heart valves. PLoS One. 2011;6(e29758) doi: 10.1371/journal.pone.0029758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock JD, Huk DJ, Ediriweera HN, Lincoln J. Sox9 transcriptionally represses Spp1 to prevent matrix mineralization in maturing heart valves and chondrocytes. PLoS One. 2011;6(e26769) doi: 10.1371/journal.pone.0026769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Cheek J, Sakthivel B, Aronow BJ, Yutzey KE. Shared gene expression profiles in developing heart valves and osteoblast progenitor cells. Physiol Genomics. 2008;35:75–85. doi: 10.1152/physiolgenomics.90212.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, et al. Expression of functional Toll-like receptors 2 and 4 in human aortic valve interstitial cells: potential roles in aortic valve inflammation and stenosis. Am J Physiol Cell Physiol. 2008;294:29–35. doi: 10.1152/ajpcell.00137.2007. [DOI] [PubMed] [Google Scholar]
- Dyer LA, Wu Y, Moser M, Patterson C. BMPER-induced BMP signaling promotes coronary artery remodeling. Developmental Biology. 2014;386:385–394. doi: 10.1016/j.ydbio.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eslami A, Lujan J. Western blotting: sample preparation to detection. J Vis Exp. 2010. [DOI] [PMC free article] [PubMed]
- Garside VC, Chang AC, Karsan A, Hoodless PA. Co-ordinating Notch, BMP, and TGF-beta signaling during heart valve development. Cell Mol Life Sci. 2013;70:2899–2917. doi: 10.1007/s00018-012-1197-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Scherptong RW, et al. Morphogenesis of outflow tract rotation during cardiac development: the pulmonary push concept. Dev Dyn. 2012;241:1413–1422. doi: 10.1002/dvdy.23833. [DOI] [PubMed] [Google Scholar]
- Mohler ER, Adam 3rd LP, McClelland P, Graham L, Hathaway DR. Detection of osteopontin in calcified human aortic valves. Arterioscler Thromb Vasc Biol. 1997;17:547–552. doi: 10.1161/01.atv.17.3.547. [DOI] [PubMed] [Google Scholar]