

Abstract
Previous experiments using genetic and pharmacological manipulations have provided strong evidence that etomidate impairs synaptic plasticity and memory by modulating α5-subunit containing GABAA receptors (α5-GABAARs). Because α5-GABAARs mediate tonic inhibition (TI) in hippocampal CA1 pyramidal cells and etomidate enhances TI, etomidate enhancement of TI in pyramidal cells has been proposed as the underlying mechanism (Martin et al., 2009). Here we tested this hypothesis by selectively removing α5-GABAARs from pyramidal neurons (CA1–pyr–α5–KO) and comparing the ability of etomidate to enhance TI and block LTP in fl–α5 (WT), global–α5–KO (gl–α5–KO), and CA1–pyr–α5–KO mice. Etomidate suppressed LTP in slices from WT and CA1–pyr–α5–KO but not gl–α5–KO mice. There was a trend toward reduced TI in both gl–α5–KO and CA1–pyr–α5–KO mice, but etomidate enhanced TI to similar levels in all genotypes. The dissociation between effects of etomidate on TI and LTP in gl–α5–KO mice indicates that increased TI in pyramidal neurons is not the mechanism by which etomidate impairs LTP and memory. Rather, the ability of etomidate to block LTP in WT and CA1–pyr–α5–KO mice, but not in gl–α5–KO mice, points toward α5-GABAARs on nonpyramidal cells as the essential effectors controlling plasticity in this in vitro model of learning and memory.
Keywords: GABAA receptor, learning and memory, LTP, mechanisms of anesthesia
Introduction
The mechanisms by which general anesthetics produce a variety of endpoints, including hypnosis, immobility, and amnesia, remain incompletely understood. Recognizing that a unitary mechanism is unlikely, a major goal of current research in this area is to relate specific molecular- and cellular-level targets to their network- and behavioral-level consequences.
Of the many candidate targets that have been considered, GABAA receptors (GABAARs) are perhaps the most universally recognized and extensively studied. They comprise a family of heteropentameric ligand-gated anion channels that are composed of five subunits, selected from a number of subfamilies (α1–α6, β1–β3, γ1–γ3, δ, π, and ρ), most commonly two α subunits, two β subunits, and one γ subunit (Olsen and Sieghart, 2008). These receptors are modulated by a wide range of anesthetic agents, some of which act on additional molecular targets and others that are relatively specific (Jones et al., 1992; Jones and Harrison, 1993; Uchida et al., 1995). This latter group includes etomidate, an imidazole derivative that is used clinically to induce general anesthesia. This drug has also become an important experimental compound in mechanistic studies of general anesthesia because of its relative target specificity for GABAARs that incorporate the β2 or β3 subunit (Jones et al., 1992; Uchida et al., 1995; Forman, 2011).
Like many other general anesthetics, etomidate produces amnesia at a fraction of the concentration that produces the other endpoints of anesthesia (Rudolph and Antkowiak, 2004). Substantial evidence indicates that it does so by enhancing the activity of α5-subunit containing GABAARs (α5-GABAARs; Cheng et al., 2006; Orser, 2007; Martin et al., 2009). Although they are sparsely expressed elsewhere in the brain, α5-GABAARs are enriched in the hippocampus, in which they are found at extrasynaptic sites and give rise to a persistent conductance termed “tonic inhibition” (McKernan et al., 1991; Sieghart and Sperk, 2002; Caraiscos et al., 2004b). Because etomidate enhances tonic inhibition (Cheng et al., 2006) and changes in tonic inhibition can alter dendritic integration and LTP (Martin et al., 2009), a causal link has been proposed.
Our recent finding that the effects of etomidate on tonic inhibition in pyramidal cells can be dissociated from its effects on memory in vivo and synaptic plasticity in vitro challenges this notion (Zarnowska et al., 2015). Studying mice that carry a mutation in β3-GABAARs that renders them insensitive to etomidate, we found that the drug no longer enhanced tonic inhibition, but it did still impair fear conditioning to context (a hippocampus-dependent memory task) and it blocked LTP. One possible explanation for this dissociation is that etomidate might act primarily by modulating α5-GABAARs found elsewhere on pyramidal neurons, such as at GABAA,slow synapses, a subset of which use α5-GABAARs (Zarnowska et al., 2009; Capogna and Pearce, 2011). Alternatively, etomidate might impair memory by targeting α5-GABAARs found on nonpyramidal cells, as described recently for oriens-lacunosum moleculare (O-LM) interneurons (Salesse et al., 2011). We report here the results of experiments designed to distinguish between these two possibilities.
Materials and Methods
All experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Research Council) and were approved by the University of Wisconsin–Madison Institutional Animal Care and Use Committee (Madison, WI), the McLean Hospital Institutional Animal Care and Use Committee (Belmont, MA), and the Cantonal Veterinary Office (Zurich, Switzerland). All efforts were made to minimize the suffering of animals and to reduce the number of animals used.
Generation and breeding of experimental mice
To generate both global and conditional Gabra5 knock-out (KO) mice, we began by creating a floxed α5 allele (Gabra5tm2.1Uru) in C57BL/6N ES cells (Eurogentec). A 7.0 kb NheI–SpeI fragment was used as homologous DNA. Exons 4 (68 bp) and 5 (221 bp) of the Gabra5 gene were flanked with two loxP sites, inserted 1.2 kb apart into the original BamHI and EcoRI sites (Fig. 1). Selectable markers were removed in vitro. The floxed α5 allele was backcrossed onto C57BL/6J mice (The Jackson Laboratory) for at least 10 generations. These mice carrying two floxed Gabra5 alleles (fl–α5) served as pseudo-wild-type mice, but for the sake of readability, they are referred to hereafter as wild-type (WT) mice.
Figure 1.
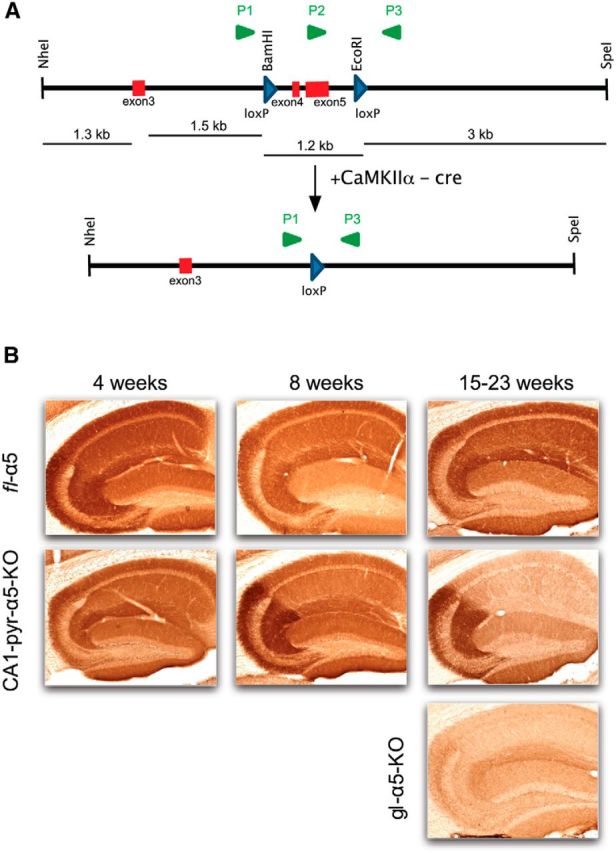
GABAAR α5 subunit KO mice. A, Generation of the floxed and conditional KO alleles. P1–P3, PCR primers used to detect floxed and KO alleles (loxP sites and primers not drawn to scale). B, Immunohistochemical detection of the GABAAR α5 subunit in the hippocampus of conditional and global α5 KO mice. Top row, fl–α5 mice (pseudo-WT) at 4, 8, and 15 weeks of age. Middle row, CA1–pyr–α5–KO mice at 4, 8, and 21 weeks of age. Bottom row, gl–α5–KO mice at 23 weeks of age. Note that CA1–pyr–α5–KO mice display apparently normal staining for the α5 subunit at 4 weeks of age, with progressive reductions at 8 and 21 weeks of age in the CA1 region driven by the expression of the CaMKIIα–Cre transgene starting in the third postnatal week (Tsien et al., 1996).
Global α5 KO (gl–α5–KO) mice were generated by excising the loxP-flanked exons 4 and 5 by cre–loxP-mediated recombination in vitro before injecting the ES cells into blastocysts. The gl–α5–KO allele was backcrossed onto C57BL/6J mice (The Jackson Laboratory) for at least nine generations. The experimental gl–α5–KO mice used for patch-clamp experiments were generated by homozygous crossings at McLean Hospital and shipped to University of Wisconsin–Madison at ∼22–25 d of age.
Conditional α5–KO mice in which the KO was restricted to pyramidal neurons, primarily in the CA1 area of the hippocampus (CA1–pyr–α5–KO), were generated by crossing CaMKIIα–Cre (T29–1) mice (Tsien et al., 1996) and fl–α5 mice. Essentially, female CaMKIIα–Cre/homozygous fl–α5 mice were bred at University of Wisconsin–Madison with male homozygous fl–α5 mice to obtain experimental animals, because the T29–1 transgene has a lower rate of germ-line recombination in the female than in the male germ line. All offspring were genotyped for the presence or absence of Cre and of the α5 KO allele to exclude undesired germ-line recombination. Mice were genotyped using DNA template from tail tips, amplified by PCR using specific primers: P1, TTTAGTGTGGGTGGTGATAGGT; P2, CTTCCACAACGGCAAGAAGTCC; and P3, CCACAGATACCCAGATGAATGTG (Fig. 1).
Immunohistochemistry
Mice were anesthetized deeply with pentobarbital (200 mg/kg) and perfused transcardially with ice-cold PBS, followed by 4% paraformaldehyde containing 15% picric acid. The brain was removed and postfixed for 4–5 h before being placed into a 30% sucrose solution for 24 h. Forty-micrometer-thick sections were collected into PBS and were kept at −20°C in an antifreeze solution until staining.
Tissue sections were washed in 50 mm Tris-buffered saline and were then incubated overnight in a Tris solution containing 0.2% Triton X-100, 2% normal goat serum, and the primary antibody (guinea pig anti-α5, 1:3000; courtesy of Dr. Jean-Marc Fritschy, University of Zurich, Zurich, Switzerland) at 4°C. Sections were then washed in Tris-buffered saline, placed in the secondary antibody solution (goat anti-guinea pig, 1:500 in 0.2% Triton X-100, 2% normal goat serum) for 30 min at room temperature, incubated in the avidin–peroxidase complex solution (Vectastain Elite ABC kit; Vector Laboratories), and washed three more times using Tris-buffered saline. Sections were reacted with DAB (0.5 mg/ml final concentration) catalyzed by hydrogen peroxide (1%) and were washed in ice-cold PBS to reduce background. Slide-mounted sections were left to air dry overnight, dehydrated with ethanol, and coverslipped.
The region-specific expression level of α5-GABAARs was quantified by optical density using NIH ImageJ version 1.48 in two fl–α5 (WT) mice aged 8 weeks, three WT mice aged 15–16 weeks, four CA1–pyr–α5–KO mice ages 8 weeks, and four CA1–pyr–α5–KO mice aged 21 weeks. For each tissue section, three regions of interest, corresponding to the molecular layers of the dentate gyrus, the CA3, and the CA1 subfields, were outlined manually. Optical density was taken as the mean gray level within the specified area, expressed on a 0–255 darkness scale, where 0 indicates white and 255 indicates black. To determine whether the expression level was altered within a given region in CA1–pyr–α5–KO mice aged 8 and 15–16 weeks, the mean optical density was compared with the same region in WT mice. Because no differences were observed in WT mice at 8 versus 15–16 weeks of age, these values were combined into a single WT group for statistical comparisons between genotypes.
Electrophysiology
Long-term potentiation.
Transverse 500-μm-thick hippocampal slices were prepared from male mice aged 8 weeks (45–80 d old, 62 ± 8 d, mean ± SD, 41 mice) or 16 weeks (110–121 d old, 115 ± 6 d, mean ± SD, 3 mice). The preparation procedure was detailed previously (Zarnowska et al., 2015). Slices were recovered in carbogenated (95% O2/5% CO2), artificial CSF (aCSF) containing the following (in mm): 127 NaCl, 1.9 KCl, 1.2 KH2PO4, 26 NaHCO3, 1.4 MgSO4 · 7H2O, 2.2 CaCl2 · 2H2O, 2.5 ascorbic acid, and 15 glucose, maintained at room temperature (21–23°C) until transfer to the experimental chamber. Extracellular recordings were obtained using a 16-channel linear array recording electrode (50 μm separating recording sites; NeuroNexus Technologies) inserted orthogonal to the hippocampal layers in the middle of the hippocampal CA1 region. Slices were bathed in aCSF at a flow rate of 2.5–3.0 ml/min at a temperature of 30 ± 0.5°C. Field EPSPs (fEPSPs) were evoked electrically by a tungsten stereotrode stimulating electrode (0.5 MΩ; World Precision Instruments) placed in the stratum radiatum to activate Schaffer collateral/commissural fibers. Recorded signals were digitized at 10 kHz (Digidata 1440A; Molecular Devices), amplified 1000×, bandpass filtered between 1 and 3000 Hz (Lynx-8 amplifiers; Neuralynx), and acquired using pClamp software (version 10.2; Molecular Devices). Stimuli 0.1 ms in duration were delivered using a constant-current stimulus isolator (model A365D; World Precision Instruments) at 0.03 Hz using a stimulus intensity (“baseline”) adjusted to evoke responses below half-maximal fEPSP amplitude. Baseline stimulus amplitude ranged from 30–150 μA and was typically between 30 and 90 μA. The recording electrode site used for LTP analysis was selected by choosing the site with the largest amplitude of fEPSP in response to a baseline stimulus and for input–output analysis of population spike (PS) amplitude as the site with a negative-going deflection superimposed on an upward fEPSP.
Recordings were performed in slices incubated in either drug-free aCSF or aCSF preincubated with etomidate for at least 1 h (Benkwitz et al., 2007). After a stable baseline period, defined as a <10% change in fEPSP amplitude over 30 min, LTP was evoked using a theta burst stimulus (TBS) protocol consisting of three stimulus trains separated by 1 min. Each train consisted of five bursts of 10 pulses at 100 Hz, delivered every 300 ms (i.e., bursts at ∼3.33 Hz), at baseline stimulus intensity. LTP was expressed as a percentage of the pre-tetanus baseline, calculated as the average fEPSP slope over the last 10 min of recording divided by the average fEPSP slope measured during the 10 min before TBS, times 100.
Tonic and phasic inhibition.
Horizontal 350-μm-thick hippocampal slices were prepared from male mice aged 7–9 weeks (64 ± 1.5 d, mean ± SD, 19 mice, experiments at 30°C in gl–α5–KO, and CA1–pyr–α5–KO and fl–α5 littermates) or 3–5 weeks (32.5 ± 1.5 d, mean ± SD, 11 mice, experiments at room temperature, gl–α5–KO and fl– α5 littermates only; data not shown). The preparation procedure was detailed previously (Zarnowska et al., 2015). In short, an ice-cold oxygenated N-methyl-d-glucamine-based cutting solution was used for transcardiac perfusion and slicing of the brain. Slices recovered while submerged in warmed (35°C) carbogenated cutting solution, which was exchanged slowly (at 5 ml/min rate) with warmed (35°C) carbogenated aCSF containing the following (in mm): 130 NaCl, 2.5 KCl, 1.25 Na2HPO4, 25 NaHCO3, 2 MgSO4 · 7H2O, 2 CaCl2 · 2H2O, 10 glucose, and 2.5 sodium ascorbate, pH 7.3 (300–310 mOsm). Thereafter, the slices were maintained at room temperature (21–23°C) until transfer to the experimental chamber. Electrophysiological recordings were performed in a submerged chamber perfused by aCSF at a flow rate of 2.5–3.0 ml/min with carbogenated aCSF at 30 ± 1°C containing kynurenic acid (KA; 3 mm) and GABA (5 μm). KA and GABA were added fresh as powder to the aCSF. CA1 pyramidal cells were visualized with a 40× water-immersion objective and infrared differential interference contrast video camera installed on a BX50WI microscope (Olympus). Patch-clamp recordings were amplified and low-pass filtered at 4 kHz using a MultiClamp 700B amplifier (Molecular Devices) and then digitized at 10 kHz using a Digidata 1322A (Molecular Devices). Borosilicate glass pipettes (1.5 mm outer diameter × 0.86 mm inner diameter; Sutter Instruments) were pulled to tip diameters of ∼1 μm using a horizontal puller (P-97; Sutter Instruments) and filled with an intracellular solution containing 90 mm CsCl, 30 mm KCl, 5 mm NaCl, 10 mm Na-HEPES, 5 mm EGTA, 4 mm Mg2ATP 2, 0.4 mm Na3GTP, 10 mm Na2 phosphocreatine, and 4 mm QX-314, pH adjusted with 1 m CsOH to 7.3 (290 ± 5 mOsm). The resistances of the pipettes filled with internal solution were 5–7 MΩ. Whole-cell voltage-clamp recordings were performed at a holding potential of −60 mV. Cell capacitance and membrane resistance were measured using the membrane test algorithm provided in the acquisition software (Clampex 10.3; Molecular Devices). Series resistance was not compensated but was monitored, and recordings were discontinued if it increased >25% through the course of an experiment. Uncompensated series resistance varied between 8 and 18 MΩ.
The effects of etomidate on tonic and phasic inhibition were measured in slices that had been preincubated in etomidate for at least 1 h (Benkwitz et al., 2007). Tonic inhibition was assessed by applying the noncompetitive GABAAR antagonist picrotoxin (PTX; 100 μm) and measuring the change in holding current. Holding current was derived from the Gaussian fit to an all-points amplitude histogram. Histograms (1 pA bin width) were constructed using 1 min of data before PTX application and 20 s of data after the attainment of a steady-state PTX effect, which required ∼2 min. Before the application of PTX, histograms were skewed toward larger negative values, reflecting the presence of inward sIPSCs. To exclude these sIPSCs in the measurement of the holding current, only the upper half of the distribution was fit to the Gaussian function. The resulting parameters were used to simulate a symmetric Gaussian curve for purposes of illustration. The difference between the peak values of the Gaussian fits before and after addition of PTX was used as the measure of the tonic current.
To detect sIPSCs, the search protocol threshold was set in Mini Analysis Program version 6 (Synaptosoft) at three times the root mean square noise level, which was typically 3–6 pA. For each cell, the averaged frequency and amplitude characteristics of sIPSCs were computed automatically. For each cell, at least 40 sIPSCs were averaged, normalized, and characterized by their 10–90% rise and weighted (τw) decay time. The sIPSCs used for averaging were selected based on the presence of a stable baseline level and the lack of spontaneous events during the deactivation phase. These events were aligned at the time of half-maximal amplitude of the rising phase. The decay phases of averaged fast sIPSCs were fitted to biexponential functions using a Simplex fitting algorithm Mini Analysis Program version 6. Weighted decay time constant of decay was calculated using the formula τw = A1/(A1 + A2) × τ1 + A2/(A1 + A2) × τ2.
Data analysis
Data analysis was performed in Clampfit 10.3 (Molecular Devices), Origin 9.0 (OriginLab), custom-written R programming language scripts (R Foundation for Statistical Computing, Vienna, Austria), GraphPad Prism version 5.04 (GraphPad Software), and Mini Analysis Program version 6 (Synaptosoft) software.
Statistics
Data are presented as mean ± SEM unless indicated otherwise, with n specifying the number of mice, slices, or cells. To assess region-specific changes in α5-GABAAR expression levels, a two-tailed t test was used to compare optical density in each of the hippocampal subfields in CA1–pyr–α5–KO mice at 8 and 15–16 weeks of age versus WT. Two-way ANOVA was used to test the effects of etomidate and genotype on tonic and synaptic currents. Subsequent post hoc comparisons were made using Tukey's test, with p values adjusted for multiple comparisons. For each comparison, the q ratio was calculated using the formula q = sqrt(2) × D/SED, where D is the difference between the two means and SED is the standard error of that difference (computed from all the data). For experiments assessing tonic inhibitory current under control conditions, a one-sample t test was used to test the null hypothesis that the shift in baseline current during PTX application was equal to zero. For experiments assessing the effects of etomidate on LTP, a one-tailed Student's t test was used to compare LTP in the presence versus absence of etomidate, because etomidate has been found in multiple studies to sometimes decrease, but never increase, LTP (Cheng et al., 2006; Martin et al., 2010; Zurek et al., 2014; Zarnowska et al., 2015). The critical value for statistical significance was set at p < 0.05. All reported significant findings have survived correction for multiple comparisons using the Benjamini–Hochberg procedure (Benjamini and Hochberg, 1995).
Chemicals
Etomidate [(R)-1-(1-phenylethyl)-1H-imidazole-5-carboxylic acid ethyl ester] was purchased from Tocris Bioscience as a powder, dissolved in DMSO, and kept in aliquots at 50 mm concentration. For each experiment, an aliquot was thawed and diluted appropriately in recording aCSF. All salts were obtained from Sigma-Aldrich. KA was obtained from Abcam, Na-HEPES was from ChemCruz Biochemicals, and CaCl2 · 2H2O was from Thermo Fisher Scientific.
Results
Expression of the GABAAR α5-subunit is reduced in the CA1 area of the hippocampus of gl–α5–KO and CA1–pyr–α5–KO mice
To determine whether the genetic modifications achieved the intended results—i.e., whether GABAAR α5 subunits were eliminated from all areas (gl–α5–KO) or selectively (CA1–pyr–α5–KO)—we assessed the presence of the GABAAR α5 subunit at ages ranging from 4 to 21 weeks by immunohistochemistry (Fig. 1). In the fl–α5 (WT) mice, dense staining was present throughout the hippocampus at all ages. In the gl–α5–KO mice, staining was reduced greatly in all regions at 4 weeks of age. In the CA1–pyr–α5–KO mice, staining was preserved throughout the hippocampus at 4 weeks of age, modestly reduced in the CA1 region at 8 weeks of age (p = 0.019 vs WT, n = 4 CA1–pyr–α5–KO and 5 WT mice, two-tailed t test), and strongly reduced at 15 weeks of age (p = 0.004 vs WT, n = 4 CA1–pyr–α5–KO and 5 WT mice, two-tailed t test). However, even at 15 weeks of age, there was significantly greater expression in the CA1 region in the CA1–pyr–α5–KO mice compared with gl–α5–KO mice (p = 0.006, n = 4 CA1–pyr–α5–KO and 1 gl–α5–KO mice, z test). No significant changes in expression were seen in the DG or CA3 regions compared with WT. This time course of changes restricted to the CA1 region matches previous reports of the age-dependent expression of CaMKIIα promoter-driven changes in NMDAR expression in CA1 pyramidal neurons of Cre (T29–1) mice (Tsien et al., 1996).
Etomidate impairs LTP in slices from WT but not gl–α5–KO mice
We investigated the effects of etomidate (0.5 and 1 μm) on LTP in slices from gl–α5–KO and fl–α5 (WT) mice (Fig. 2A,B,D). In the absence of etomidate, TBS induced LTP in both genotypes (WT: 157 ± 13%, n = 10, one-sample t test, t = 4.37, p = 0.009; gl–α5–KO: 149 ± 9%, n = 10, one-sample t test, t = 5.53, p = 0.0002). Etomidate reduced LTP in slices from WT mice (0.5 μm etomidate: 117 ± 8%, n = 5, one-tailed t test, t = 2.57, p = 0.012; 1 μm etomidate: 110 ± 10%, one-tailed t test, n = 9, t = 2.92, p = 0.005) but not gl–α5–KO mice (165 ± 8%, n = 9, one-tailed t test, t = 1.36, p = 0.90). These observations replicate previous reports that etomidate impairs LTP in WT but not gl–α5–KO mice (Cheng et al., 2006).
Figure 2.

LTP is suppressed by etomidate in fl–α5 and CA1-pyr-α5–KO mice but not gl–α5–KO mice. A, Etomidate (Etom) suppressed LTP in brain slices from fl–α5 mice. A TBS was delivered at time 0. Control (Ctrl) experiments were performed in drug-free conditions. Etomidate experiments were performed in the continuous presence of etomidate at the indicated concentrations, in brain slices that had been equilibrated with etomidate for at least 1 h before initiating the recording. Data points indicate mean ± SEM. Inset traces show representative fEPSPs recorded before TBS (thick lines) and 60 min after TBS (thin lines). B, Etomidate did not suppress LTP in brain slices from gl–α5–KO mice. C, Etomidate suppressed LTP in brain slices from CA1–pyr–α5–KO mice 8 weeks (gray circles) and 16-weeks-old (gray squares). D, Summary of LTP results. Bars show fEPSP slope (mean ± SEM) during the last 11 min (50–60 min) of recording. *p < 0.05, **p < 0.01.
Etomidate enhances residual tonic inhibition in WT and gl–α5–KO mice
We also investigated the effect of etomidate on tonic inhibition in CA1 pyramidal neurons, in brain slices bathed in aCSF supplemented with 5 μm GABA and held at 30°C (Fig. 3). Tonic current was present in both WT (16.8 ± 2.3 pA, n = 9, one-sample t test, t = 7.386, p < 0.0001) and gl–α5–KO (10.4 ± 2.2 pA, n = 5, one-sample t test, t = 4.758, p = 0.009) mice, although there was a trend toward reduced amplitude in the gl–α5–KO versus WT mice (62 ± 15%, Tukey's test, q = 2.064, p = 0.7). Nevertheless, etomidate increased tonic currents to comparable levels in both genotypes (WT, 0.5 μm: 36.4 ± 2.9 pA, n = 9; gl– α5–KO, 0.5 μm: 30.8 ± 5.3 pA, n = 6, Tukey's test, q = 1.894, p = 0.76).
Figure 3.

Etomidate potentiates tonic inhibition in fl–α5 and KO mice. A–C, Spontaneous inhibitory currents recorded in CA1 pyramidal neurons of brain slices prepared from fl–α5 (A), gl–α5–KO (B), and CA1–pyr–α5–KO (C) mice, in the absence (Ctrl) and presence of etomidate (Etom; 0.5 and 1 μm). Bars above traces represent application of the noncompetitive GABAAR channel blocker PTX (100 μm). D, Summary bar plot of amplitudes (mean ± SEM) of tonic inhibition measured in the three genotypes under different conditions. **p < 0.005, ***p < 0.0005, ****p < 0.0001; n = 5–9 cells per group.
These results showing that tonic inhibition is present (although perhaps at a reduced level) in gl–α5–KO mice and that it is enhanced to an extent that is comparable with WT are somewhat surprising given previous indications that α5-GABAARs underlie tonic inhibition in CA1 pyramidal cells (McKernan et al., 1991; Sieghart and Sperk, 2002; Caraiscos et al., 2004b).
To test whether GABA supplementation and experimental temperature might have influenced our results (Houston et al., 2012; Bright and Smart, 2013), we repeated these experiments at room temperature and without GABA added to the aCSF. As before, tonic current was present in both WT and gl–α5–KO mice (WT: 11.3 ± 2.3 pA, n = 7, one-sample t test, t = 0.499, p = 0.002; gl–α5–KO: 5.5 ± 1.1 pA, n = 8, one-sample t test, t = 0.359, p = 0.01), again with a trend toward smaller-amplitude tonic current in gl–α5–KO versus WT mice (Tukey's test, q = 1.707, p = 0.63). In the presence of etomidate (0.5 μm), tonic currents were enhanced in slices from both genotypes (WT: 224 ± 81%, n = 7, Tukey's test, q = 3.675, p = 0.07; gl–α5–KO: 294 ± 82%, n = 8, Tukey's test, q = 3.056, p = 0.005), with no difference between genotypes in percentage increase (Tukey's test, q = 2.287, p = 0.39).
We conclude that global knock-out of the Gabra5 gene might reduce, but does not eliminate, tonic inhibition in CA1 pyramidal cells. Furthermore, the tonic inhibition that remains is enhanced by etomidate to an equal or greater extent compared with WT. These conclusions are independent of experimental temperature and whether 5 μm GABA is added to the perfusate. This dissociation between the enhancement of tonic current by etomidate and its ability to impair LTP in gl–α5–KO mice indicates that etomidate suppresses LTP through a mechanism other than enhancement of tonic current in CA1 pyramidal neurons.
Etomidate impairs LTP in slices from CA1–pyr–α5–KO mice
The evidence presented above indicates that etomidate suppresses LTP by targeting α5-GABAARs but not those that give rise to tonic inhibition in pyramidal neurons. Therefore, we considered two other possibilities: (1) etomidate might impair LTP by modulating other forms of α5-GABAAR-medidated inhibition on CA1 pyramidal neurons, such as GABAA,slow, a slowly decaying (∼50 ms) synaptic response that is mediated in part by α5-GABAARs (Pearce, 1993; Banks et al., 1998; Zarnowska et al., 2009); and (2) etomidate might impair LTP by targeting α5-GABAARs present on nonpyramidal cells, such as those that underlie slow synaptic inhibition on O-LM interneurons (Chamberland and Topolnik, 2012).
To distinguish between these two possibilities, we tested whether removing α5 subunits from pyramidal neurons (CA1–pyr–α5–KO) but not interneurons would interfere with the ability of etomidate to suppress LTP (Fig. 2C). In the absence of etomidate, TBS produced robust LTP in CA1–pyr–α5–KO mice (140 ± 8%, n = 8, one-sample t test, t = 4.92, p = 0.0009), as it had in WT and gl–α5–KO mice. However, unlike gl–α5–KO mice, etomidate reduced the amplitude of LTP in CA1–pyr–α5–KO mice (120 ± 6%, n = 8, one-tailed t test, t = 1.92, p = 0.0384).
The ability of etomidate to reduce LTP in CA1–pyr–α5–KO mice thus supports the second possibility, i.e., that etomidate acts via a mechanism that is independent of pyramidal neuron α5-GABAARs. However, because the expression of CaMKIIα is developmentally regulated and at 8 weeks of age the elimination of α5 subunits may not have been complete, we performed an additional set of experiments using CA1–pyr–α5–KO mice that were aged 16 weeks, an age at which CaMKIIα–Cre has reached full expression (Tsien et al., 1996) and that our immunohistochemical results indicated that α5 subunit knock-out was complete (Fig. 1B). In these older animals, etomidate similarly blocked LTP (99 ± 9%, n = 5, one-tailed t test, t = 3.40, p = 0.0073). Therefore, we conclude that etomidate blocks LTP independently of α5-GABAARs on pyramidal neurons.
Characterization of inhibition in pyramidal cells of CA1–pyr–α5–KO mice
Although the results presented above point toward nonpyramidal cells as the target of etomidate, we examined other aspects of inhibition onto CA1 pyramidal cells of CA1–pyr–α5–KO mice to test for any unexpected changes that might have occurred and influenced our results.
Tonic current in CA1 pyramidal neurons in CA1–pyr–α5–KO mice under control conditions was similar to levels seen in WT and gl–α5–KO mice (12.1 ± 1.1 pA, Tukey's test, q = 1.789 vs WT, p = 0.80, q = 0.552 vs gl–α5–KO, p = 0.99; Fig. 3). There was a trend toward an increase in the amplitude of tonic inhibition in the presence of 0.5 μm etomidate (Tukey's test, q = 3.476, p = 0.2), to a level not different from that in gl–α5–KO mice under the same conditions (Tukey's test, CA1–pyr–α5–KO vs gl–α5–KO, q = 3.058, p = 0.43). Characteristics of fast synaptic inhibition, including IPSC frequency, amplitude, 10–90% rise time, and weighted time constant of deactivation (τdeact), were not different between genotypes under drug-free conditions (p = 0.3–0.9; Table 1), and etomidate prolonged τdeact to a similar extent in all genotypes (WT: 129 ± 13%, Tukey's test, q = 2.219, p = 0.623; gl–α5–KO: 132 ± 31%, Tukey's test, q = 3.088, p = 0.269; CA1–pyr–α5–KO: 126 ± 13%, Tukey's test, q = 2.192, p = 0.635; Table 1). Because the recording conditions for these experiments produced high rates of ongoing fast sIPSCs with substantial baseline noise attributable to tonic current, we were unable to separate and characterize slow sIPSCs, which occur infrequently in vitro even under optimized conditions (Zarnowska et al., 2009). Therefore, we are unable to provide a detailed analysis of this inhibitory component. Nevertheless, we did not observe any overt changes in GABAA,slow IPSCs, such as a strong increase in frequency or amplitude, that might have influenced our ability to induce LTP in these mice.
Table 1.
Average characteristics of sIPSCs recorded from CA1 pyramidal neurons
| Condition | Frequency (s−1) | Amplitude (pA) | 10–90% Rise time (ms) | Weighted τdecay (ms) |
|---|---|---|---|---|
| fl–α5 | ||||
| Control | 14.1 ± 1.4 | 41.8 ± 3.2 | 0.56 ± 0.07 | 6.6 ± 0.5 |
| 0.5 μm Etomidate | 19.1 ± 3.1 | 39.0 ± 3.8 | 0.63 ± 0.06 | 8.5 ± 0.8 |
| gl–α5–KO | ||||
| Control | 18.2 ± 4.2 | 46.4 ± 4.7 | 0.63 ± 0.05 | 8.2 ± 1.0 |
| 0.5 μm Etomidate | 14.6 ± 2.6 | 42.6 ± 3.7 | 0.56 ± 0.05 | 10.8 ± 2.1 |
| CA1–pyr–α5–KO | ||||
| Control | 11.8 ± 1.2 | 37.8 ± 3.6 | 0.55 ± 0.07 | 7.2 ± 0.5 |
| 0.5 μm Etomidate | 16.6 ± 2.6 | 41.6 ± 3.8 | 0.56 ± 0.06 | 9.1 ± 0.9 |
n = 5–9 cells per group.
Thus, we conclude that characteristics of tonic and synaptic inhibition are similar in pyramidal neurons of gl–α5–KO and CA1–pyr–α5–KO mice, so these factors cannot account for differences in the ability of etomidate to suppress LTP in the two genotypes.
Input–output characteristics in WT, global α5–KO, and CA1–pyr–α5–KO mice
We also considered whether the genetic alterations in the mutant mice we studied might have altered other circuit properties in ways that could have influenced their susceptibility to etomidate block of LTP, in which case the mechanism by which etomidate blocks LTP in the three genotypes may not be the same. Thus, we measured input–output relationships for fEPSP strength (Fig. 4A) and PS amplitude (Fig. 4B) to assess average excitatory synaptic strength and overall circuit responsiveness. We also examined the relationship between fEPSP strength and PS amplitude (Fig. 4C) to assess intrinsic pyramidal neuron excitability. In all cases, these relationships were similar in the three genotypes, across a range of stimulus intensities that included the stimulus intensities used to assess and induce LTP (30–90 μA). Focusing on this low range of stimulus intensities, the one measure that may have differed between genotypes was the PS amplitude in response to stimuli between 50 and 100 μA. However, in this case, the PS amplitude was greater in CA1–pyr–α5–KO mice than in gl–α5–KO or WT mice, and this higher level of “circuit responsiveness” would be expected to make CA1–pyr–α5–KO mice resistant to the effect of etomidate, rather than more sensitive than gl–α5–KO, as we observed (Fig. 2). Moreover, this change would be expected to bias the system toward stronger LTP under drug-free conditions, which was also not observed (Fig. 2), nor was there an increase in intrinsic excitability, as shown by overlapping fEPSP/PS ratios in the three genotypes (Fig. 4C). Thus, the lack of any differences between genotypes in basic circuit characteristics in a way that might explain the sensitivity of CA1–pyr–α5–KO mice to suppression of LTP by etomidate supports the conclusion that etomidate acts by the same mechanism in WT and CA1–pyr–α5–KO mice to block LTP.
Figure 4.
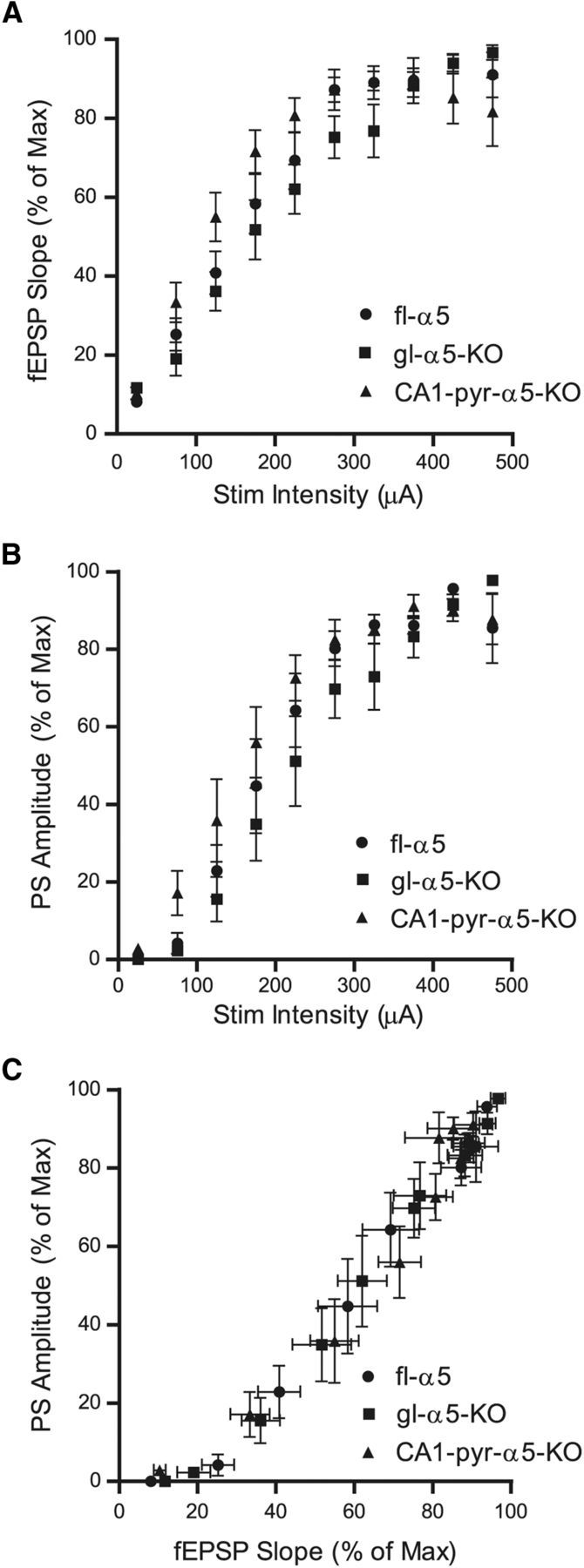
Characterization of baseline responses in fl–α5 and KO mice. A, B, Input–output relationships for fEPSP slope (A) and PS amplitude (B), obtained over a range of stimulus intensities in brain slices prepared from fl–α5 (n = 7), gl–α5–KO (n = 6), and CA1–pyr–α5–KO (n = 8) mice. The slopes of fEPSPs recorded in the stratum radiatum and amplitudes of PSs recorded near the stratum pyramidale were normalized to the maximum values attained for each brain slice. The stimulus intensities applied to each brain slice varied; therefore, values were sorted and averaged in 50 μA bins. C, Input–output relationship between fEPSP slope and PS amplitude for each mouse strain. Data are shown as mean ± SEM.
Discussion
The results presented above support the now well established concept that α5-GABAARs serve to constrain hippocampus-dependent learning and memory and that modulation of these receptors underlies anesthetic-induced amnesia. However, rather than α5-GABAARs on pyramidal neurons, which until now have been considered the most likely anesthetic targets, our results point to α5-GABAARs on non-pyramidal cells—most likely inhibitory interneurons—as the essential effectors controlling plasticity in this in vitro model of learning and memory. This conclusion is based on the finding that removing α5 subunits from hippocampal pyramidal cells (CA1–pyr–α5–KO) did not reproduce the effect of removing them from all cells (gl–α5–KO).
Whereas clinically relevant concentrations of etomidate suppressed LTP in brain slices from fl–α5 (WT) mice (Fig. 2A), brain slices from gl–α5–KO mice were resistant (Fig. 2B), as reported previously by other investigators (Cheng et al., 2006). When combined with other experiments showing that selective pharmacological or genetic manipulation of α5-GABAARs alters learning and memory in vivo as well as LTP in vitro (Collinson et al., 2002; Martin et al., 2009, 2010), the accumulated evidence points strongly toward a causal role for α5-GABAARs in suppression of LTP and memory by etomidate. Our finding that etomidate suppresses LTP in CA1–pyr–α5–KO but not in gl–α5–KO mice (Fig. 2) demonstrates that α5-GABAARs on pyramidal neurons are dispensable and indicates that α5-GABAARs on nonpyramidal cells are the critical targets.
The prevalent concept that enhancement of tonic inhibition in pyramidal neurons mediates the ability of etomidate to suppress synaptic plasticity was based on the previous studies cited above, plus immunocytochemical evidence that α5 subunits are found at extrasynaptic sites on pyramidal neuron dendrites (Brünig et al., 2002; Crestani et al., 2002), pharmacological evidence that tonic inhibition in pyramidal neurons is mediated by α5-GABAARs (Caraiscos et al., 2004b; Prenosil et al., 2006), and electrophysiological evidence that tonic inhibition is enhanced by etomidate and other anesthetics that impair memory (Caraiscos et al., 2004a). Together, these findings led to the intuitively appealing notion that these drugs impair memory by increasing membrane conductance, making dendrites “leaky” and preventing the depolarization necessary to relieve Mg2+ block of NMDARs and thereby impairing synaptic plasticity.
Given this strong and well reasoned narrative, we were surprised to discover the dissociation between enhancement of tonic current and block of LTP in β3-N265M mice, which carry a mutation in the GABAAR β3 subunit that make these receptors insensitive to etomidate (Jurd et al., 2003). In those mice, etomidate failed to increase tonic current in pyramidal neurons yet it still blocked LTP (Zarnowska et al., 2015). Our present experiments in gl–α5–KO mice revealed a second, but converse, dissociation: etomidate increased tonic current in pyramidal neurons, yet it failed to block LTP. We presume that tonic current was not completely eliminated in the α5–KO mice because of compensatory substitution by other etomidate-sensitive GABAARs. The most likely candidate is GABAARs that incorporate δ subunits, which in many other cell types are also found at extrasynaptic sites and mediate tonic current (Sperk et al., 1997; Nusser and Mody, 2002). Indeed, δ-GABAARs have been shown to undergo compensatory upregulation in hippocampal CA3 neurons of α5–KO mice (Glykys and Mody, 2006). It is also possible that the tonic current was carried by residual α5 subunits, but this explanation seems less likely given the strong reduction in immunohistochemical staining that we observed (Fig. 1B). In either case, the results present a clear dissociation between effects of etomidate on tonic current and LTP and indicate that other mechanisms must be considered. The proposal that etomidate impairs LTP by targeting nonpyramidal cells provides a ready explanation for this lack of correlation.
What might these nonpyramidal targets be? In the CA1 region, GABAergic inhibitory neurons and glial cells comprise the two major classes of nonpyramidal cells. Combined pharmacological and electrophysiological experiments have shown that α5 subunits do exist on O-LM interneurons, in which they mediate slowly decaying IPSCs (Salesse et al., 2011; Chamberland and Topolnik, 2012). Other types of interneurons also express slow GABAAR-mediated inhibition (Banks et al., 2000; Price et al., 2005; Fuentealba et al., 2008), but whether α5-GABAARs contribute at those synapses is unknown. Glial cells also express inhibitory receptors, including α5-GABAARs (Song et al., 2012; Renzel et al., 2013). Treatment of neuronal cultures with conditioned medium collected from cultured astrocytes that had been treated with etomidate has been found to enhance tonic inhibition in hippocampal neurons (Zurek et al., 2014), and glia can influence synaptic plasticity (McCall et al., 1996; Allen, 2014). Therefore, glia also represent a plausible target.
Because interneurons are generally thought to reduce pyramidal cell excitability through feedforward and feedback inhibitory pathways, it is somewhat counterintuitive to propose that anesthetic enhancement of α5-GABAAR-mediated inhibition onto interneurons might impair LTP, because this would instead increase pyramidal neuron excitability by removing the constraining influence of inhibition, as shown, for example, by the increase in burst firing that is seen when dendrite-targeting interneurons are silenced using cell-type-specific pharmacogenetic or optogenetic methods in vitro and in vivo (Lovett-Barron et al., 2012; Royer et al., 2012). Indeed, it has long been recognized that LTP is more easily elicited, and more robust, in the presence of GABAAR antagonists such as PTX.
One way to reconcile these potentially conflicting views is through disinhibitory circuits. In this scenario, interneurons that inhibit other interneurons create positive feedback loops that support the induction of LTP. O-LM cells, which are activated by pyramidal neurons, may play an important role in this regard. In addition to inhibiting the distal apical dendrites of CA1 pyramidal neurons, O-LM cells also inhibit interneurons located in the stratum radiatum that counterbalance excitatory input by targeting mid-apical dendrites in a classical feedforward inhibitory arrangement (Leão et al., 2012). By inhibiting these SR interneurons, O-LM cells allow unopposed excitatory input from the Schaffer collateral pathway to more effectively depolarize the pyramidal neurons, in turn leading to stronger firing of O-LM cells and further disinhibition, in a positive feedback loop that ultimately provides sufficient depolarization to initiate the cascade of events that leads to LTP. By enhancing the activity of α5-GABAARs that are present on O-LM cells, anesthetics might interrupt this positive feedback loop, thereby allowing conventional feedback and feedforward inhibitory influences (which are also enhanced by GABAergic anesthetics) to quench circuit activation and prevent LTP. This suggestion is in line with an emerging recognition that interneurons can control memory through either direct inhibition of pyramidal cells or disinhibitory circuits (Freund and Gulyás, 1997; Leão et al., 2012; Pi et al., 2013; Groen et al., 2014; Lovett-Barron et al., 2014; Wolff et al., 2014). It is interesting to note that this proposed mechanism, wherein anesthetics engage the circuit elements that control memory formation under natural conditions, is reminiscent of the proposal that certain anesthetics may “hijack” endogenous sleep circuitry to produce sedation and unconsciousness (Nelson et al., 2003; Lu et al., 2008).
If interruption of essential disinhibitory circuitry is the mechanism by which etomidate exerts its control of LTP, our recent finding that this drug suppresses LTP through β2-subunit-containing GABAARs (Zarnowska et al., 2015) provides a clear test of relevance for candidate interneurons and synapses that underlie this process. Biochemical and electrophysiological evidence supports the existence of α5β2- and α5β3-containing GABAARs in the CA1 region of hippocampus (Belelli et al., 1997; Sanna et al., 1997; Ju et al., 2009), and β2- and α5-containing GABAARs mediate a portion of GABAA,slow synaptic currents (Benkwitz et al., 2007; Zarnowska et al., 2009). However, the relative sparsity of α5β2-GABAARs compared with α5β3-GABAARs (Burgard et al., 1996; Sur et al., 1998; Caraiscos et al., 2004b) suggests that a limited subset of α5-mediated inhibitory processes plays a critical role in anesthetic impairment of memory circuits. Identifying those processes will be a useful step toward furthering our understanding of the mechanisms of this essential general anesthetic action.
Footnotes
This work was supported by National Institute of General Medical Sciences/National Institutes of Health Grants GM101497 (R.A.P.) and R01GM086448 (U.R.). We thank Drs. Horst Bluethmann (Roche, Basel, Switzerland) and Birgit Ledermann (University of Zurich, Zurich, Switzerland) for their help in the generation of the mutant alleles, Dr. Susumu Tonegawa (Howard Hughes Medical Institute/Massachusetts Institute of Technology, Cambridge, MA) for providing the CamKIIα–Cre (T29-1) mice, and Mark Perkins (University of Wisconsin–Madison, Madison, WI) for expert technical support.
U.R. has received compensation for professional services from Concert Pharmaceuticals. The other authors declare no competing financial interests.
References
- Allen NJ. Astrocyte regulation of synaptic behavior. Annu Rev Cell Dev Biol. 2014;30:439–463. doi: 10.1146/annurev-cellbio-100913-013053. [DOI] [PubMed] [Google Scholar]
- Banks MI, Li TB, Pearce RA. The synaptic basis of GABAA,slow. J Neurosci. 1998;18:1305–1317. doi: 10.1523/JNEUROSCI.18-04-01305.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks MI, White JA, Pearce RA. Interactions between distinct GABA(A) circuits in hippocampus. Neuron. 2000;25:449–457. doi: 10.1016/S0896-6273(00)80907-1. [DOI] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ, Peters JA, Wafford K, Whiting PJ. The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid. Proc Natl Acad Sci U S A. 1997;94:11031–11036. doi: 10.1073/pnas.94.20.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300. [Google Scholar]
- Benkwitz C, Liao M, Laster MJ, Sonner JM, Eger EI, 2nd, Pearce RA. Determination of the EC50 amnesic concentration of etomidate and its diffusion profile in brain tissue: implications for in vitro studies. Anesthesiology. 2007;106:114–123. doi: 10.1097/00000542-200701000-00020. [DOI] [PubMed] [Google Scholar]
- Bright DP, Smart TG. Methods for recording and measuring tonic GABAA receptor-mediated inhibition. Front Neural Circuits. 2013;7:193. doi: 10.3389/fncir.2013.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünig I, Scotti E, Sidler C, Fritschy JM. Intact sorting, targeting, and clustering of gamma-aminobutyric acid A receptor subtypes in hippocampal neurons in vitro. J Comp Neurol. 2002;443:43–55. doi: 10.1002/cne.10102. [DOI] [PubMed] [Google Scholar]
- Burgard EC, Tietz EI, Neelands TR, Macdonald RL. Properties of recombinant gamma-aminobutyric acid A receptor isoforms containing the alpha 5 subunit subtype. Mol Pharmacol. 1996;50:119–127. [PubMed] [Google Scholar]
- Capogna M, Pearce RA. GABA A,slow: causes and consequences. Trends Neurosci. 2011;34:101–112. doi: 10.1016/j.tins.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Caraiscos VB, Newell JG, You-Ten KE, Elliott EM, Rosahl TW, Wafford KA, MacDonald JF, Orser BA. Selective enhancement of tonic GABAergic inhibition in murine hippocampal neurons by low concentrations of the volatile anesthetic isoflurane. J Neurosci. 2004a;24:8454–8458. doi: 10.1523/JNEUROSCI.2063-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraiscos VB, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Rosahl TW, Wafford KA, MacDonald JF, Orser BA. Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 2004b;101:3662–3667. doi: 10.1073/pnas.0307231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberland S, Topolnik L. Inhibitory control of hippocampal inhibitory neurons. Front Neurosci. 2012;6:165. doi: 10.3389/fnins.2012.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng VY, Martin LJ, Elliott EM, Kim JH, Mount HTJ, Taverna FA, Roder JC, Macdonald JF, Bhambri A, Collinson N, Wafford KA, Orser BA. alpha 5GABA(A) receptors mediate the amnestic but not sedative-hypnotic effects of the general anesthetic etomidate. J Neurosci. 2006;26:3713–3720. doi: 10.1523/JNEUROSCI.5024-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinson N, Kuenzi FM, Jarolimek W, Maubach KA, Cothliff R, Sur C, Smith A, Otu FM, Howell O, Atack JR, McKernan RM, Seabrook GR, Dawson GR, Whiting PJ, Rosahl TW. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J Neurosci. 2002;22:5572–5580. doi: 10.1523/JNEUROSCI.22-13-05572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestani F, Keist R, Fritschy JM, Benke D, Vogt K, Prut L, Blüthmann H, Möhler H, Rudolph U. Trace fear conditioning involves hippocampal alpha5 GABA(A) receptors. Proc Natl Acad Sci U S A. 2002;99:8980–8985. doi: 10.1073/pnas.142288699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman SA. Clinical and molecular pharmacology of etomidate. Anesthesiology. 2011;114:695–707. doi: 10.1097/ALN.0b013e3181ff72b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Gulyás AI. Inhibitory control of GABAergic interneurons in the hippocampus. Can J Physiol Pharmacol. 1997;75:479–487. doi: 10.1139/y97-33. [DOI] [PubMed] [Google Scholar]
- Fuentealba P, Begum R, Capogna M, Jinno S, Márton LF, Csicsvari J, Thomson A, Somogyi P, Klausberger T. Ivy cells: a population of nitric-oxide-producing, slow-spiking GABAergic neurons and their involvement in hippocampal network activity. Neuron. 2008;57:917–929. doi: 10.1016/j.neuron.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Mody I. Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABA A receptor alpha5 subunit-deficient mice. J Neurophysiol. 2006;95:2796–2807. doi: 10.1152/jn.01122.2005. [DOI] [PubMed] [Google Scholar]
- Groen MR, Paulsen O, Pérez-Garci E, Nevian T, Wortel J, Dekker MP, Mansvelder HD, van Ooyen A, Meredith RM. Development of dendritic tonic GABAergic inhibition regulates excitability and plasticity in CA1 pyramidal neurons. J Neurophysiol. 2014;112:287–299. doi: 10.1152/jn.00066.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston CM, McGee TP, Mackenzie G, Troyano-Cuturi K, Rodriguez PM, Kutsarova E, Diamanti E, Hosie AM, Franks NP, Brickley SG. Are extrasynaptic GABAA receptors important targets for sedative/hypnotic drugs? J Neurosci. 2012;32:3887–3897. doi: 10.1523/JNEUROSCI.5406-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Harrison NL. Effects of volatile anesthetics on the kinetics of inhibitory postsynaptic currents in cultured rat hippocampal neurons. J Neurophysiol. 1993;70:1339–1349. doi: 10.1152/jn.1993.70.4.1339. [DOI] [PubMed] [Google Scholar]
- Jones MV, Brooks PA, Harrison NL. Enhancement of gamma-aminobutyric acid-activated Cl− currents in cultured rat hippocampal neurones by three volatile anaesthetics. J Physiol. 1992;449:279–293. doi: 10.1113/jphysiol.1992.sp019086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YH, Guzzo A, Chiu MW, Taylor P, Moran MF, Gurd JW, MacDonald JF, Orser BA. Distinct properties of murine alpha 5 gamma-aminobutyric acid type a receptors revealed by biochemical fractionation and mass spectroscopy. J Neurosci Res. 2009;87:1737–1747. doi: 10.1002/jnr.21991. [DOI] [PubMed] [Google Scholar]
- Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J. 2003;17:250–252. doi: 10.1096/fj.02-0611fje. [DOI] [PubMed] [Google Scholar]
- Leão RN, Mikulovic S, Leão KE, Munguba H, Gezelius H, Enjin A, Patra K, Eriksson A, Loew LM, Tort AB, Kullander K. OLM interneurons differentially modulate CA3 and entorhinal inputs to hippocampal CA1 neurons. Nat Neurosci. 2012;15:1524–1530. doi: 10.1038/nn.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barron M, Turi GF, Kaifosh P, Lee PH, Bolze F, Sun XH, Nicoud JF, Zemelman BV, Sternson SM, Losonczy A. Regulation of neuronal input transformations by tunable dendritic inhibition. Nat Neurosci. 2012;15:423–430. S1–S3. doi: 10.1038/nn.3024. [DOI] [PubMed] [Google Scholar]
- Lovett-Barron M, Kaifosh P, Kheirbek MA, Danielson N, Zaremba JD, Reardon TR, Turi GF, Hen R, Zemelman BV, Losonczy A. Dendritic inhibition in the hippocampus supports fear learning. Science. 2014;343:857–863. doi: 10.1126/science.1247485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Nelson LE, Franks N, Maze M, Chamberlin NL, Saper CB. Role of endogenous sleep-wake and analgesic systems in anesthesia. J Comp Neurol. 2008;508:648–662. doi: 10.1002/cne.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Oh GH, Orser BA. Etomidate targets alpha5 gamma-aminobutyric acid subtype A receptors to regulate synaptic plasticity and memory blockade. Anesthesiology. 2009;111:1025–1035. doi: 10.1097/ALN.0b013e3181bbc961. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Zurek AA, MacDonald JF, Roder JC, Jackson MF, Orser BA. Alpha5GABAA receptor activity sets the threshold for long-term potentiation and constrains hippocampus-dependent memory. J Neurosci. 2010;30:5269–5282. doi: 10.1523/JNEUROSCI.4209-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MA, Gregg RG, Behringer RR, Brenner M, Delaney CL, Galbreath EJ, Zhang CL, Pearce RA, Chiu SY, Messing A. Targeted deletion in astrocyte intermediate filament (Gfap) alters neuronal physiology. Proc Natl Acad Sci U S A. 1996;93:6361–6366. doi: 10.1073/pnas.93.13.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan RM, Quirk K, Prince R, Cox PA, Gillard NP, Ragan CI, Whiting P. GABAA receptor subtypes immunopurified from rat brain with alpha subunit-specific antibodies have unique pharmacological properties. Neuron. 1991;7:667–676. doi: 10.1016/0896-6273(91)90379-E. [DOI] [PubMed] [Google Scholar]
- National Research Council. Guide for the care and use of laboratory animals. Ed 8. Washington, DC: National Academies Press; 2011. [Google Scholar]
- Nelson LE, Lu J, Guo T, Saper CB, Franks NP, Maze M. The alpha2-adrenoceptor agonist dexmedetomidine converges on an endogenous sleep-promoting pathway to exert its sedative effects. Anesthesiology. 2003;98:428–436. doi: 10.1097/00000542-200302000-00024. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Mody I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J Neurophysiol. 2002;87:2624–2628. doi: 10.1152/jn.2002.87.5.2624. [DOI] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W. International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev. 2008;60:243–260. doi: 10.1124/pr.108.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orser BA. Lifting the fog around anesthesia. Sci Am. 2007;296:54–61. doi: 10.1038/scientificamerican0607-54. [DOI] [PubMed] [Google Scholar]
- Pearce RA. Physiological evidence for two distinct GABAA responses in rat hippocampus. Neuron. 1993;10:189–200. doi: 10.1016/0896-6273(93)90310-N. [DOI] [PubMed] [Google Scholar]
- Pi HJ, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ, Kepecs A. Cortical interneurons that specialize in disinhibitory control. Nature. 2013;503:521–524. doi: 10.1038/nature12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenosil GA, Schneider Gasser EM, Rudolph U, Keist R, Fritschy JM, Vogt KE. Specific subtypes of GABAA receptors mediate phasic and tonic forms of inhibition in hippocampal pyramidal neurons. J Neurophysiol. 2006;96:846–857. doi: 10.1152/jn.01199.2005. [DOI] [PubMed] [Google Scholar]
- Price CJ, Cauli B, Kovacs ER, Kulik A, Lambolez B, Shigemoto R, Capogna M. Neurogliaform neurons form a novel inhibitory network in the hippocampal CA1 area. J Neurosci. 2005;25:6775–6786. doi: 10.1523/JNEUROSCI.1135-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzel R, Sadek AR, Chang CH, Gray WP, Seifert G, Steinhäuser C. Polarized distribution of AMPA, but not GABAA, receptors in radial glia-like cells of the adult dentate gyrus. Glia. 2013;61:1146–1154. doi: 10.1002/glia.22505. [DOI] [PubMed] [Google Scholar]
- Royer S, Zemelman BV, Losonczy A, Kim J, Chance F, Magee JC, Buzsáki G. Control of timing, rate and bursts of hippocampal place cells by dendritic and somatic inhibition. Nat Neurosci. 2012;15:769–775. doi: 10.1038/nn.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci. 2004;5:709–720. doi: 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- Salesse C, Mueller CL, Chamberland S, Topolnik L. Age-dependent remodelling of inhibitory synapses onto hippocampal CA1 oriens-lacunosum moleculare interneurons. J Physiol. 2011;589:4885–4901. doi: 10.1113/jphysiol.2011.215244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna E, Murgia A, Casula A, Biggio G. Differential subunit dependence of the actions of the general anesthetics alphaxalone and etomidate at gamma-aminobutyric acid type A receptors expressed in Xenopus laevis oocytes. Mol Pharmacol. 1997;51:484–490. [PubMed] [Google Scholar]
- Sieghart W, Sperk G. Subunit composition, distribution and function of GABA(A) receptor subtypes. Curr Top Med Chem. 2002;2:795–816. doi: 10.2174/1568026023393507. [DOI] [PubMed] [Google Scholar]
- Song J, Zhong C, Bonaguidi MA, Sun GJ, Hsu D, Gu Y, Meletis K, Huang ZJ, Ge S, Enikolopov G, Deisseroth K, Luscher B, Christian KM, Ming GL, Song H. Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature. 2012;489:150–154. doi: 10.1038/nature11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperk G, Schwarzer C, Tsunashima K, Fuchs K, Sieghart W. GABA(A) receptor subunits in the rat hippocampus I: immunocytochemical distribution of 13 subunits. Neuroscience. 1997;80:987–1000. doi: 10.1016/S0306-4522(97)00146-2. [DOI] [PubMed] [Google Scholar]
- Sur C, Quirk K, Dewar D, Atack J, McKernan R. Rat and human hippocampal alpha5 subunit-containing gamma-aminobutyric acidA receptors have alpha5 beta3 gamma2 pharmacological characteristics. Mol Pharmacol. 1998;54:928–933. doi: 10.1124/mol.54.5.928. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/S0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Uchida I, Kamatchi G, Burt D, Yang J. Etomidate potentiation of GABAA receptor gated current depends on the subunit composition. Neurosci Lett. 1995;185:203–206. doi: 10.1016/0304-3940(95)11263-V. [DOI] [PubMed] [Google Scholar]
- Wolff SB, Gründemann J, Tovote P, Krabbe S, Jacobson GA, Müller C, Herry C, Ehrlich I, Friedrich RW, Letzkus JJ, Lüthi A. Amygdala interneuron subtypes control fear learning through disinhibition. Nature. 2014;509:453–458. doi: 10.1038/nature13258. [DOI] [PubMed] [Google Scholar]
- Zarnowska ED, Keist R, Rudolph U, Pearce RA. GABAA receptor alpha5 subunits contribute to GABAA, slow synaptic inhibition in mouse hippocampus. J Neurophysiol. 2009;101:1179–1191. doi: 10.1152/jn.91203.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnowska ED, Rodgers FC, Oh I, Rau V, Lor C, Laha KT, Jurd R, Rudolph U, Eger EI, 2nd, Pearce RA. Etomidate blocks LTP and impairs learning but does not enhance tonic inhibition in mice carrying the N265M point mutation in the beta3 subunit of the GABAA receptor. Neuropharmacology. 2015;93:171–178. doi: 10.1016/j.neuropharm.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurek AA, Yu J, Wang DS, Haffey SC, Bridgwater EM, Penna A, Lecker I, Lei G, Chang T, Salter EW, Orser BA. Sustained increase in alpha5GABAA receptor function impairs memory after anesthesia. J Clin Invest. 2014;124:5437–5441. doi: 10.1172/JCI76669. [DOI] [PMC free article] [PubMed] [Google Scholar]