

Highlights
-
•
MeCP2 is upregulated in the dorsal root ganglia (DRG) from a spared nerve injury model of neuropathic pain.
-
•
miR-19a- and miR-301-mediated regulation of MeCP2 resulted in Bdnf alteration.
-
•
MeCP2 T158A mice have decreased mechanical sensitivity.
-
•
Bdnf is downregulated in the DRG in Mecp2-null mice and MeCP2 T158A mice.
Abbreviations: ATF3, activating transcription factor 3; BDNF, brain derived neurotrophic factor; CFA, complete Freund’s adjuvant; DRG, dorsal root ganglia; L4/L5, 4th or 5th lumbar vertebra; MeCP2, methyl-CpG-binding protein 2; MeCP2 T158A/Y, male MeCP2 T158A knock in mouse; RTT, Rett syndrome; SNI, spared nerve injury; T158A, threonine 158 conversion to alanine; TrkB, tropomyosin receptor kinase B; 3′UTR, three prime untranslated region; −/Y, male Mecp2-null mouse; +/Y, male wild-type littermate control for either MeCP2 T158A knock in mouse or Mecp2-null mouse
Keywords: MeCP2, miRNA, Neuropathic pain, BDNF
Abstract
Nerve injury induces chronic pain and dysregulation of microRNAs in dorsal root ganglia (DRG). Several downregulated microRNAs are predicted to target Mecp2. MECP2 mutations cause Rett syndrome and these patients report decreased pain perception. We confirmed MeCP2 upregulation in DRG following nerve injury and repression of MeCP2 by miRNAs in vitro. MeCP2 regulates brain-derived neurotrophic factor (BDNF) and downregulation of MeCP2 by microRNAs decreased Bdnf in vitro. MeCP2 T158A mice exhibited reduced mechanical sensitivity and Mecp2-null and MeCP2 T158A mice have decreased Bdnf in DRG. MeCP2-mediated regulation of Bdnf in the DRG could contribute to altered pain sensitivity.
1. Introduction
Chronic pain is a prevalent, disabling health condition affecting more than 100 million people in the United States [1]. Neuropathic pain caused by injury or dysfunction of the nervous system can result in peripheral and central sensitization, a state of hyperexcitability due to reduction in threshold and an amplification in the responsiveness of nociceptors [2]. Elucidation of signaling pathways underlying pain hypersensitivity is crucial in improving our understanding of molecular mechanisms that drive plasticity in the nervous system promoting the development and maintenance of chronic pain states [3]. Animal models of neuropathic pain have been pivotal in the exploration of molecular mechanisms of pain underlying nerve injury [4]. Our miRNA profiling study in the dorsal root ganglion (DRG) from a nerve injury model identified 63 miRNAs differentially regulated compared to sham controls [5]. Bioinformatic prediction indicates 15 downregulated miRNAs are predicted to target the 3′ untranslated region (3′UTR) of Mecp2. Translational repression of MeCP2 mediated by binding of miRNAs to the 3′UTR has been reported [6–10].
MeCP2 binds methylated DNA, and together with co-repressors or transcriptional activators, mediates downstream changes in gene expression either directly or by altering chromatin structure [11,12]. Mutations in the X-linked methyl-CpG-binding protein 2 (MECP2) gene cause Rett syndrome (RTT) [13,14]. Decreased pain perception is commonly reported in children with RTT [15,16]. The abnormal sensitivity can be as high as 75%, with a potential relationship between hyposensitivity and specific mutations in the MECP2 gene [15]. The observations from RTT patients indicate that decreased functional MeCP2 contributes either directly or indirectly to reduced pain sensitivity. Since reduced pain sensitivity observed in RTT patients results from a decrease in functional MeCP2 protein, we postulated that a decrease in miRNAs that bind and repress translation of MeCP2 will cause an increase in MeCP2 levels and thus contribute to hypersensitivity.
MeCP2 can mediate downstream transcriptional changes of a large number of genes and depending on its interacting protein partners and target genes, MeCP2 can act either as an activator or repressor [12,17]. MeCP2 is a known regulator of Bdnf [18], a neurotrophin important in both neuronal development and peripheral pain mechanisms [19]. BDNF levels in the hypothalamus correlated with MeCP2 levels, with lower levels in Mecp2-null mice and higher levels in MECP2 overexpressing mice compared to controls [12]. The role of BDNF in both inflammatory and neuropathic pain is well established [20–23]. Here we investigated if miRNAs downregulated in a rodent model of neuropathic pain that modulate MeCP2 expression, can induce changes in Bdnf levels in Neuro-2a cells.
Several mouse models have been generated for investigating MeCP2 function [24] including alteration of the endogenous Mecp2 gene, or introduction of the human MECP2 gene with RTT-associated mutations. One of the most common mutations observed in RTT is in amino acid T158, located at the C terminus of the methyl-CpG binding domain of MeCP2. It has been reported that 70.6% of patients with this mutation have decreased pain sensitivity [15]. The phenotype of MeCP2 T158A knockin mice resembles developmental symptoms found in RTT patients [25]. MeCP2 T158A mice showed a reduction in MeCP2 binding to methylated DNA and a decrease in MeCP2 protein stability. Female Mecp2+/− mice and a conditional mouse allele that expresses 50% of the wild-type level of MeCP2 had a slower reaction to a conductive heat stimulus [26,27]. We sought to assess the pain sensitivity of MeCP2 T158A mice as well as expression of Bdnf to test our hypothesis that MeCP2 plays a role in mediating pain sensitivity and confirm the functional implication of a mutation in the methyl binding domain.
Thus here we sought to determine whether miRNAs downregulated after nerve injury regulate MeCP2 and hence modulate Bdnf expression, contributing to hypersensitivity. We used MeCP2 T158A mice to determine if a point mutation in the methyl binding domain can attenuate pain sensitivity, and we measured expression of Bdnf in the DRG from MeCP2 T158A mice and Mecp2-null mice to determine the regulatory role of MeCP2 on Bdnf in the DRG.
2. Materials and methods
2.1. Cell culture, transfection and luciferase reporter assay
HEK293 and Neuro-2a cells obtained from American Type Culture Collection (ATCC) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum at 37 °C in 5% CO2. For the reporter assay HEK293 cells were co-transfected with precursor miRNA or anti-miRNA plasmid (GeneCopoeia) and luciferase reporter plasmid containing the 3′UTR of Mecp2 using Lipofectamine 2000 (Life Technologies) for 48 h. The ∼8.5 kb 3′UTR sequence of Mecp2 was cloned downstream of the luciferase reporter gene as 4 constructs of ∼2 kb fragment each (GeneCopoeia). The fragment with the miRNA target sequence of interest (1–2210 bp or 6360–8591 bp) was co-transfected with corresponding miRNA. The Luc-Pair Duo-Luciferase assay (GeneCopoeia) was used to measure firefly and renilla luciferase according to the manufacturer’s instructions. Firefly luciferase measurements normalized to renilla was used as a transfection control. For monitoring changes in endogenous MeCP2, Neuro-2a cells were transfected with precursor miRNA or anti-miRNA plasmid (GeneCopoeia) using X-tremeGENE HP (Roche) for 48 h.
2.2. Western blot
Protein from Neuro-2a cells or DRG was isolated using radioimmunoprecipitation assay buffer (Thermo Scientific). For western blotting, 20 μg protein lysates were resolved by a 4–12% SDS–PAGE gel, transferred to the nitrocellulose membrane. The membranes were probed with MeCP2 antibody [25] at 1:3000 dilution overnight. Chemiluminescence was detected using FluorChem M System (Protein Simple). The membrane was also probed with goat anti-rabbit GAPDH-HRP (1:2000 dilution, Santa Cruz) as a loading control. Quantification was determined using UN-SCAN-IT software, MeCP2 expression was normalized to GAPDH.
2.3. Immunocytochemistry
Neuro-2a cells grown on 12mm glass coverslips were transfected with miRNA precursor plasmids with GFP using X-tremeGENE HP DNA transfection reagent for 48 h. Cells were fixed in 4% formaldehyde and blocked in 10% normal goat serum followed by a 3 h incubation in 1:200 anti-MeCP2 antibody (mentioned above). Anti-Rabbit-IgG Atto 647N secondary antibody (Sigma) was used for detection of MeCP2. Coverslips were mounted using Vectashield Mounting Medium with DAPI (Vector Laboratories). Images were acquired using the 60× objective on the Olympus 1 × 81 confocal microscope and Fluoview FV10-ASW software. Image J was used for quantification. MeCP2 levels were normalized to DAPI staining of the nucleus. Transfected (GFP positive) and untransfected (GFP negative) cells were then compared.
2.4. Quantitative RT-PCR
RNA was purified from Neuro-2a cells, DRG collected from spared nerve injury (SNI) model, sham control, Mecp2-null, MeCP2 T158A and the corresponding wild-type littermate mice using the mirVana RNA isolation kit (Life technologies). cDNA synthesis and qRT-PCR were performed as previously described [28]. The Assay ID for the Taqman primer probes used were Mm01193535_M1 (Mecp2) and Mm04230607_s1 (Bdnf). Gapdh was used as the normalizer and a t-test was used to perform statistical analysis.
2.5. Animal model of neuropathic pain
The care and use of all mice were approved by the Institutional Animal Care & Use Committee of Drexel University College of Medicine. The SNI model was generated using 8-week old C57BL/6 male mice (Taconic) as previously described [29,30]. Briefly, mice were anesthetized with isoflurane during surgery. The common peroneal and tibial nerves of the left paw were ligated, 2–4 mm of the nerve was sectioned and removed distal to ligation. Sham mice underwent the same surgical procedures as the SNI group without ligation and sectioning. Development of mechanical hypersensitivity was assessed using von Frey filaments [30] and the Mann–Whitney U test was used to calculate significance. L4 and L5 DRG on the ipsilateral side of surgery were collected 4 weeks post SNI surgery at 12 weeks of age.
2.6. Behavior studies using MeCP2 T158A mice
Male MeCP2 T158A mice and wild-type littermates were purchased from Jackson laboratories (Bar Harbor, Maine). Thermal and mechanical sensitivity were determined using the Hargreaves’ method [31] and von Frey filaments respectively. Behavior testing and tissue collection was conducted at 12 weeks of age. Mann–Whitney U test was used for statistical analysis.
3. Results
3.1. Chronic neuropathic pain induced MeCP2 upregulation in DRG
MeCP2 has been predicted to play a role in pain and modulation of MeCP2 has been observed in various inflammatory and neuropathic pain models [32,33]. To investigate whether MeCP2 expression is altered in a neuropathic pain state, we generated an SNI model. We confirmed increased mechanical sensitivity and data 4 weeks post-surgery on 12 week old mice is shown in Fig. 1a. Though there was a trend, the increase in Mecp2 mRNA in DRG after nerve injury was not significant (Fig. 1b). However, there was a significant increase in MeCP2 protein in the SNI model (Fig. 1c). This increased expression of MeCP2 in a neuropathic pain state could in part be mediated by the reduced expression of miRNAs predicted to target Mecp2 in the DRG [5]. We therefore sought to determine the regulatory role of a selected subset of miRNAs on MeCP2 expression.
Fig. 1.

MeCP2 expression in DRG 4 weeks after SNI or sham surgery in 12 weeks old mice. (A) von Frey filaments used to assess paw withdrawal threshold confirmed that mice were hypersensitive 4 weeks post SNI surgery (n = 10). Significance was determined using Mann–Whitney U test p value <0.001. (B) Taqman analysis of Mecp2 mRNA showed a trend toward increased expression that was not significant in SNI model compared to sham control (n = 7). Gapdh was used as the normalizer. (C) Western blot analysis of MeCP2 expression in DRG showed a significant increase in SNI model, determined using Student t-test, p value *<0.05. Tissue from 9 animals was pooled into 3 samples (n = 3). Protein expression was normalized to GAPDH.
3.2. Confirmation of miRNAs predicted to target Mecp2 3′UTR
To experimentally validate the bioinformatics prediction of miRNAs targeting the Mecp2 3′UTR, a luciferase reporter assay was performed. We tested a subset of miRNAs that were decreased in the DRG from a nerve injury model based on seed sequence complementarity, varying positions throughout the 3′UTR, the presence of multiple binding sites, and those not predicted to target Bdnf mRNA. Reduction in luciferase expression and therefore, miRNA binding to the Mecp2 3′UTR was validated for miR-19a, miR-301 and miR-132 (Fig. 2), while miR-17 and miR-181 did not reduce luciferase activity (data not shown).
Fig. 2.
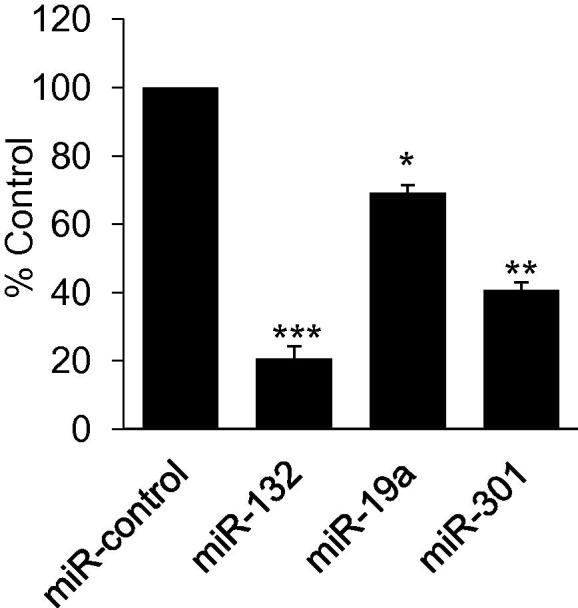
miRNA binding to MeCP2 3′UTR. Luciferase assay showing miR-132, miR-19a, and miR-301 binding to the 3′UTR of Mecp2. The luciferase activity was measured 24 h after miRNA transfection. The data expressed as percentage of control is the average of 3 independent experiments. Statistically significant difference from control was calculated using one way ANOVA and Student t-test, p value **<0.01, ***<0.001.
3.3. MeCP2 expression is modulated by miRNAs
To further evaluate regulation of MeCP2 expression from miRNA binding, Neuro-2a cells were transfected with precursor miRNAs or antagomirs that are inhibitors of endogenous miRNAs. Our data indicate that transfection of Neuro-2a cells with miR-19a, miR-301 and miR-132 did not decrease Mecp2 mRNA (Fig. 3a) but reduced MeCP2 protein as shown in western blot (Fig. 3b) and immunocytochemistry of endogenous MeCP2 in cells transfected with individual miRNAs (Fig. 3d–f). The antagomirs of miR-301 and miR-132 increased MeCP2 expression (Fig. 3c). We did not observe an increase in MeCP2 protein after the transfection of miR-19a inhibitor which could be due to a low endogenous level of miR-19a in Neuro-2a cells, the inhibition of which would not lead to the modulation of MeCP2 expression. Since we did not observe significant changes in Mecp2 mRNA after over expressing miRNAs, we conclude that Mecp2 regulation by miRNAs tested is mediated by translational repression and not through RNA degradation.
Fig. 3.

Regulation of MeCP2 by miRNAs. (A) Taqman analysis of Mecp2 in Neuro-2a cells 48 h after miRNA transfection showed no significant reduction in mRNA (n = 3). (B) Western blot analysis after transfection of Neuro-2a cells with miR-301, miR-132 and miR-19a significantly reduced MeCP2 protein (n = 3). (C) MeCP2 expression increased when cells were transfected with anti-miR (miRNA inhibitors) for miR-301 and miR-132; (n = 3, representative blot shown). (D and E) Immunocytochemisty and quantification for MeCP2 in Neuro-2a cells transfected with miR-132. (F and G) Immunocytochemistry and quantification for MeCP2 in Neuro-2a cells transfected with miR-19a. (H and I) Immunocytochemistry and quantification for MeCP2 in Neuro-2a cells transfected with miR-301. MeCP2 levels were normalized to DAPI staining of the nucleus and the graph represents MeCP2 expression relative to untransfected cells. Representative images are shown, n = 3. Significance was determined using Student t-test, p value *<0.05, **<0.001 ***<0.001.
3.4. miRNA-mediated decrease of MeCP2 lead to concomitant reduction in Bdnf
To determine if miRNA mediated regulation of MeCP2 affects expression of the MeCP2 target genes, we measured Bdnf mRNA after transfecting Neuro-2a cells with miR-19a, miR-301 and miR-132. Fig. 4a shows that there was reduction in Bdnf transcripts after overexpressing miRNAs. Additionally, inhibiting endogenous miR-132 and miR-301 resulted in increased Bdnf transcript (Fig. 4b). The miRNAs used in this study were confirmed to target the Mecp2 3′UTR, but are not predicted to bind the Bdnf 3′UTR. Thus, the reduced expression of Bdnf can be a direct consequence of miRNA mediated regulation of MeCP2.
Fig. 4.

MeCP2 mediated changes in Bdnf transcripts. (A) miRNA-mediated reduction of MeCP2 expression decreased Bdnf mRNA in Neuro-2a cells 48 h after miRNA transfection as determined by Taqman analysis. (B) Antagomirs, inhibitors of endogenous miRNAs, increased Bdnf mRNA. Gapdh was used as the normalizer, n = 3. Significance determined using Student t-test, p value *<0.05, **<0.001.
3.5. MeCP2 T158A mice have reduced mechanical sensitivity
Mice expressing 50% of the wild-type level of MeCP2 and female Mecp2+/− mice have reduced thermal sensitivity [26,34]. We wanted to further investigate the effect of MeCP2 mutations on pain behavior by assessing nociception in MeCP2 T158A mice. We observed that MeCP2 T158A mice have reduced mechanical sensitivity compared to wild-type littermate control mice (Fig. 5a). We found no significant changes in thermal sensitivity as determined by Hargreaves’ method between MeCP2 T158A and wild-type littermate control mice (data not shown). These results indicate a role for MeCP2 in mediating mechanical sensitivity.
Fig. 5.

MeCP2 T158A knockin mice have decreased mechanical sensitivity and lower Bdnf. (A) Mechanical sensitivity measured by von Frey filaments showed an increase in withdrawal threshold in 12 weeks old MeCP2 T158A mice, indicating hyposensitivity compared to wild-type littermate controls (n = 5 for MeCP2 T158A mice and n = 7 for wild-type littermate). Statistically significant difference was determined using the Mann–Whitney U test; there was a significant reduction in withdrawal threshold in the ipsilateral paw p value *<0.02. (B) DRG from Mecp2-null mice and (C) MeCP2 T158A mice has significantly lower Bdnf mRNA compared to wild-type littermates, (n = 6). (D) Bdnf was also lower in MeCP2 T158A mice compared to wild-type littermates after SNI surgery. Significance determined using Student t-test, p value *<0.05 (n = 3 for both MeCP2 T158A mice and wild-type littermate).
3.6. Bdnf is downregulated in the DRG of Mecp2-null mice and MeCP2 T158A mice
Several studies using mouse models have shown a direct correlation between MeCP2 and Bdnf expression in the brain indicating that MeCP2 regulates Bdnf [18]. We examined Bdnf expression in DRG from Mecp2-null mice. Fig. 5b shows a reduction in Bdnf mRNA levels in Mecp2-null mice compared to control. Thus MeCP2 mediated regulation of Bdnf expression in DRG appears to be similar to that observed in brain. We then investigated Bdnf levels in DRG from MeCP2 T158A mice to determine if a loss of function mutation would yield a similar result (Fig. 5c). We observed a decrease in Bdnf mRNA in the DRG of MeCP2 T158A mice as was previously reported in the brain [25]. These results indicate that in DRG, the role of MeCP2 as an activator is important for precise regulation of Bdnf and alteration of Bdnf in DRG may be a contributing factor leading to aberrant pain sensitivity in RTT.
3.7. Bdnf is decreased in MeCP2 T158A SNI model compared to wild-type SNI model mice
To determine how changes in MeCP2 in the DRG affect pain behavior, we generated an SNI model using MeCP2 T158A mice and wild-type littermate controls. Unfortunately, conducting mechanical and thermal sensitivity assessment post-SNI surgery was not possible in the MeCP2 T158A mouse; post-surgical over grooming behavior resulted in injuries compared to wild-type littermates. We collected the DRG from MeCP2 T158A mice 4-weeks post SNI and from wild-type SNI mice, and measured Bdnf transcripts. After nerve injury, MeCP2 T158A mice have decreased Bdnf compared to wild-type littermate control mice (Fig. 5d). Bdnf levels did not differ significantly between SNI and sham MeCP2 T158A mice (data not shown). This suggests that in the absence of fully functional MeCP2, the upregulation of Bdnf is impaired even after nerve injury, again suggesting the regulatory role of MeCP2 on Bdnf.
4. Discussion
The role of MeCP2 in the development and function of the central nervous system is well established and neuronal dysfunction contributes to symptoms associated with RTT [14,35]. Previous studies investigating the role of MeCP2 in pain have shown differences in expression pattern of MeCP2 in DRG and spinal cord from various rodent models of pain. An increase in MeCP2 has been associated with the development of neuropathic pain in the chronic constriction injury model in rats 14 days after injury [36]. MeCP2 is known to play an important role in signaling in the superficial dorsal horn upon induction of peripheral inflammation by injecting complete Freund’s adjuvant (CFA) in rat ankle joint. This study showed that phosphorylation of MeCP2 led to the release of DNA-bound MeCP2, relieving the repression on a few MeCP2 target genes in lamina I neurons. These genes were upregulated rapidly in this model of inflammatory pain [37]. MeCP2 expression was increased in the superficial dorsal horn 7 days following CFA injection in the ankle joint but decreased 7 days following SNI [38]. This study also showed that MeCP2 levels were decreased in activating transcription factor 3 (ATF3)-positive neurons in DRG after SNI [38]. Another report showed an increase in Mecp2 transcripts in the spinal cord 2 h after peripheral injection of formalin [9]. Upregulation of MeCP2 in the central nucleus of the amygdala was reported in the CFA model [39]. Differences in models, tissues, and time points can influence gene expression and so we performed qPCR and western blot analysis for MeCP2 using DRG from a mouse SNI model of neuropathic pain 4 weeks after surgery. We observed a significant increase of MeCP2 protein in DRG.
MeCP2 can regulate microRNAs (miRNAs) [40] and dysregulation of miRNAs was observed in the cerebella of Mecp2-null mice [41]. miRNA-mediated alteration of MeCP2 was reported to alter pain threshold in wild-type mice. miR-124a was downregulated in spinal cord after peripheral noxious stimulation with formalin and this was associated with an increase in Mecp2 mRNA [9]. Intravenous administration of a miRNA-124a inhibitor enhanced the nociceptive behavior and miRNA-124a mimic significantly reduced formalin-induced inflammatory pain. In addition to confirming that Mecp2 is a target for miR-124a, this study showed administration of miR-124a mimic induced a decrease in Mecp2 mRNA and miR-124a inhibitor enhanced MeCP2 levels in the spinal cord [9]. In another study, intrathecal miR-124 treatment reversed persistent hyperalgesia induced by carrageenan and prevented the development of mechanical allodynia in the SNI model of chronic neuropathic pain [42]. Thus future studies investigating the role of miRNAs validated to target Mecp2 will be beneficial in confirming their role in pain.
Our previous study investigating miRNA changes in DRG identified several miRNAs predicted to target Mecp2 3′UTR. The miRNAs were downregulated after nerve injury and because of the expected inverse correlation in expression between miRNAs and genes they regulate, we selected a subset of 5 miRNAs for further studies. Of these, miR-19a, miR-301 and miR-132 were confirmed to bind and regulate Mecp2 by translational repression. miR-132 is a previously validated miRNA for MeCP2 [6] and we included it as a positive control. This reduction of MeCP2 protein resulted in decreased Bdnf, indicating that activation by MeCP2 is an important mechanism of regulation for normal expression of BDNF. Our findings that inhibition of miRNA-mediated repression increased MeCP2 and Bdnf is similar to what was reported for miR-132 in cultured rat neurons [6].
Alterations in BDNF expression is a common phenomenon in both RTT and pain states, albeit in reverse direction. BDNF is an important modulator of sensory neurotransmission in nociceptive pathways both at spinal and supraspinal levels and has an important role in the development of central sensitization that underlies many forms of hyperalgesia [19]. Many of the biological effects of BDNF are mediated by the high affinity postsynaptic receptor Tropomyosin receptor kinase B (TrkB). TrkB signaling contributes to both the induction and maintenance of injury induced persistent pain and Trk inhibitors have been developed for the treatment of pain [43]. In addition to correlation of BDNF levels in the hypothalamus with MeCP2 levels, with lower levels in Mecp2-null mice and higher levels in MECP2 overexpressing mice compared to controls [12], disruption of total BDNF levels and secretion in Mecp2-null neurons indicate that functional MeCP2 is important for BDNF expression [44]. BDNF-related therapies have shown a reversal of cardiorespiratory deficits in Mecp2-null mice and is being pursued for the amelioration of RTT-like neurological symptoms in mouse models [18]. Our qPCR studies showed that Bdnf levels in the DRG were downregulated in both Mecp2-null and MeCP2 T158A mice. Thus the reduction in endogenous Bdnf could be one of the contributing factors leading to decreased pain sensitivity. After SNI surgery, lower levels of Bdnf in MeCP2 T158A mice compared to wild-type mice further indicates that increased Bdnf in the DRG after nerve injury is in part mediated by MeCP2. A recent study investigating the role of MeCP2 in pain regulation and morphine reward showed that MeCP2 and histone dimethyltransferase G9a induce a transcriptional de-repression of Bdnf in central nucleus of the amygdala, promoting pain behavior through BDNF upregulation [39]. Thus in addition to direct activation of Bdnf, MeCP2 in its role as a transcriptional repressor of G9a, further enhance BDNF levels in chronic pain.
Our studies showed that mice with the loss of function mutation, T158, have reduced mechanical sensitivity compared to wild-type littermates. Mecp2Flox/y mice expressing 50% of the wild-type level of MeCP2 and female Mecp2+/− mice have reduced thermal sensitivity in a hot plate test [26,34] but there are no reports to date investigating their mechanical sensitivity. Our assessment of thermally induced nociception using the Hargreaves’ method showed no differences in MeCP2 T158A mice and wild-type littermates. Differences in the thermal sensitivity could be due to the specific mutation of MeCP2 or the method of measuring thermal sensitivity. Since BDNF release in dorsal horn in neuropathic pain model is associated with thermal hyperalgesia [45,46], we postulate that other modes of action may be playing a role in mediating mechanical sensitivity in T158A mice. Studies investigating function of DRG neurons in naïve animals have shown that delivering BDNF directly to DRGs induced mechanical allodynia. Conversely, after nerve injury direct administration of anti-BDNF antibody to injured DRG attenuated mechanical allodynia [47]. MeCP2 and BDNF are expressed in microglia and astrocytes in addition to neurons and additional studies are needed to determine the contribution of these different cell types in modulating aberrant pain sensation in MeCP2 T158A mice. A recent study investigated the role of miR-183, a miRNA that was significantly downregulated in the ipsilateral L5 DRG after SNL [48]. Intrathecal administration of miR-183 attenuated mechanical allodynia and this was associated with the downregulation of BDNF transcripts in DRG [48]. This study demonstrated that downregulation of BDNF in DRG was correlated with attenuated mechanical allodynia in SNL model of neuropathic pain. Our data shows that mutations in Mecp2 can reduce mechanical pain sensitivity. Since MeCP2 can mediate downstream transcriptional changes of a large number of genes, we cannot rule out the contribution of other dysregulated genes (due to T158A mutation in methyl binding domain of MeCP2) to aberrant pain sensitivity in MeCP2 T158A mice.
In conclusion, our results indicate that miRNA-mediated regulation of MeCP2 can contribute to the mechanical hypersensitivity observed in neuropathic pain models by altering BDNF in the DRG.
Neuropathic pain induces MeCP2 and BDNF expression in the DRG; conversely, RTT mouse models lacking or expressing reduced levels of MeCP2 have decreased Bdnf that can contribute to mechanical hyposensitivity. Further investigations including in vivo delivery of miRNAs targeting Mecp2 followed by characterization of pain behavior and investigation of MeCP2 expression and downstream targets in naïve mice, RTT and pain models can provide insights on therapeutic options for both pain and RTT.
Author contributions
S.A. conceived and supervised the study; M.M. and S.A. designed experiments; M.M. and Y.T. performed experiments; M.M. analyzed the data; Z.Z. provided reagents and critical input regarding the study design; M.M. and S.A. wrote the manuscript; Z.Z made manuscript revisions.
Acknowledgments
This work was supported by grants from Commonwealth Universal Research Enhancement (CURE), Pennsylvania Department of Health and Rita Allen Foundation to Seena Ajit.
The funding agency had no role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
References
- 1.Gereau R.W., Sluka K.A., Maixner W., Savage S.R., Price T.J., Murinson B.B., Sullivan M.D., Fillingim R.B. A pain research agenda for the 21st century. J. Pain. 2014;15:1203–1214. doi: 10.1016/j.jpain.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.von Hehn C.A., Baron R., Woolf C.J. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron. 2012;73:638–652. doi: 10.1016/j.neuron.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basbaum A.I., Bautista D.M., Scherrer G., Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mogil J.S., Davis K.D., Derbyshire S.W. The necessity of animal models in pain research. Pain. 2010;151:12–17. doi: 10.1016/j.pain.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 5.von Schack D. Dynamic changes in the microRNA expression profile reveal multiple regulatory mechanisms in the spinal nerve ligation model of neuropathic pain. PLoS ONE. 2011;6:e17670. doi: 10.1371/journal.pone.0017670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein M.E., Lioy D.T., Ma L., Impey S., Mandel G., Goodman R.H. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 2007;10:1513–1514. doi: 10.1038/nn2010. [DOI] [PubMed] [Google Scholar]
- 7.Wada R., Akiyama Y., Hashimoto Y., Fukamachi H., Yuasa Y. MiR-212 is downregulated and suppresses methyl-CpG-binding protein MeCP2 in human gastric cancer. Int. J. Cancer. 2010;127:1106–1114. doi: 10.1002/ijc.25126. [DOI] [PubMed] [Google Scholar]
- 8.Han K. Human-specific regulation of MeCP2 levels in fetal brains by microRNA miR-483-5p. Genes Dev. 2013;27:485–490. doi: 10.1101/gad.207456.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kynast K.L., Russe O.Q., Möser C.V., Geisslinger G., Niederberger E. Modulation of central nervous system–specific microRNA-124a alters the inflammatory response in the formalin test in mice. Pain. 2013;154:368–376. doi: 10.1016/j.pain.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 10.Kuhn D.E. Chromosome 21-derived microRNAs provide an etiological basis for aberrant protein expression in human down syndrome brains. J. Biol. Chem. 2010;285:1529–1543. doi: 10.1074/jbc.M109.033407. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Bird A. The methyl-CpG-binding protein MeCP2 and neurological disease. Biochem. Soc. Trans. 2008;36:575–583. doi: 10.1042/BST0360575. [DOI] [PubMed] [Google Scholar]
- 12.Chahrour M., Jung S.Y., Shaw C., Zhou X., Wong S.T., Qin J., Zoghbi H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samaco R.C., Neul J.L. Complexities of Rett syndrome and MeCP2. J. Neurosci. 2011;31:7951–7959. doi: 10.1523/JNEUROSCI.0169-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guy J., Cheval H., Selfridge J., Bird A. The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- 15.Downs J., Geranton S.M., Bebbington A., Jacoby P., Bahi-Buisson N., Ravine D., Leonard H. Linking MECP2 and pain sensitivity: the example of Rett syndrome. Am. J. Med. Genet. A. 2010;152A:1197–1205. doi: 10.1002/ajmg.a.33314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peters S.U., Hundley R.J., Wilson A.K., Warren Z., Vehorn A., Carvalho C.M.B., Lupski J.R., Ramocki M.B. The behavioral phenotype in MECP2 duplication syndrome: a comparison with idiopathic autism. Autism Res. 2013;6:42–50. doi: 10.1002/aur.1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zachariah R.M., Rastegar M. Linking epigenetics to human disease and Rett syndrome: the emerging novel and challenging concepts in MeCP2 research. Neural Plast. 2012;2012:415825. doi: 10.1155/2012/415825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li W., Pozzo-Miller L. BDNF deregulation in Rett syndrome. Neuropharmacology. 2014;76:737–746. doi: 10.1016/j.neuropharm.2013.03.024. (Part C) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merighi A., Salio C., Ghirri A., Lossi L., Ferrini F., Betelli C., Bardoni R. BDNF as a pain modulator. Prog. Neurobiol. 2008;85:297–317. doi: 10.1016/j.pneurobio.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 20.Ha S.O., Kim J.K., Hong H.S., Kim D.S., Cho H.J. Expression of brain-derived neurotrophic factor in rat dorsal root ganglia, spinal cord and gracile nuclei in experimental models of neuropathic pain. Neuroscience. 2001;107:301–309. doi: 10.1016/s0306-4522(01)00353-0. [DOI] [PubMed] [Google Scholar]
- 21.Lin Y.-T., Ro L.-S., Wang H.-L., Chen J.-C. Up-regulation of dorsal root ganglia BDNF and trkB receptor in inflammatory pain: an in vivo and in vitro study. J. Neuroinflammation. 2011;8:126. doi: 10.1186/1742-2094-8-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Obata K., Noguchi K. BDNF in sensory neurons and chronic pain. Neurosci. Res. 2006;55:1–10. doi: 10.1016/j.neures.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Uchida H., Matsushita Y., Ueda H. Epigenetic regulation of BDNF expression in the primary sensory neurons after peripheral nerve injury: implications in the development of neuropathic pain. Neuroscience. 2013;240:147–154. doi: 10.1016/j.neuroscience.2013.02.053. [DOI] [PubMed] [Google Scholar]
- 24.Calfa G., Percy A.K., Pozzo-Miller L. Experimental models of Rett syndrome based on Mecp2 dysfunction. Exp. Biol. Med. (Maywood) 2011;236:3–19. doi: 10.1258/ebm.2010.010261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goffin D. Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat. Neurosci. 2012;15:274–283. doi: 10.1038/nn.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samaco R.C. A partial loss of function allele of methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum. Mol. Genet. 2008;17:1718–1727. doi: 10.1093/hmg/ddn062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Samaco R.C., McGraw C.M., Ward C.S., Sun Y., Neul J.L., Zoghbi H.Y. Female Mecp2+/− mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum. Mol. Genet. 2012 doi: 10.1093/hmg/dds406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capasso K. Effect of histone deacetylase inhibitor JNJ-26481585 in pain. J. Mol. Neurosci. 2014:1–9. doi: 10.1007/s12031-014-0391-7. [DOI] [PubMed] [Google Scholar]
- 29.Decosterd I., Woolf C.J. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 30.Gao R., Gao X., Xia J., Tian Y., Barrett J.E., Dai Y., Hu H. Potent analgesic effects of a store-operated calcium channel inhibitor. Pain. 2013;154:2034–2044. doi: 10.1016/j.pain.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bennett J.G. Animal Models of Pain. In Methods in Pain Research. CRC Press; 2001. [Google Scholar]
- 32.Geranton S.M. Targeting epigenetic mechanisms for pain relief. Curr. Opin. Pharmacol. 2012;12:35–41. doi: 10.1016/j.coph.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 33.Crow M., Denk F., McMahon S. Genes and epigenetic processes as prospective pain targets. Genome Med. 2013;5:12. doi: 10.1186/gm416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Samaco R.C., McGraw C.M., Ward C.S., Sun Y., Neul J.L., Zoghbi H.Y. Female Mecp2(+/−) mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum. Mol. Genet. 2013;22:96–109. doi: 10.1093/hmg/dds406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Na E.S., Monteggia L.M. The role of MeCP2 in CNS development and function. Horm. Behav. 2011;59:364–368. doi: 10.1016/j.yhbeh.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y., Liu C., Guo Q.-L., Yan J.-Q., Zhu X.-Y., Huang C.-S., Zou W.-Y. Intrathecal 5-azacytidine inhibits global DNA methylation and methyl-CpG-binding protein 2 expression and alleviates neuropathic pain in rats following chronic constriction injury. Brain Res. 2011;1418:64–69. doi: 10.1016/j.brainres.2011.08.040. [DOI] [PubMed] [Google Scholar]
- 37.Geranton S.M., Morenilla-Palao C., Hunt S.P. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J. Neurosci. 2007;27:6163–6173. doi: 10.1523/JNEUROSCI.1306-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tochiki K.K., Cunningham J., Hunt S.P., Geranton S.M. The expression of spinal methyl-CpG-binding protein 2, DNA methyltransferases and histone deacetylases is modulated in persistent pain states. Mol. Pain. 2012;8:14. doi: 10.1186/1744-8069-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z., Tao W., Hou Y.Y., Wang W., Kenny P.J., Pan Z.Z. MeCP2 repression of G9a in regulation of pain and morphine reward. J. Neurosci. 2014;34:9076–9087. doi: 10.1523/JNEUROSCI.4194-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szulwach K.E. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J. Cell Biol. 2010;189:127–141. doi: 10.1083/jcb.200908151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu H. Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18161–18166. doi: 10.1073/pnas.1005595107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willemen H., Huo X.-J., Mao-Ying Q.-L., Zijlstra J., Heijnen C., Kavelaars A. MicroRNA-124 as a novel treatment for persistent hyperalgesia. J. Neuroinflammation. 2012;9:143. doi: 10.1186/1742-2094-9-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang T., Yu D., Lamb M.L. Trk kinase inhibitors as new treatments for cancer and pain. Expert Opin. Ther. Pat. 2009;19:305–319. doi: 10.1517/13543770902721261. [DOI] [PubMed] [Google Scholar]
- 44.Wang H., Chan S.-A., Ogier M., Hellard D., Wang Q., Smith C., Katz D.M. Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in Mecp2 null mice. J. Neurosci. 2006;26:10911–10915. doi: 10.1523/JNEUROSCI.1810-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miletic G., Miletic V. Increases in the concentration of brain derived neurotrophic factor in the lumbar spinal dorsal horn are associated with pain behavior following chronic constriction injury in rats. Neurosci. Lett. 2002;319:137–140. doi: 10.1016/s0304-3940(01)02576-9. [DOI] [PubMed] [Google Scholar]
- 46.Pezet S., McMahon S.B. Neurotrophins: mediators and modulators of pain. Annu. Rev. Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- 47.Zhou X.F., Deng Y.S., Xian C.J., Zhong J.H. Neurotrophins from dorsal root ganglia trigger allodynia after spinal nerve injury in rats. Eur. J. Neurosci. 2000;12:100–105. doi: 10.1046/j.1460-9568.2000.00884.x. [DOI] [PubMed] [Google Scholar]
- 48.Lin C.-R., Chen K.-H., Yang C.-H., Huang H.-W., Sheen-Chen S.-M. Intrathecal miR-183 delivery suppresses mechanical allodynia in mononeuropathic rats. Eur. J. Neurosci. 2014;39:1682–1689. doi: 10.1111/ejn.12522. [DOI] [PubMed] [Google Scholar]