

Abstract
Following its activation by PINK1, parkin is recruited to depolarized mitochondria where it ubiquitinates outer mitochondrial membrane proteins, initiating lysosomal-mediated degradation of these organelles. Mutations in the gene encoding parkin, PARK2, result in both familial and sporadic forms of Parkinson's disease (PD) in conjunction with reductions in removal of damaged mitochondria. In contrast to what has been reported for other PARK2 mutations, expression of the Q311X mutation in vivo in mice appears to involve a downstream step in the autophagic pathway at the level of lysosomal function. This coincides with increased PARIS expression and reduced expression of a reciprocal signaling pathway involving the master mitochondrial regulator peroxisome proliferator-activated receptor-gamma coactivator (PGC1α) and the lysosomal regulator transcription factor EB (TFEB). Treatment with rapamycin was found to independently restore PGC1α-TFEB signaling in a manner not requiring parkin activity and to abrogate impairment of mitochondrial quality control and neurodegenerative features associated with this in vivo model. Losses in PGC1α-TFEB signaling in cultured rat DAergic cells expressing the Q311X mutation associated with reduced mitochondrial function and cell viability were found to be PARIS-dependent and to be independently restored by rapamycin in a manner requiring TFEB. Studies in human iPSC-derived neurons demonstrate that TFEB induction can restore mitochondrial function and cell viability in a mitochondrially compromised human cell model. Based on these data, we propose that the parkin Q311X mutation impacts on mitochondrial quality control via PARIS-mediated regulation of PGC1α-TFEB signaling and that this can be independently restored via upregulation of TFEB function.
SIGNIFICANCE STATEMENT Mutations in PARK2 are generally associated with loss in ability to interact with PINK1, impacting on autophagic initiation. Our data suggest that, in the case of at least one parkin mutation, Q311X, detrimental effects are due to inhibition at the level of downstream lysosomal function. Mechanistically, this involves elevations in PARIS protein levels and subsequent effects on PGC1α-TFEB signaling that normally regulates mitochondrial quality control. Treatment with rapamycin independently restores PGC1α-TFEB signaling in a manner not requiring parkin activity and abrogates subsequent mitochondrial impairment and neuronal cell loss. Taken in total, our data suggest that the parkin Q311X mutation impacts on mitochondrial quality control via PARIS-mediated regulation of PGC1α-TFEB signaling and that this can be independently restored via rapamycin.
Keywords: autophagy, mitochondria, parkin, Parkinson's, PGC1α, TFEB
Introduction
Degradation of damaged mitochondria via lysosomal autophagy is a key cellular pathway in the maintenance of mitochondrial homeostasis, disruption of which contributes to the progressive DAergic SNpc cell loss associated with Parkinson's disease (PD) (Pickrell and Youle, 2015). Interventions that catalyze this process therefore constitute a likely therapeutic avenue for the condition. Mutations in PARK2, encoding the ubiquitin E3 ligase parkin, are associated with both familial and sporadic forms of PD (Kitada et al., 1998). A subset of these mutations act via formation of insoluble parkin-containing inclusions and subsequent decreases parkin activity that can impact on lysosomal removal of damaged mitochondria contributing to mitochondrial dysfunction; this has also been reported to occur in wild-type (WT) parkin in association with elevated oxidative or nitrosative stress (Cookson, 2003; Marx et al., 2003; Winklhofer et al., 2003; Chung et al., 2004; Yao et al., 2004; LaVoie et al., 2005; Wang et al., 2005; Wong et al., 2007: Siddiqui, 2012; Bao et al., 2012; Sunico et al., 2013; Vandiver et al., 2013). We previously reported that oxidative reductions in parkin solubility within DAergic cell lines stably expressing WT parkin resulting in reduced parkin solubility, and E3 ligase activity reduced the ability of cells to clear damaged mitochondria (Siddiqui et al., 2012). These effects were found to be reversed by rapamycin, a known inducer of autophagy. This coincided with a restoration of lost mitochondrial function, although the exact mechanisms involved were not explored. To obtain a better mechanistic understanding of the processes underlying these events and whether they are translatable in a chronic age-related in vivo model more fully emulating the human condition, we examined the impact of rapamycin treatment in a mouse line expressing the C-terminally truncated Q311X mutation selectively within DAergic neurons (Lu et al., 2009). Expression of the Q311X mutation in vitro in human SY5Y cells has been reported to result in reduced parkin solubility and E3 ligase activity resulting in increased levels of parkin substrates (Sriram et al., 2005; Wang et al., 2005; Corti and Brice, 2013; Winklhofer, 2014). Given parkin's proposed role in lysosomal mitochondrial degradation, we set out to explore whether the parkin Q311X mutation results in reduced mitochondrial function in effected nigral tissues, whether this was reversible via rapamycin treatment, and whether this in turn prevents neurodegenerative features associated with this model. Interestingly, data from these studies suggest, that rather than interfering with parkin's ability to be recruited to damaged mitochondria by PINK1, the Q311X parkin mutation results in a late-stage blockage in autophagy at the level of the autophagolysosome (APL) as a consequence of reduced lysosomal function. Mechanistically, this appears to be due to PARIS-dependent losses in PGC1α-transcription factor EB (TFEB) signaling that are independently restored by rapamycin in a manner requiring TFEB. Our results suggest that parkin, through its effects on PARIS, plays a key role in overall mitochondrial homeostasis through cellular regulation of the PGC1α-TFEB pathway.
Materials and Methods
Mice and rapamycin treatment.
Parkin Q311X mice were a generous gift from William Yang (University of California–Los Angeles) (Lu et al., 2009). Mice for experiments were generated by crossing homozygous Q311X mutants with WT FVB mice to generate heterozygous offspring with respect to the parkin Q311X transgene. Nontransgenic littermates were used as negative controls. Animals were divided into four experimental groups: control-fed nontransgenics (Tgs), n = 13; rapamycin-fed non-Tgs, n = 13; control-fed Tgs, n = 16; and rapamycin-fed Tgs, n = 17. Animals were a mixed population of males and females, 16–17 months of age at the time of testing.
Parkin Q311X mutant mice and their WT littermates were fed either normal chow or chow containing microencapsulated rapamycin at a concentration of 2.24 mg per kg body weight/d as previously described starting at 13 months of age for a 12 week period (Harrison et al., 2009; Spilman et al., 2010). All mice were given ad libitum access to food and water for the duration of the experiment. Mice were housed according to standard animal care protocols on a 12 h light/dark cycle and maintained in a pathogen-free environment in the Vivarium facility of the Buck Institute under an approved Buck Institutional Animal Care and Use Committee protocol.
Stereological SNpc TH+ cell counts.
Midbrain sections (40 μm) were cut on a sliding microtome and collected in PBS. Free-floating sections were permeabilized and blocked in PBS containing 0.4% Triton X-100, 4% normal goat serum at room temperature for 30 min. Sections were then incubated in rabbit polyclonal anti-tyrosine hydroxylase (TH) antibody (1:1000 dilution, Millipore Bioscience Research Reagents) in PBS containing 0.2% Triton X-100, 2% normal goat serum overnight at 4°C. Sections were then washed with PBS containing 0.2% Triton X-100, 1% normal goat serum followed by incubation for 1 h in biotinylated goat anti-rabbit antibody (1:1000; Jackson ImmunoResearch Laboratories) in PBS containing 0.2% Triton X-100, 1.5% goat serum, followed by washing and visualization via incubation in biotin-streptavidin-HRP complex (ABC kit; Vector Laboratories), followed by incubation with 3,3′-diaminobenzidine per the manufacturer's instructions (Sigma). Sections were mounted on glass slides and allowed to air-dry overnight. Unbiased stereological cell counts were performed using the optical fractionator method to count TH+ neurons in the SNpc in every fourth section in a total of 8–10 sections at 100× objective using Stereoinvestigator version 6.0 software (MicroBrightField). Results were verified by Nissl staining.
Western blot analyses.
Striatal tissue lysates were homogenized in buffer containing 50 mm Tris/HCl, pH 8.0, 150 mm NaCl, 0.25% sodium deoxycholate, and 1 mm EDTA followed by centrifugation at 10,000 × g for 10 min. Supernatant was taken and protein concentration estimated via the Bradford assay. Aliquots of 25 μg protein were boiled at 95°C in Lamelli buffer for 5 min and loaded onto a 10% SDS-PAGE gel. Following electrophoresis, bands were transferred to PVDF membranes. Membranes were next incubated in primary antibodies (goat anti-cathepsin, 1:500 dilution, Santa Cruz Biotechnology; mouse anti-p62, 1:500 dilution, MBL Biosciences; mouse anti-α-synuclein, 1:1000 dilution, BD Transduction; rabbit anti-ubiquitin, 1:1000 dilution, DAKO; rabbit anti-Ub-K63, 1:250 dilution, Millipore; mouse anti-β-actin, 1:1000 dilution, Invitrogen; anti-tubulin, 1:50,000 dilution, Invitrogen; mouse-anti-PARIS, 1:500 dilution, Millipore) for 24 h, 4°C. This was followed by incubation in appropriate HRP-conjugated secondary antibodies (anti-rabbit, anti-mouse, or anti-goat, 1:2500 dilution, Santa Cruz Biotechnology) for 1 h at room temperature. Immunoblots were developed using enhanced chemiluminescence reagent and analyzed using a ChemDoc system from Bio-Rad.
Fluorescent immunocytochemistry.
Sections (40 μm) were prepared from 4% PFA-fixed tissues and incubated in 5% BSA solution for 1 h. This was followed by incubation in primary antibody (rabbit anti-LC3, 1:500 dilution, Novus Biological; mouse anti-VDAC, 1:1000 dilution, Millipore; mouse anti-p62, 1:500 dilution, Novus Biological; chicken anti-TH, 1:1000 dilution, Novus Biological; rabbit-anti-TFEB, 1:500 dilution, Novus Biological; mouse anti-α-synuclein, 1:500 dilution, BD Biosciences) for 24 h at 4°C. This was followed by incubation in appropriate HRP-conjugated secondary antibody (anti-rabbit, 1:500–1000; anti-mouse, 1:500–1000 dilution; or anti-chicken, 1:1000 dilution, AlexaFluor) for 1 h at room temperature.
Proteinase K treatment of striatal tissue lysates and SNpc sections.
For Western blot analyses, whole striatal tissue lysates were prepared as described above. Aliquots of 25 μg protein were incubated in 0–10 μg/ml of proteinase K (pro-K) for 10 min before gel loading. For detection of pro-K-resistant α-synuclein via SNpc ICC, 40 μm sections from 4% PFA-fixed tissues were washed twice in 0.1% Tween 20 and then incubated for 30 min in Tris buffer saline, 0.1% Tween 20. Sections were then incubated for 60 min at 55°C in 5 μg/ml pro-K in TBS, then washed 3× in TBS. Tissues were next incubated in 3 m guanidine thiocyanate in 10 mm Tris-HCl solution for 10 min as previously described (Lu et al., 2009). Sections were processed for ICC as above.
Pole test.
In the pole test, animals were placed on top of a rough-surfaced wooden pole (50 cm in length and 1 cm in diameter) and allowed to descend to the base of the pole (Lieu et al., 2013, 2014). Mice were initially habituated and trained the day before testing. On testing day, animals were placed head-up on the top of the pole. The time it took for the animal to turn its head downward (movement initiation) and descend the entire length of the pole was taken. The best performance for each animal over five consecutive trials was subsequently recorded.
Open field analysis.
To monitor spontaneous activity, mice were placed in a TruScan open field chamber and allowed to explore freely over a 10 min period using a TruScan photobeam apparatus (Colbourn Instruments), as previously described by our laboratory (Lieu et al., 2013, 2014). The number of moves over a 10 min time period was as determined using TruScan software.
Confocal microscopy and 3D reconstruction for quantitation of puncta.
Sections processed as above were used for imaging. Image Z-stacks were collected using a Zeiss 710 confocal microscope with a 63× objective. Lasers were set at 488 and 561 nm excitation wavelengths to optimize visualization while excluding overlap and bleed-through. Image stacks were run for 3D reconstruction using Imaris (version 7.1.0; Bitplane). For nuclear TFEB, puncta-positive nuclei were counted where >2 puncta were present: 12 sections per group, n = 4 per condition. For iPSCs, following fixation, TFEB puncta outside of the nucleus were quantified as above.
Electron microscopy (EM) and quantitation of APG:APL ratios and mitochondrial volume.
SNpc tissue was dissected and immediately fixed in 2% PFA and 0.2% glutaraldehyde in 0.1 m sodium phosphate buffer, pH 7.4, for 2 h at room temperature; 50-nm-thick sections were cut using an Ultracut S ultramicrotome (Leica). Sections were collected on carbon-coated Formvar grids using a mixture of 1.8% methylcellulose and 2.3 m sucrose, fixed with 1% glutaraldehyde, counterstained with uranyl acetate, and embedded in methylcellulose–uranyl acetate. Sections were viewed on a Phillips Tecnai 12 transmission EM. A uniform random approach to imaging was used by choosing one section of 152 serial sections at random and imaging a subset of evenly spaced sections thereafter. Individual sections were imaged, selected by stepping across each grid in a predetermined x/y meander. Two sets of images were collected: one to analyze APG (autophagosomes) and AL (autolysosomes) and another for mitochondrial volume. Classification of autophagic vacuoles according to their luminal content was performed by a blinded independent observer using one single-category allocation for each vesicle. Autophagic vacuoles (vesicles <0.5 m) were classified as APGs when they met two or more of the following criteria: double membranes (complete or at least partially visible), absence of ribosomes attached to the cytosolic side of the membrane, luminal density similar to cytosol, and identifiable organelles or regions of organelles in their lumen. Vesicles of similar size but with a single membrane (or <40% of the membrane visible as double), luminal density lower than the surrounding cytosol, mulltiple single membrane-limited vesicles containing light or dense amorphous material were classified as ALs. Images were collected at 2.6 × 104 at a pixel dimension of 5.7 nm × 5.7 nm. Images were analyzed using the volume estimator Cavalieri probe available in MicroBrightfield's stereo-investigator software.
Parkin E3 ligase assay.
In vitro ubiquitination reactions were performed as previously described (Siddiqui et al., 2012). Immunoprecipitated parkin from striatal tissue lysates was mixed with 100 ng of E1 ubiquitin-activating enzyme (Calbiochem), 250 ng of E2 ubiquitin-conjugating enzyme (UbcH7, Boston Biochem), and 10 μg of ubiquitin (Sigma) in ubiquitination buffer (2 mm ATP, 50 mm Tris, 2.5 mm MgCl2 at pH 7.5) for 2 h at room temperature. Auto-ubiquitination of parkin was then detected by anti-ubiquitin antibody via Western blot analysis (1:1000 dilution, DAKO).
qRT-PCR.
RNA was isolated using the pure RNA tissue kit (Roche). RNA quality and concentration were determined by measuring absorbance at 260/280 nm. cDNA was synthesized using purified RNA (5 μg), GO script reverse transcriptase (Promega), and random hexamers. Gene expression was determined via qRT-PCR in a Light Cycler 480 Sequence Detection system (Roche Applied Science). Forward and reverse primers for each gene of interest were designed (Operon) using the Roche Universal Probe Library Assay Design Center; each gene (“assay”) of interest was represented by a UPL probe (Universal Probe Library, Roche). At least three technical replicates were performed. Primer sets used were as follows: mouse TFEB forward, GAGCTGGGAATGCTGATCC; reverse, GGGACTTCTGCAGGTCCTT; mouse PGC 1α forward, GAAAGGGCCAAACAGAGAGA; reverse, GTAAATCACACGGCGCTCTT; mouse TFAM forward, CAAAGGATGATTCGGCTCAG; reverse, AAGCTGAATATATGCCTGCTTTTC; mouse Nrf1 forward, TGGAGTCCAAGATGCTAATGG; reverse, GCGAGGCTGGTTACCACA; human PGC1α forward, TGAGAGGGCCAAGCAAAG; reverse, ATAAATCACACGGCGCTCTT; human Nrf1 forward, CCATCTGGTGGCCTGAAG, reverse, GTAGTGCCTGGGTCCATGA; human CTSD forward, GAGGATGAGGCCGCTAGA, reverse, CAACAAGGTGGGTTTTGTCC; human GLA forward, TGGAAAATTTGGCAGATGGT; reverse, AAAGAGGCCACTCACAGGAG. Data were normalized to actin. Cycling conditions consisted of 45 cycles of 95°C, 30 s, 62°C, 30 s, 72°, 25 s. Fold-change was determined by using the 2−Δ ΔCT method; all reactions were performed in triplicate.
Analyses of mitochondrial complex I (CI) activity.
Rotenone-sensitive NADH dehydrogenase activity was measured in immunoisolated DAergic striatal synaptosomal fractions isolated as previously described (Chinta et al., 2007). Activity was quantitated by measuring 2,6-dichlorophenolindophenol reduction following addition of 200 μm NADH, 200 μm decylubiquinone, 2 mm KCN, and 0.002% 2,6-dichlorophenolindophenol in the presence and absence of 2 μm rotenone. Readings were recorded using a Spectra Max. Values were normalized/protein using Bio-Rad reagent.
Generation and analysis of DAergic cell lines expressing human WT versus Q311X mutant parkin.
DAergic N27 cells were transfected with p3xFLAG-CMV-7.1 plasmid vectors containing an insertion of either full-length human parkin cDNA or the Q311X truncation mutation (replicating that expressed in the transgenic mouse model used in these studies) via Lipofectamine 3000 reagent (Invitrogen) The p3xFLAG-CMV-7.1Q3111X plasmid was generated by PCR cloning the parkin fragment (nucleotide 1–930) into the Hind1-EcoR1 sites of the FLAG vector followed by sequence verification (Eurofin). Transfection efficiency was determined via Western blot analysis using an anti-mouse 3X-FLAG antibody (Sigma-Aldrich). Mitochondrial function was monitored by measurement of mitochondrial membrane potential via fluorescent TMRM assay and cell viability via MTT assay 24 h after transfection.
PARIS and TFEB knockdown via siRNA and rapamycin treatment in parkin WT versus Q311X cells.
To interrogate whether reductions in PARIS levels restore mitochondrial function and cell viability in mutant Q311X versus WT parkin-expressing cell lines, cells were transfected with either PARIS or scrambled (Scr) siRNA using reagents from Invitrogen 24 h before subsequent parkin plasmid transfections. Reductions in PARIS protein levels were verified via Western blot analysis; TFEB and PGC1α expression via RT-PCR and mitochondrial function and cell viability was also performed 24 h following plasmid transfections. To test whether functional neuroprotective effects of rapamycin requires TFEB, cells were transfected with either TFEB or scrambled Scr siRNA using reagents from Invitrogen. Twenty-two hours following siRNA transfections, 2 μm rapamycin was added to the cells. Nontoxic rapamycin concentrations were initially determined via cell viability analyses using the MTT assay (Siddiqui et al., 2012). WT or Q311X parkin transfections were performed 2 h later; all subsequent analyses were performed 24 h after transfection. Reductions in TFEB protein levels were verified via Western blot analysis; TFEB and PGC1α expression via RT-PCR, mitochondrial function, and cell viability were monitored in the absence and presence of rapamycin.
iPSC-derived DAergic neurons and treatments.
Human iPSC-derived DOPA cells were purchased from XCell Science. Cells were treated with 0.4 μm rotenone for 24 h. Cells were pretreated with rapamycin (50 μm) or trehalose (100 mm) for 1 h before rotenone addition. Cell viability in all treatment groups was monitored via the MTT assay. Effects of ±100 mm trehalose on TFEB activity via quantitative ICC of extranuclear nuclear puncta, TFEB target gene expression via RT-PCR (PGC1α, cathepsin D, Nfr1, GLA), and mitochondrial function via measurement of ATP levels via luminescence assay and MPP by TMRM assay were monitored.
Statistics.
Two-way ANOVA followed by Tukey's multiple-comparison test were performed to analyze statistical significance of in vivo data and one-way ANOVA followed by Tukey's multiple-comparison test for in vitro studies and in human iPSC-derived neurons, all run using PRISM software.
Results
Age-related neurodegenerative effects in chronic parkin Q311X mutant mouse model are reversed by rapamycin feeding
The role of parkin in mitochondrial quality control is traditionally thought to involve interaction with the PINK1 protein (Pickrell and Youle, 2015). Following activation of PINK1 by mitochondrial depolarization or other mitochondrial stress-related events, PINK1 recruits parkin to the outer mitochondrial membrane. Parkin, acting in its capacity as an E3 ligase, ubiquitinates select outer mitochondrial membrane proteins, including the mitofusins (Mfn1/2) and Miro1 (Narendra et al., 2010, 2012). This results in interaction of ubiquitin-tagged mitochondria with the microtubule-associated light chain protein 3 (LC3) complex via the autophagic adaptor protein p62/SQSTM1 (p62 Sequestome, hereafter referred to as p62) to form APGs. APGs in turn fuse with lysosomes to form APLs. Intra-autophagolysosomal components, including mitochondria, are then degraded by lysosomal hydrolases.
Parkin Q311X mutant mice used in our studies have been reported by 16 months of age and display several parkinsonian neuropathologies, including SNpc DAergic cell loss, accumulation of insoluble (proteinase K-resistant) α-synuclein, and hypokinetic motor deficits (Lu et al., 2009). Given the established role of parkin in lysosomal mitochondrial degradation, we hypothesized that age-related neurodegenerative effects associated with this particular parkin mutant mouse model would be reversed by treatment with rapamycin, an established inducer of autophagy, coinciding with restoration of lost autophagic functions.
Rapamycin was administered in the chow to parkin Q311X mutant mice and their littermate controls for a period of 3 months, starting at 13–14 months of age. Control diet consisted of normal feed. To assess whether chronic rapamycin administration resulted in abrogation of age-related neurodegeneration, we first examined DAergic SNpc cell numbers via stereological TH+ cell counts. As previously reported (Lu et al., 2009), by 16 months of age, parkin Q311X mutants displayed a significant (∼40%) decrease in DAergic cell numbers in the SNpc that was significantly prevented by chronic rapamycin feeding (Fig. 1).
Figure 1.
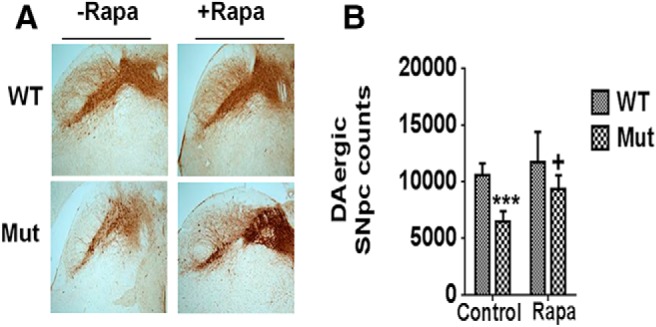
Losses in DAergic SNpc cell numbers in parkin Q311X mutants are restored by rapamycin (Rapa) feeding. A, Representative immunocytochemistry of SNpc from parkin mutant mice (Mut) versus WT littermate controls on control or rapamycin diet (Rapa) probed with antibody against TH (n = 7 per condition). B, Stereological quantitation of DAergic SNpc cell numbers. ***p < 0.001 versus WT. +p < 0.05 versus Mut.
We next assessed the effects of rapamycin administration on reported increases in SNpc pro-K-insoluble α-synuclein levels in the parkin Q311X mutant mice by both Western blot and immunocytochemistry (Lu et al., 2009) (Fig. 2). Western blot analysis demonstrated a significant elevation in the levels of higher molecular weight insoluble α-synuclein oligomers (∼150 kDa) in the parkin mutants that were significantly reduced by chronic rapamycin treatment (Fig. 2A,B). This was found to coincide with immunocytochemical data demonstrating increased levels of punctate pro-K-resistant α-synuclein immunostaining in the parkin Q311X mutant SNpc that was abrogated by rapamycin treatment, along with retention of TH+ cell numbers as previously demonstrated (Figs. 1, 2C).
Figure 2.

Increased levels of pro-K-insoluble α-synuclein in parkin Q311X mutants are abrogated by rapamycin feeding. A, Striatal tissue lysates from parkin mutant mice (Mut) versus WT littermate controls on control or rapamycin diet (Rapa) were incubated with 0–10 μg/ml pro-K and run on Western blots probed with antibody against α-synuclein. B, Densometric quantitation of monomeric (∼17 kDa) versus large oligomeric (∼150 kDa) α-synuclein levels in 5 μg/ml pro-K-treated samples (red circled on Western blot). ***p < 0.001 versus WT. +++p < 0.001 versus Mut. Three separate experiments were performed in triplicate. C, Representative confocal SN immunocytochemistry from Mut versus WT littermates on control or Rapa diet treated with 5 μg/ml pro-K (Lu et al., 2009) and probed with antibody against TH (red) or α-synuclein (Syn, green). Blue represents nuclear DAPI staining. Original magnification, ×63.
Next, effects of rapamycin feeding on locomotor behavior was assessed in the parkin Q311X mutants via both open field analysis and the pole test (Fig. 3). As previously reported by ourselves and others, parkin Q311X mutants displayed significant reductions in locomotor activities that were significantly prevented by chronic rapamycin feeding (Fig. 3) (Lu et al., 2009; Lieu et al., 2013, 2014).
Figure 3.
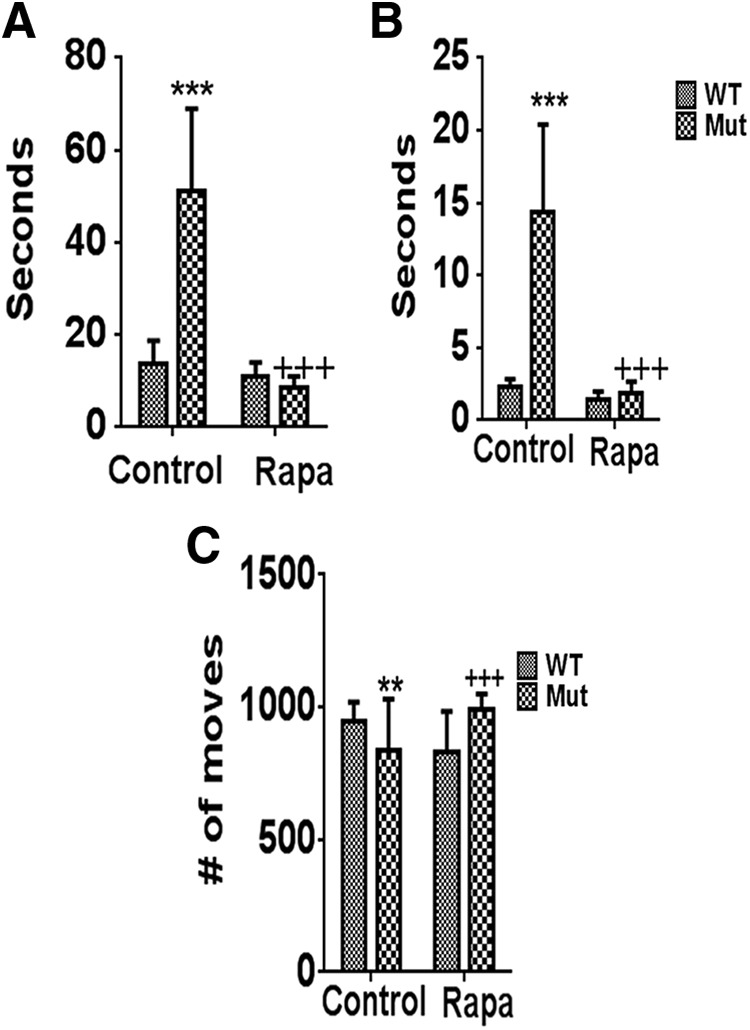
Losses in motor function in parkin Q311X mutants are abrogated by rapamycin feeding. Motor coordination and balance were assessed in parkin mutant mice (Mut) versus WT littermate controls on control or rapamycin diet (Rapa) via the pole test as previously described by our laboratory (Lieu et al., 2013). Time to turn downward on the pole (A) and to descend to the base (B) was measured over 5 consecutive trials. ***p < 0.001 versus WT. +++p < 0.001 versus Mut. C, Spontaneous activity was measured in Mut versus WT littermates on control or Rapa diet as previously described in these animals by Lu et al. (2009) using a Tru Scan photobeam apparatus (Colbourn Instruments) in five separate trials. **p < 0.01 versus WT. +++p < 0.001 versus Mut.
Parkin Q311X mutant mice display a late-stage block in autophagy at the level of lysosomal function
We next examined the impact of both parkin Q311X mutation and rapamycin on autophagic function in our four cohorts. We first looked at LC3 accumulation. During initiation of autophagy, LC3 is recruited to APG membranes where it interacts with p62; p62 acts as an adaptor to link ubiquitin-tagged mitochondria with APG membranes. These then form mature APGs that engulf the ubiquitin-labeled mitochondria. Given that APG formation has been reported to occur downstream of parkin-mediated mitochondrial tagging and recruitment, we predicted that we would see reduced levels of accumulation of LC3 puncta within effected parkin mutant SNpc tissues. Surprisingly, we observed significant increased numbers of total LC3 puncta within the parkin Q311X mutant SNpc compared with WT littermate controls (Fig. 4A). Many of these puncta were found to colocalize with the mitochondrial VDAC marker (Fig. 4B,C). Numbers of LC3 puncta were further increased following rapamycin treatment, in both parkin Q311X mice and WT littermate controls.
Figure 4.

Increased levels of the autophagic marker LC3 within mitochondria in parkin Q311X mutant SNpc tissues. A, Quantitation of LC3 puncta as assessed via 3D reconstruction of collected Z-stacks using Imaris (version 7.1.0, Bitplane) (Costes et al., 2004). Values are total puncta-positive pixels. ***p < 0.001 versus WT. +++p < 0.001 versus Mut on control versus rapamycin diet (Control vs Rapa). Data are from three or four separate tissue samples per animal performed in triplicate. B, Quantitation of LC3 puncta colocalized with mitochondrial VDAC assessed as in A. ***p < 0.0001 versus WT. +++p < 0.0001 versus Mut. Data are from three or four separate tissue samples per animal performed in triplicate. C, Representative confocal SN immunocytochemistry from parkin mutant mice (Mut) versus WT littermate controls on control or rapamycin diet (Rapa) probed with antibody against either the mitochondrial marker VDAC (red) or the autophagic marker LC3 (green); merged (yellow). Original magnification ×63. n = 4 animals per condition. Insets, Higher magnification of single representative cells for merged images in each experimental condition.
We reasoned that observed increases in LC3 puncta in the SNpc of Q311X mutant mice could either be a consequence of a compensatory increases in autophagy or an inhibition downstream of APG formation resulting in impaired flux. To address this issue, we performed quantitation of p62 levels in our cohorts (Geisler et al., 2010a); p62 not only targets ubiquitinated mitochondria to APGs but, following their fusion with lysosomes, is itself degraded by resultant APLs. Sustained increases in its levels would therefore be consistent with a downstream inhibition in autophagy. We observed significant increases in levels of p62 puncta formation within parkin Q311X mutant SNpc versus WT controls (Fig. 5A,B). Levels were significantly reduced by rapamycin feeding. Levels of striatal p62 protein itself were also shown to be elevated in the parkin Q311X mutant and reduced by rapamycin (Fig. 5C).
Figure 5.
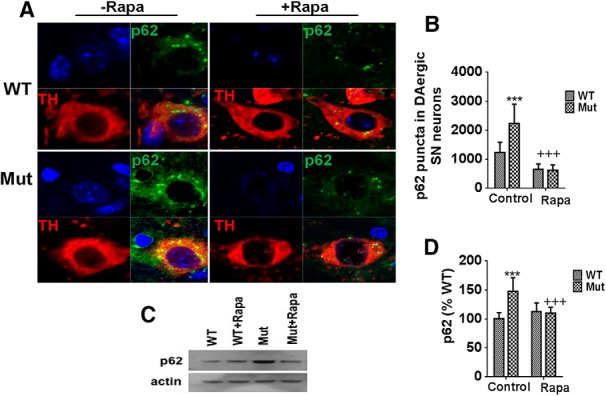
The autophagic protein p62 remains elevated in parkin Q311X mutant SNpc tissues. A, Representative SN immunocytochemistry from parkin mutant mice (Mut) versus WT littermate controls on control or rapamycin diet (Rapa) probed with antibody against p62 (top right, green) or the dopaminergic marker TH (bottom left, red). Yellow represents merged (bottom right). Blue represents nuclear DAPI (top left). n = 4 animals per condition. B, Densitometric quantitation of p62 puncta colocalized with TH. ***p < 0.001 versus WT. +++p < 0.001 versus Mut. Data are from 10–12 separate tissue sections per animal performed in triplicate. C, Representative Western blots of striatal tissue extracts isolated from parkin mutant mice (Mut) versus WT littermate controls on control or rapamycin diet (Rapa) probed with antibody against p62 (actin used as loading control). D, Quantitation of p62 levels. ***p < 0.001 versus WT. +++p < 0.001 versus Mut. Data are from three or four separate tissue samples per animal performed in triplicate.
To to see whether there was a blockage at the level of APL formation, we next performed EM quantitation to verify relative numbers of APG versus APLs (Fig. 6A–C). EM images of SNpc sections demonstrated a significant increase in the ratio of normal APGs to APLs in the parkin Q311X mutant mice versus littermate controls (Fig. 6D). Parkin Q311X mutants also displayed along with increased numbers of APLs containing undigested mitochondria. These data suggest a reduction in numbers of functional APLs. Both of these morphological events were found to be reversed by chronic rapamycin administration.
Figure 6.
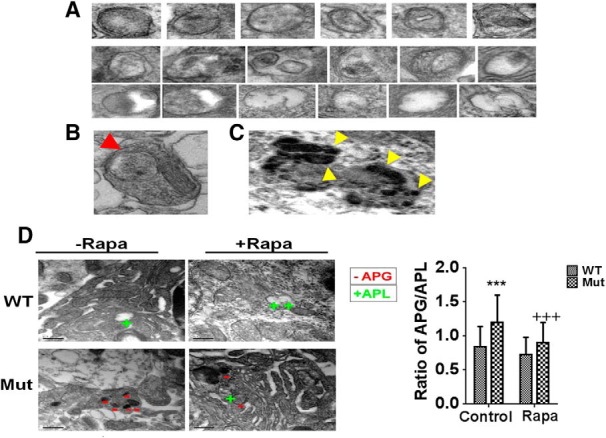
EM analysis of parkin Q311X mutant SNpc tissues demonstrate increased APG:APL ratios that are abrogated by rapamycin feeding. A, Representative high-magnification EM images prepared from SNpc tissue sections of APGs (top) and ALs (bottom) at different stages of maturation used for morphometric quantification. Most APGs were identified in the soma, but in some instances they were also observed in the neuronal processes (red arrow) (B). Mature lysosomes were not included in the morphometric analysis, identified by the presence of lipofuscin-like deposits in their lumen (yellow arrows; ∼5% total APL) (C). D, Representative images and quantitation of APG:APL ratios analyzed by Cavaleiri estimation for large datasets. ***p < 0.001 versus WT. +++p < 0.05 versus Mut. Representative APGs are marked with − and APLs with +.
Cathepsin D is the principal lysosomal endopeptidase responsible for degradation of both proteins and mitochondria (Jones et al., 1982; Hossain et al., 2001; Qiao et al., 2008). Along with reduced numbers of functional APLs, we also noted significant reductions in levels of mature activated cathepsin D within striatal tissues from the parkin Q311X mutant, suggesting a defect in lysosomal function (Fig. 7). Cathepsin D levels were found to be near that of WT littermate controls in the presence of rapamycin feeding.
Figure 7.
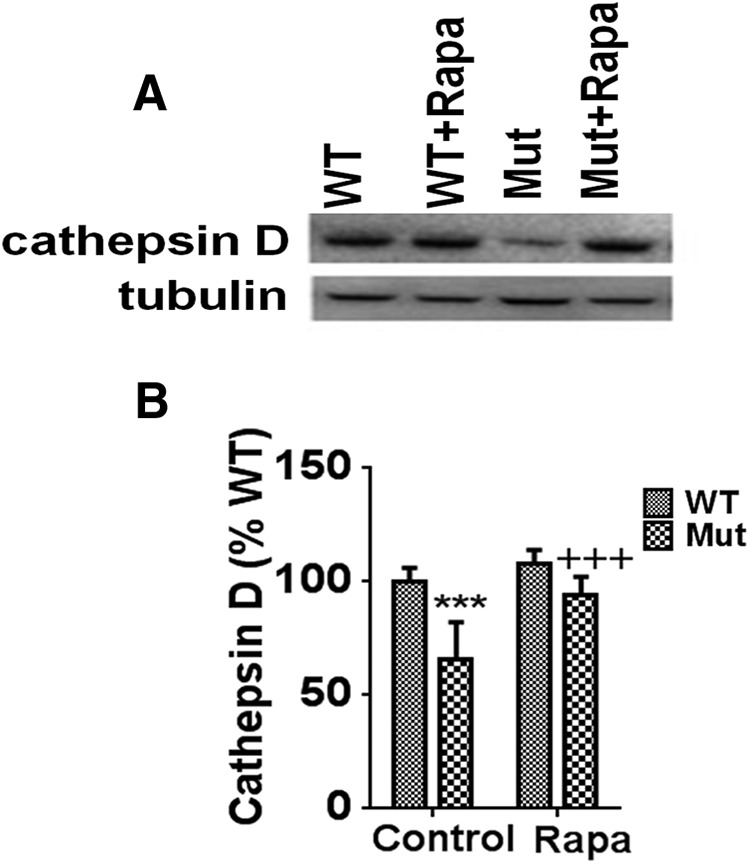
Levels of the lysosomal activity marker cathepsin D are downregulated in parkin Q311X mutants and restored by rapamycin. A, Representative Western blots of striatal tissue extracts isolated from parkin mutant mice (Mut) versus WT littermate controls on control or rapamycin diet (Rapa) probed with antibody against cathepsin D (tubulin used as loading control). n = 3 animals per condition. B, Densitometric quantitation of cathepsin D levels (reported as % WT). ***p < 0.001 versus WT. +++p < 0.001 versus Mut. Data are from three separate striatal tissue samples performed in triplicate.
Parkin Q311X mutants display reduced expression of TFEB, which is restored by rapamycin independent of parkin E3 ligase activity
Cathepsin D is one of a suite of genes whose levels are controlled by the master lysosomal transcription factor, TFEB (Sardiello et al., 2009; Settembre et al., 2012). Impaired TFEB activity has been observed in both the human PD SNpc and in a rat α-synuclein overexpression model of the disease (Decressac et al., 2013). In the latter, treatment with a rapamycin derivative was found to restore TFEB activity and autophagic clearance of α-synuclein aggregates and to rescue DAergic SNpc cell loss. Based on these data, we hypothesized that observed reductions in functional APLs and activated cathepsin D levels in the parkin Q311X mutants could be due to reduced lysosomal biogenesis as a result of losses in TFEB activity that are restorable by rapamycin treatment. Rapamycin treatment was indeed found to elicit significant increases in nuclear TFEB localization within SNpc cells of the parkin mutants, although to a somewhat lesser degree than in WT littermate controls (Fig. 8A,B). Rapamycin's ability to reestablish TFEB activity in the parkin Q311X mutants was not however associated with restoration of compromised parkin E3 ligase activity, suggesting that this is a parkin-independent event (Fig. 8C). Interestingly, reduced TFEB activity in the parkin Q311X mutants was found to coincide with reductions in expression of TFEB and its downstream TFEB target genes TFAM and Nrf1, all restored by rapamycin treatment (Fig. 8D–G). Together, these data suggest that parkin Q311X mutant mice have reductions in the master lysosomal transcription factor TFEB that can be restored by rapamycin in a manner independent of parkin E3 ligase activity.
Figure 8.

Reductions in TFEB activity in parkin Q311X mutants are restored by rapamycin in a manner independent of E3 ligase activity. A, Representative confocal SN immunocytochemistry from Mut versus WT littermate controls on control or rapa (Rapa) diet probed with antibody against TFEB (red) versus nuclear DAPI staining (blue). Original magnification, ×63. B, Quantitation of nuclear TFEB puncta as assessed via 3D reconstruction of collected Z-stacks using Imaris (version 7.1.0, Bitplane) (Costes et al., 2004). Data are total number per section from four total (three separate experiments performed in triplicate). C, In vitro ubiquitination reactions were performed by mixing immunoprecipitated parkin from striatal tissue lysates with E1 ubiquitin-activating enzyme, the E2 ubiquitin-conjugating enzyme UbcH7, and ubiquitin in ubiquitination buffer. Ubiquitination of parkin was then detected by anti-parkin/anti-ubiquitin antibody and quantitated by densitometry. ***p < 0.001 versus WT. Three separate experiments were performed in triplicate. D, Relative TFEB expression as assessed by RT-PCR from mRNA extracted from striatal tissues of parkin mutant mice (Mut) versus WT littermate controls on control or rapamycin diet (Rapa); actin was used as normalizing control. *p < 0.01 versus WT. +++p < 0.001 versus Mut. Three separate experiments were performed in triplicate. E, Relative Nrf1 expression as assessed by RT-PCR from mRNA extracted from striatal tissues of parkin mutant mice (Mut) versus WT littermate controls on control or rapamycin diet (rapa); actin was used as normalizing control. **p < 0.01 versus WT. +p < 0.05 versus Mut. Three separate experiments were performed in triplicate. F, Relative TFAM expression as assessed by RT-PCR from mRNA extracted from striatal tissues of parkin mutant mice (Mut) versus WT littermate controls on control or rapamycin diet (rapa); actin was used as normalizing control. **p < 0.01 versus WT. ++p < 0.01 versus Mut. Three separate experiments were performed in triplicate.
Parkin Q311X mutants display increased levels of the parkin substrate PARIS, which coincides with reductions in PGC1α expression
TFEB has recently been demonstrated to participate in a positive transcriptional feedback loop with the master mitochondrial transcriptional regulator PGC1α (Roczniak-Ferguson et al., 2012; Settembre et al., 2012; Tsunemi et al., 2012). Upregulation of PGC1α-TFEB signaling was shown to prevent proteotoxic stress associated with a mouse model of Huntington's disease. PGC1α expression itself is indirectly regulated by parkin, via parkin's ability to limit protein levels of the transcriptional repressor PARIS (Shin et al., 2011). PARIS has been shown by this same group to be upregulated in nigrostriatal neurons in the context of human PD, whereas PGC1α and its target genes, including Nrf1, have been reported to be underexpressed in the brains of PD patients (Zheng et al., 2010). We therefore next asked whether losses in parkin E3 ligase activity in the parkin Q311X mutant mice could be driving reductions in PGC1α-TFEB signaling via increases in PARIS protein levels (Shin et al., 2011). The Q311X mutant protein has previously been demonstrated to bind less avidly to PARIS than WT protein, coinciding with buildup of PARIS levels (Shin et al., 2011). PARIS protein levels were indeed found to be elevated in SNpc tissues from parkin Q311X mutants versus controls (Fig. 9A,B). They were also not reduced by rapamycin treatment (data not shown). Additionally, along with alterations in TFEB and its target genes (Fig. 8), PGC1α expression was found to also be reduced in Q311X mutants and restored in the presence of chronic rapamycin feeding (Fig. 9) (Shin et al., 2011).
Figure 9.
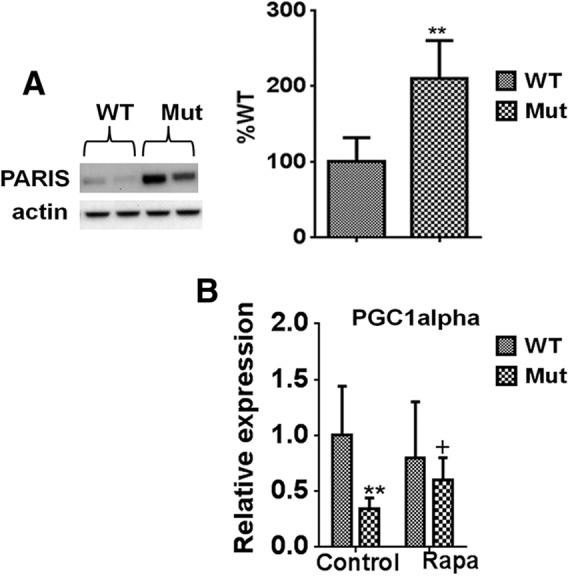
Q311X mutation results in elevations in PARIS levels corresponding with reductions in PGC1α expression. A, Representative Western blots and densitometric quantiation of striatal tissue extracts isolated from parkin mutant mice (Mut) versus WT littermate controls probed with antibody against PARIS (actin used as loading control). n = 3 animals per condition (reported as % WT). **p < 0.01 versus WT. B, Relative PGC1α expression as assessed by RT-PCR from mRNA extracted from striatal tissues of parkin mutant mice (Mut) versus WT littermate controls on control or rapamycin diet (Rapa); actin was used as normalizing control. **p < 0.01 versus WT. +p < 0.05 versus Mut. Three separate experiments were performed in triplicate.
Mitochondrial defects in the parkin Q311X mutants are prevented by rapamycin treatment
We postulated that reductions in PGC1α-TFEB signaling as a consequence of PARIS elevation would in turn coincide with reductions in mitochondrial quality control contributing in turn to the neurodegenerative effects observed in the parkin Q311X mutant mouse model. To assess mitochondrial “health” in these animals, we first measured mitochondrial volume via EM. Mitochondrial volume fractions were found to be reduced in the parkin Q311X mutants and restored following rapamycin treatment (Fig. 10A). We next assessed activity of mitochondrial CI, abnormalities in which have been observed in association with PD (Schapira et al., 1990). CI activity was also reduced in the parkin Q311X mutants and restored by rapamycin (Fig. 10B).
Figure 10.
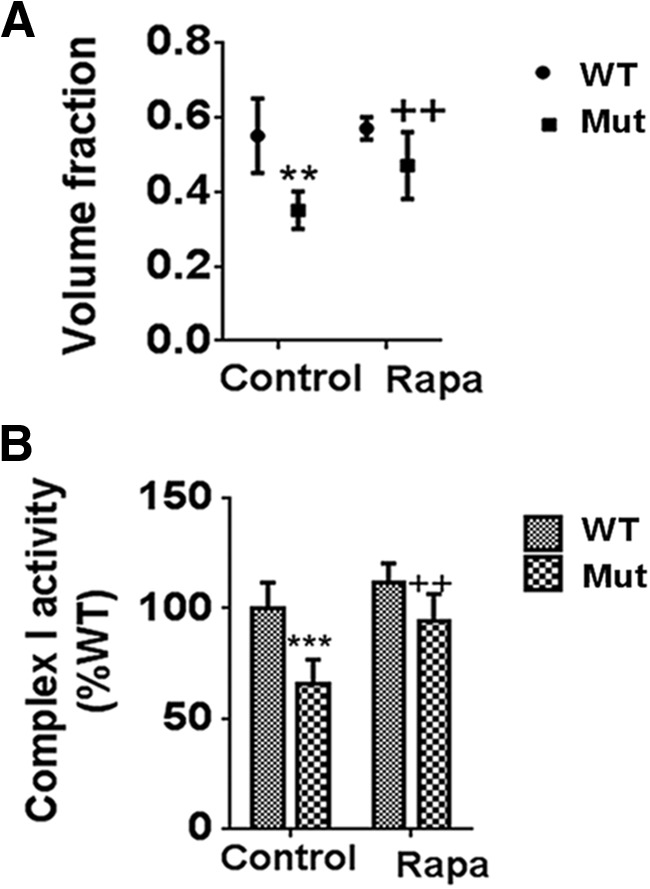
Compromises in mitochondrial volume and function in parkin Q311X mutants are restored by rapamycin feeding. A, Measurement of mitochondrial volume by EM. **p < 0.01 versus WT. ++p < 0.01 versus Mut. B, Mitochondrial CI activity measured in isolated striatal DAergic synaptosomes as described previously (Chinta et al., 2007). ***p < 0.001 versus WT. ++p < 0.01 versus Mut. Data are from three separate synaptosomal preparations performed in triplicate.
The detrimental effects of parkin Q311X expression in DAergic cells on PGC1α-TFEB signaling are PARIS-dependent and coincide with effects on mitochondrial function and cell viability
To directly demonstrate that the deterimental effects of Q311X expression within DAergic cells on PGC1α-signaling impacting on mitochondrial health and cell viability is PARIS-dependent, we looked at the impact of PARIS knockdown in WT versus Q311X-FLAG-tagged transfected cultured DAergic neurons. As expected, Q311X mutant-transfected cells display a band corresponding to the correct size for the truncated mutant protein (Fig. 11A). Reductions in PARIS expression levels in these cells were found to result in increased PGC1α-TFEB expression and restoration of lost mitochondrial function and cell viability associated with Q311X versus WT expression (Fig. 11B–E).
Figure 11.

PARIS siRNA knockdown in DAergic cells expressing WT versus Q311X parkin. A, Representative Western blot of cellular extracts isolated from N27 cells transfected with either human WT or Q311X-FLAG-tagged plasmid probed with antibody against FLAG (actin used as loading control). B, Representative Western blot and quantitation of cellular extracts isolated from N27 cells transfected with PARIS siRNA versus scrambled (Scr) siRNA probed with antibody against PARIS (actin used as loading control). Expressed as % Scr control. ***p < 0.001 versus Scr siRNA. C, Relative PGC1α and TFEB expression levels as assessed by RT-PCR from mRNA extracted from WT versus Q311X-transfected cells transfected with either PARIS siRNA or Scr siRNA control. **p < 0.01 versus WT+Scr siRNA. ++p < 0.01 versus Mut+Scr siRNA. ***p < 0.001 versus WT+Scr siRNA. +++p < 0.001 versus Mut+Scr siRNA. D, Mitochondrial activity as monitored by TMRM fluorescence expressed as % WT+Scr siRNA. **p < 0.001 versus WT+Scr siRNA. +++p < 0.001 versus Mut+Scr siRNA. E, Measurement of cellular viability via MTT assay expressed as % WT+Scr siRNA. ***p < 0.001 versus WT+Scr siRNA. ++p < 0.01 versus Mut+Scr siRNA.
Rapamycin restoration of mitochondrial function and cell viability in Q311X-expressing cells requires expression of TFEB
To assess whether rapamycin's ability to protect against deterimental effects of Q311X expression within DAergic cells requires TFEB, we looked at the impact of TFEB knockdown in these same cells in the context of rapamycin treatment. Reductions in TFEB expression were found to abrogate rapamycin-mediated increases effects on both mitochondrial function and cell viability in the Q311X-expressing mutants; TFEB was also found to impact on PGC1α expression (Fig. 12A–E).
Figure 12.

Rapamycin treatment ± TFEB siRNA in DAergic cells expressing WT versus Q311X parkin. A, Representative Western blot and quantitation of cellular extracts isolated from N27 cells transfected with TFEB siRNA versus scrambled (Scr) siRNA probed with antibody against TFEB (actin used as loading control). Expressed as % Scr control. ***p < 0.001 versus Scr. B, Relative TFEB expression levels as assessed by RT-PCR from mRNA extracted from WT versus Q311X-transfected cells ± rapamycin (Rapa) treatment ± TFEB siRNA versus Scr siRNA control. ***p < 0.001 versus WT+Scr. **p < 0.01 versus WT+Scr. ++p < 0.01 versus Mut+Scr. #p < 0.05 WT+Scr+Rapa. −p < 0.05 versus Mut+Scr+Rapa. n = 3. C, Mitochondrial activity as monitored by TMRM fluorescence expressed as % WT+Scr siRNA control. ***p < 0.001 versus WT+Scr siRNA. +p < 0.05 versus Mut+Scr siRNA. #p < 0.05WT+Scr+Rapa. −p < 0.05 versus Mut+Scr+RAPA. n = 3 per condition. D, Measurement of cellular viability via MTT assay expressed as % WT+Scr siRNA control. ***p < 0.001 versus WT+Scr siRNA. +++p < 0.001 versus Mut+Scr siRNA. #p < 0.05 WT+Scr+Rapa. −p < 0.05 versus Mut+Scr+Rapa. n = 3 per condition. E, Relative PGC1α expression levels as assessed by RT-PCR from mRNA extracted from WT versus Q311X-transfected cells transfected with either TFEB siRNA or Scr siRNA control. ***p < 0.001 versus WT+Scr. ++p < 0.01 versus Mut+Scr. n = 3.
TFEB activation is neuroprotective in mitochondrially compromised human iPSC-derived DAergic neurons
To evaluate the effects of TFEB activation in a model more closely resembling the human disease condition, we chose to assess the effects of TFEB in human DAergic cultures differentiated from iPSCs. We elected to look at effects in these cells following rotenone administration, as this has previously been shown to result in elevated LC3 and p62 levels in human SY5Y cells similar to what we observe in vivo in our parkin mouse mutant in conjunction with reduced mitochondrial quality control (Mader et al., 2012). Both rapamycin and the select TFEB-inducing agent trehalose were found to result in protection against mitochondrial neurotoxicity in the iPSC-derived DAergic neurons (Fig. 13A,B). Furthermore, TFEB activation via trehalose was found to coincide with abrogation of lost mitochondrial function as assessed by ATP levels and mitochondrial membrane potential (Fig. 13C–G) in conjunction with reversal of rotenone-mediated inhibition of several TFEB target genes (PGC1α, cathepsin D, NRF1, GLA) (Fig. 13H–K).
Figure 13.
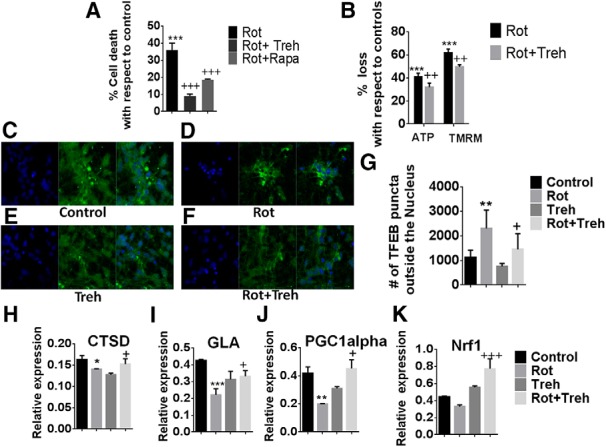
TFEB activation prevents losses in mitochondrial function and cell viability in mitochondrially compromised human iPSC-derived DAergic neurons. A, Measurement of cellular viability via MTT assay in iPSCs differentiated into DAergic cells treated with rotenone ± trehalose (Treh) or rapamycin (Rapa), expressed as % cell death versus untreated control. ***p < 0.001 versus untreated control. +++p < 0.001 versus rotenone alone. n = 3 per condition. B, Mitochondrial activity as monitored by ATP levels and TMRM fluorescence for measurement of mitochondrial membrane potential, expressed as % loss versus untreated control. ***p < 0.001 versus untreated control. ++p < 0.01 versus rotenone alone. n = 3 per condition. Activation of TFEB via measurement of cytoplasmic ICC in (C) untreated controls, (D) iPSC-derived neurons treated with rotenone (Rot) alone, (E) treatment with Treh alone, and (F) combined Rot/Treh. G, Quantitation reported as % TFEB puncta outside the nucleus. **p < 0.01 versus untreated control. +p < 0.05 versus Rot alone. TFEB target gene expression via RT-PCR for (H) cathepsin D (CTSD), (I) galactosidase α (GLA), (J) PGC1α, and (K) Nrf1. *p < 0.05 versus untreated control. **p < 0.01 versus untreated control. ***p < 0.001 versus untreated control. +p < 0.05 for Rot alone. +++p < 0.0001 for Rot alone. n = 3.
Discussion
A large body of work in cultured mammalian cells demonstrates that parkin via its interaction with PINK1 mediates initiation of lysosomal autophagy, particularly under conditions of damage or stress (Narendra et al., 2008; Geisler et al., 2010b; Matsuda and Tanaka, 2010; Vives-Bauza et al., 2010). Following its activation by PINK1, parkin ubiquitinates damaged mitochondria, labeling them for transport to the autophagophore and triggering their subsequent engulfment by the APG. The APG then fuses with lysosomes to form APLs, resulting in hydrolytic mitochondrial degradation (Pickrell and Youle, 2015). Mutations in the gene encoding parkin, PARK2, have been shown to result in increased levels of damaged mitochondria in affected brain tissues. Here we report that, in contrast to what has been previously reported for other mutations, in vivo expression of the parkin Q311X mutation appears to effect a downstream step in the autophagic pathway at the level of lysosomal function (Fig. 14). This in turn coincides with defects in both mitochondrial and protein quality control and increased neurodegenerative and behavioral phenotypes. Cell biology experiments demonstrate that this is a PARIS-dependent process involving disrupted regulation of a reciprocal transcriptional pathway involving PGC1α-TFEB signaling. Downregulation of PGC1α and its target genes, including TFEB, have been reported to be a key feature in recent meta-analyses of genome-wide gene expression in autopsied tissues from sporadic PD patients versus age-matched controls, suggesting that this signaling pathway constitutes an important therapeutic target for the disorder (Zheng et al., 2010).
Figure 14.

The Q311X parkin mutation impacts on mitochondrial quality control by interfering with PARIS-mediated regulation of PGC1α-TFEB signaling, which can be independently restored by rapamycin in a TFEB-dependent fashion. Our data suggest that compromise of parkin E3 ligase activity in Q311X-expressing mutants results in increased PARIS levels and reductions in master PGC1α-TFEB signaling. This impacts in turn on mitochondrial quality control via effects on both mitochondrial and lysosomal biogenesis and function. Independent induction of TFEB activity by rapamycin can restore this signaling pathway along with abrogating detrimental effects of the Q311X mutation on mitochondrial function and neuronal cell viability.
Treatment of mice carrying the human Q311X mutation with rapamycin was found to independently restore PGC1α-TFEB signaling in a manner not requiring parkin activity. This coincided with restoration of lysosomal and mitochondrial function in conjunction with abrogation of neurodegenerative-related features both in vitro and in vivo. Subsequent studies in human iPSC-derived neurons closely related to those effected in the human disease state show that TFEB induction can restore mitochondrial function and cell viability in these cells following mitochondrial compromise.
The Q311X mutation introduces a stop codon downstream of RING finger 1 (RING1) of the parkin protein, generating a C-terminal truncation that removes downstream in-between ring (IBR) and RING2 domains (Pickrell and Youle, 2015). In cultured cells, expression of this mutated form of the protein results in reduced parkin solubility and inhibition of the protein's E3 ligase activity, along with increases in parkin substrate levels (Sriram et al., 2005; Wang et al., 2005). In addition to ubiquitination of several other proteins, parkin also ubiquitinates PARIS, a Kruppel-associated box (KRAB) zinc finger transcriptional repressor, marking it for degradation by the proteosome. Q311X-mutated parkin protein has been reported to have reduced binding to PARIS, which requires both RING1 and RING2 domains for maximal binding efficiency (Shin et al., 2011). PARIS protein levels have been shown to be selectively increased in the nigrostriata in autopsied tissues from patients diagnosed with sporadic PD (Shin et al., 2011). Reductions in binding and ubiquitination of PARIS results in turn in increases in steady-state PARIS protein levels, along with increased PARIS occupancy at insulin response sequence sites within the PGC1α promoter and inhibition of subsequent PGC1α expression (Shin et al., 2011). Loss of DAergic neurons in a conditional parkin mouse knock-out model has been demonstrated to require upregulation of PARIS and to be inhibited by PGC1α expression (Shin et al., 2011). PGC1α and its target genes have been reported to be underexpressed in the brains of PD patients resulting in the identification by the Global PD Gene Expression consortium of PGC1α signaling as a therapeutic target for early intervention in PD (Zheng et al., 2010).
In addition to resulting in reduced expression of several genes directly involved in mitochondrial biogenesis and function, PARIS-mediated inhibition of PGC1α would also be predicted to result in reductions in expression of the master lysosomal regulator TFEB (Tsunemi et al., 2012). Nuclear localization of TFEB has been shown to be significantly reduced in postmortem human brains from PD patients and expression of TFEB and its targets to be reduced in a SNpc-specific fashion in animal models (Decressac et al., 2013). Loss of TFEB function has been associated with reduced lysomal function in various neurodegenerative disease models, although in general this has been attributed to reductions in the lysosomal degradation of defective proteins preventing their toxic aggregate formation (Tsunemi et al., 2012; Cortes et al., 2014); (Decressac et al., 2013; Polito et al., 2014). This is in agreement with data from this current study demonstrating that restoration of lysosomal function by rapamycin in addition to impact on mitochondrial function also prevents α-synuclein aggregation associated with the Q311X mouse model.
Treatment with the pharmacological mTOR inhibitor rapamycin was found to restore TFEB activity; previous publications suggest that TFEB activity is under the control of the mTOR pathway (Sardiello et al., 2009; Settembre et al., 2012) (Roczniak-Ferguson et al., 2012; Decressac et al., 2013). Inhibition of mTOR via rapamycin has been demonstrated to promote autophagic turnover of mitochondria in human cybrid lines carrying pathological mitochondrial DNA mutations (Gilkerson et al., 2012). We propose that, in addition to reported effects on protein quality control, TFEB activation also provides benefit via maintenance of mitochondrial homeostasis and function via the reciprocal positive feedforward PGC1α-TFEB signaling pathway affecting both mitochondrial biogenesis and lysosomal degradation. TFEB function can be independently restored via rapamycin.
In conclusion, our results suggest that parkin, through its effects on PARIS, plays a key role in overall mitochondrial dynamics in the cell by both inducing lysosomal degradation of damaged mitochondria via TFEB and promoting mitochondrial biogenesis through its effects on PGC-1α. These data further serve to validate PGC1α-TFEB signaling as a promising disease-modifying target for PD and other related disorders.
Footnotes
This work was supported by National Institutes of Health Grant AG025901 to J.K.A. We thank Dr. William Yang (University of California–Los Angeles) for providing us with the parkin Q311X mice; Monique O'Leary (Buck Institute) for assistance with rapamycin feeding; members of the Buck Morphology Core for help with EM protocols (Shannon O'Hare, Taki Ti Ko, and Cathy Vitelli); Anna Maria Cuervo (Einstein University) for assistance with in vivo EM data analyses; Rabiya Shabnam (J.K.A. laboratory) for assistance with tissue culture experiments; and Theo Tazannis for help with Western blotting and stereological cell counts.
The authors declare no competing financial interests.
References
- Bao XQ, Kong XC, Qian C, Zhang D. FLZ protects dopaminergic neuron through activating protein kinase B/mammalian target of rapamycin pathway and inhibiting RTP801 expression in Parkinson's disease models. Neuroscience. 2012;202:396–404. doi: 10.1016/j.neuroscience.2011.11.036. [DOI] [PubMed] [Google Scholar]
- Chinta SJ, Kumar MJ, Hsu M, Rajagopalan S, Kaur D, Rane A, Nicholls DG, Choi J, Andersen JK. Inducible alterations of glutathione levels in adult dopaminergic midbrain neurons result in nigrostriatal degeneration. J Neurosci. 2007;27:13997–14006. doi: 10.1523/JNEUROSCI.3885-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-Nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- Cookson MR. Parkin's substrates and the pathways leading to neuronal damage. Neuromolecular Med. 2003;3:1–13. doi: 10.1385/NMM:3:1:1. [DOI] [PubMed] [Google Scholar]
- Cortes CJ, Miranda HC, Frankowski H, Batlevi Y, Young JE, Le A, Ivanov N, Sopher BL, Carromeu C, Muotri AR, Garden GA, La Spada AR. Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat Neurosci. 2014;17:1180–1189. doi: 10.1038/nn.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti O, Brice A. Mitochondrial quality control turns out to be the principal suspect in parkin and PINK1-related autosomal recessive Parkinson's disease. Curr Opin Neurobiol. 2013;23:100–108. doi: 10.1016/j.conb.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Björklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A. 2013;110:E1817–E1826. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010a;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Treis A, Skujat D, Weber SS, Fiesel FC, Kahle PJ, Springer W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010b;6:871–878. doi: 10.4161/auto.6.7.13286. [DOI] [PubMed] [Google Scholar]
- Gilkerson RW, De Vries RL, Lebot P, Wikstrom JD, Torgyekes E, Shirihai OS, Przedborski S, Schon EA. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum Mol Genet. 2012;21:978–990. doi: 10.1093/hmg/ddr529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain S, Alim A, Takeda K, Kaji H, Shinoda T, Ueda K. Limited proteolysis of NACP/alpha-synuclein. J Alzheimers Dis. 2001;3:577–584. doi: 10.3233/jad-2001-3608. [DOI] [PubMed] [Google Scholar]
- Jones EW, Zubenko GS, Parker RR. PEP4 gene function is required for expression of several vacuolar hydrolases in Saccharomyces cerevisiae. Genetics. 1982;102:665–677. doi: 10.1093/genetics/102.4.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism [see comments] Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11:1214–1221. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- Lieu CA, Chinta SJ, Rane A, Andersen JK. Age-related behavioral phenotype of an astrocytic monoamine oxidase-B transgenic mouse model of Parkinson's disease. PLoS One. 2013;8:e54200. doi: 10.1371/journal.pone.0054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu CA, Dewey CM, Chinta SJ, Rane A, Rajagopalan S, Batir S, Kim YH, Andersen JK. Lithium prevents parkinsonian behavioral and striatal phenotypes in an aged parkin mutant transgenic mouse model. Brain Res. 2014;1591:111–117. doi: 10.1016/j.brainres.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XH, Fleming SM, Meurers B, Ackerson LC, Mortazavi F, Lo V, Hernandez D, Sulzer D, Jackson GR, Maidment NT, Chesselet MF, Yang XW. Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant alpha-synuclein. J Neurosci. 2009;29:1962–1976. doi: 10.1523/JNEUROSCI.5351-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader BJ, Pivtoraiko VN, Flippo HM, Klocke BJ, Roth KA, Mangieri LR, Shacka JJ. Rotenone inhibits autophagic flux prior to inducing cell death. ACS Chem Neurosci. 2012;3:1063–1072. doi: 10.1021/cn300145z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx FP, Holzmann C, Strauss KM, Li L, Eberhardt O, Gerhardt E, Cookson MR, Hernandez D, Farrer MJ, Kachergus J, Engelender S, Ross CA, Berger K, Schöls L, Schulz JB, Riess O, Krüger R. Identification and functional characterization of a novel R621C mutation in the synphilin-1 gene in Parkinson's disease. Hum Mol Genet. 2003;12:1223–1231. doi: 10.1093/hmg/ddg134. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Tanaka K. Uncovering the roles of PINK1 and parkin in mitophagy. Autophagy. 2010;6:952–954. doi: 10.4161/auto.6.7.13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb Perspect Biol. 2012;4:piia011338. doi: 10.1101/cshperspect.a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y, Swartzlander DB, Palmieri M, di Ronza A, Lee VM, Sardiello M, Ballabio A, Zheng H. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med. 2014;6:1142–1160. doi: 10.15252/emmm.201303671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao L, Hamamichi S, Caldwell KA, Caldwell GA, Yacoubian TA, Wilson S, Xie ZL, Speake LD, Parks R, Crabtree D, Liang Q, Crimmins S, Schneider L, Uchiyama Y, Iwatsubo T, Zhou Y, Peng L, Lu Y, Standaert DG, Walls KC, Shacka JJ, Roth KA, Zhang J. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain. 2008;1:17. doi: 10.1186/1756-6606-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui A, Hanson I, Andersen JK. Mao-B elevation decreases parkin's ability to efficiently clear damaged mitochondria: protective effects of rapamycin. Free Radic Res. 2012;46:1011–1018. doi: 10.3109/10715762.2012.662277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram SR, Li X, Ko HS, Chung KK, Wong E, Lim KL, Dawson VL, Dawson TM. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum Mol Genet. 2005;14:2571–2586. doi: 10.1093/hmg/ddi292. [DOI] [PubMed] [Google Scholar]
- Sunico CR, Nakamura T, Rockenstein E, Mante M, Adame A, Chan SF, Newmeyer TF, Masliah E, Nakanishi N, Lipton SA. S-Nitrosylation of parkin as a novel regulator of p53-mediated neuronal cell death in sporadic Parkinson's disease. Mol Neurodegener. 2013;8:29. doi: 10.1186/1750-1326-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1alpha rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra197. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandiver MS, Paul BD, Xu R, Karuppagounder S, Rao F, Snowman AM, Ko HS, Lee YI, Dawson VL, Dawson TM, Sen N, Snyder SH. Sulfhydration mediates neuroprotective actions of parkin. Nat Commun. 2013;4:1626. doi: 10.1038/ncomms2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrané J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Tan JM, Ho MW, Zaiden N, Wong SH, Chew CL, Eng PW, Lim TM, Dawson TM, Lim KL. Alterations in the solubility and intracellular localization of parkin by several familial Parkinson's disease-linked point mutations. J Neurochem. 2005;93:422–431. doi: 10.1111/j.1471-4159.2005.03023.x. [DOI] [PubMed] [Google Scholar]
- Winklhofer KF. Parkin and mitochondrial quality control: toward assembling the puzzle. Trends Cell Biol. 2014;24:332–341. doi: 10.1016/j.tcb.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Winklhofer KF, Henn IH, Kay-Jackson PC, Heller U, Tatzelt J. Inactivation of parkin by oxidative stress and C-terminal truncations: a protective role of molecular chaperones. J Biol Chem. 2003;278:47199–47208. doi: 10.1074/jbc.M306769200. [DOI] [PubMed] [Google Scholar]
- Wong ES, Tan JM, Wang C, Zhang Z, Tay SP, Zaiden N, Ko HS, Dawson VL, Dawson TM, Lim KL. Relative sensitivity of parkin and other cysteine-containing enzymes to stress-induced solubility alterations. J Biol Chem. 2007;282:12310–12318. doi: 10.1074/jbc.M609466200. [DOI] [PubMed] [Google Scholar]
- Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, Lipton SA. Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA, Grünblatt E, Moran LB, Mandel SA, Riederer P, Miller RM, Federoff HJ, Wüllner U, Papapetropoulos S, Youdim MB, Cantuti-Castelvetri I, et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med. 2010;2:52ra73. doi: 10.1126/scitranslmed.3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]