

Abstract
The endogenous dynorphin-κ opioid receptor (KOR) system encodes the dysphoric component of the stress response and controls the risk of depression-like and addiction behaviors; however, the molecular and neural circuit mechanisms are not understood. In this study, we report that KOR activation of p38α MAPK in ventral tegmental (VTA) dopaminergic neurons was required for conditioned place aversion (CPA) in mice. Conditional genetic deletion of floxed KOR or floxed p38α MAPK by Cre recombinase expression in dopaminergic neurons blocked place aversion to the KOR agonist U50,488. Selective viral rescue by wild-type KOR expression in dopaminergic neurons of KOR−/− mice restored U50,488-CPA, whereas expression of a mutated form of KOR that could not initiate p38α MAPK activation did not. Surprisingly, while p38α MAPK inactivation blocked U50,488-CPA, p38α MAPK was not required for KOR inhibition of evoked dopamine release measured by fast scan cyclic voltammetry in the nucleus accumbens. In contrast, KOR activation acutely inhibited VTA dopaminergic neuron firing, and repeated exposure attenuated the opioid response. This adaptation to repeated exposure was blocked by conditional deletion of p38α MAPK, which also blocked KOR-induced tyrosine phosphorylation of the inwardly rectifying potassium channel (GIRK) subunit Kir3.1 in VTA dopaminergic neurons. Consistent with the reduced response, GIRK phosphorylation at this amino terminal tyrosine residue (Y12) enhances channel deactivation. Thus, contrary to prevailing expectations, these results suggest that κ opioid-induced aversion requires regulation of VTA dopaminergic neuron somatic excitability through a p38α MAPK effect on GIRK deactivation kinetics rather than by presynaptically inhibiting dopamine release.
SIGNIFICANCE STATEMENT Kappa opioid receptor (KOR) agonists have the potential to be effective, nonaddictive analgesics, but their therapeutic utility is greatly limited by adverse effects on mood. Understanding how KOR activation produces dysphoria is key to the development of better analgesics and to defining how the endogenous dynorphin opioids produce their depression-like effects. Results in this study show that the aversive effects of κ receptor activation required arrestin-dependent p38α MAPK activation in dopamine neurons but did not require inhibition of dopamine release in the nucleus accumbens. Thus, contrary to the prevailing view, inhibition of mesolimbic dopamine release does not mediate the aversive effects of KOR activation and functionally selective κ opioids that do not activate arrestin signaling may be effective analgesics lacking dysphoric effects.
Keywords: depression, dopamine, dynorphin, GIRK, kappa opioid receptor, stress
Introduction
Stress exposure has profound effects on mood and drug abuse risk (Kendler et al., 1999; Brady and Sinha, 2005). Uncontrolled, sustained stressful experiences increase depressive and anxiety disorders in humans and in animal models of these behaviors (Neumann et al., 2011; Kormos and Gaszner, 2013). Stress exposure also increases the rewarding valence of drugs of abuse, promotes escalation of addictive drug consumption, and stimulates relapse in abstinent individuals (Brady and Sinha, 2005; Mantsch et al., 2015). Developing proresilience medications based on an understanding of how stress controls these behaviors has the potential for a novel therapeutic advance. Key to this understanding is the analysis of how stress-evoked release of neurotransmitters affects neural circuit physiology and decision-making processes. A series of recent studies indicate that stress exposure produces its dysphoric, anxiogenic, and proaddictive effects through the activation of extrahypothalamic cortocotrophin releasing factor, which releases the endogenous dynorphin opioid neuropeptides in brain (Land et al., 2008; Mantsch et al., 2015). Dynorphin selectively activates κ opioid receptors (KOR), and rodents treated with κ receptor antagonists or genetically lacking either prodynorphin or KOR have reduced immobility in forced swim assays (typical of antidepressant drug actions), reduced social avoidance behaviors following repeated social defeat, reduced escalation of psychostimulant self-administration, and reduced reinstatement of extinguished ethanol or cocaine self-administration (Bruchas et al., 2010; Van't Veer and Carlezon, 2013; Lalanne et al., 2014). Recent clinical studies confirm that drugs having KOR antagonist activity significantly reduce depressive symptoms in humans with treatment-resistant depression and reduce nicotine consumption in dependent individuals (Ehrich et al., 2015; Saxon et al., 2015).
Pharmacological activation of KOR in humans evokes reports of dysphoria and anxiety similar to those detected in rodent models (Pfeiffer et al., 1986; Millan, 1990; Vortherms and Roth, 2006). At the circuit level, KOR activation in the serotonergic neurons of dorsal raphe nucleus (DRN), the dopaminergic neurons of the ventral tegmental area (VTA), and neurons of the nucleus accumbens (NAc) have been implicated in the aversive properties of the stress response (Bruchas et al., 2010; Van't Veer and Carlezon, 2013). These results suggest that dynorphin regulates mood by controlling serotonergic and dopaminergic inputs to the NAc, but these circuit mechanisms are not yet clear. At the molecular level, KOR activation stimulates Gi/o protein signaling that reduces neuronal activity by increasing potassium conductance, decreasing voltage activated calcium conductance, and inhibiting transmitter vesicle release (Bruchas et al., 2010). In addition, sustained KOR activation recruits arrestin-dependent signaling, which activates p38 mitogen-activated protein kinase (MAPK) signaling (Chavkin et al., 2014). Pharmacological inhibition of p38 MAPK, genetic block of KOR-dependent p38 MAPK activation by G-protein-coupled receptor kinase 3 (GRK3) deletion, or cell-specific deletion of p38α MAPK in serotonergic neurons blocks stress-induced aversion (Bruchas et al., 2011; Chavkin et al., 2014). The separation between the receptor signaling pathways controlling G-protein-mediated responses and arrestin-mediated responses have important implications for drug development in general, but within the KOR system specifically, these findings predict that a G-biased KOR agonist would have analgesic efficacy without the dysphoric properties of conventional KOR agonists (Chavkin, 2011). Finding a noneuphorigenic (thus nonaddictive) opioid analgesic has been a central goal of opioid medicinal chemistry for >100 years, but the prediction that G-biased KOR agonists that do not activate p38 MAPK signaling would not be dysphoric requires that we understand how activation of KOR either pharmacologically or by stress-induced dynorphin release produces dysphoria.
Di Chiara and Imperato (1988) originally noted that KOR agonists inhibited dopamine release measured by in vivo microdialysis in the striatum, and suggested that dysphoria was a consequence of reduced dopamine tone. This hypothesis was consistent with the prevailing view that euphoric effects of addictive drugs resulted from an elevation of dopamine tone (Wise and Bozarth, 1982). Subsequent studies by Schultz et al. (1998) provided a more nuanced view of dopamine function, suggesting that dopamine actually provides a “reward-prediction error signal,” rather than a simple “reward” signal. Thus, how KOR inhibition of dopamine release contributes to dysphoria is not clear, and reconciling our understanding of KOR activation of p38 MAPK mechanisms and regulation of dopamine release seems necessary to understand how stress-induced dysphoria is encoded in brain. In the present study, we measured KOR regulation of dopamine release using fast-scan cyclic voltammetry (FSCV), used Cre recombinase-dependent conditional genetic deletion of KOR or p38α MAPK in VTA dopamine neurons, and used conditioned place aversion behavioral assays to assess the mechanisms underlying dynorphin/KOR dependent dysphoria.
Materials and Methods
Animals.
Male C57BL/6 mice (Charles River) >50 d old were used and given ad libitum access to food and water. Conditional knock-out of p38α in dopaminergic neurons was achieved using p38α floxed (Mapk14lox/+) mice on C57BL/6 background as previously described (Bruchas et al., 2011) and crossing them with mice that express Cre recombinase under the control of the dopamine transporter (DAT, Slc6a3Cre/+), a unique marker for dopaminergic neurons (Zhuang et al., 2005). Global gene deletion of KOR (Oprk1−/−) on C57BL/6 background was accomplished as previously described (Hough et al., 2000). In some instances, conventional KOR gene knock-out mice were bred to also carry the DAT-Cre transgene. Conditional gene knock-out of KOR was achieved using a floxed KOR line generated by the Institut Clinique de la Souris, in which exon 1 of KOR was flanked by loxP sites. This exon was then selectively excised in dopaminergic neurons by crossing to the DAT-Cre line and in serotonergic neurons by crossing with the ePet-Cre line (Scott et al., 2005; Zhuang et al., 2005). All animal procedures were approved by the University of Washington Institutional Animal Care and Use Committee.
Genotyping.
Genotyping was performed as previously described (Bruchas et al., 2011). PCR screening was performed using the following primers: A3 (5′-ATGAGATGCAGTACCCTTGGAGACCAGAAG-3′) and A4 (5′-AGCCAGGGCTATACAGAGAAAAACCCTGTG-3′) for the floxed and wild-type (+) p38α alleles, giving bands of 230 and 180 bp, respectively. Primers A1 (5′-CCACAGAAGAGATGGAGCTATATGGATCTC-3′) and A4 were used to detect the null p38αΔ allele as a 420 bp PCR product. The DAT-Cre and ePet-Cre transgenes were detected using 5′-AGCGTTCGAACGCACTGATTTCG-3′ and 5′-CGCCGTAAATCAATCGATGAGTTG-3′, yielding a 330 bp band. The ROSA26-yellow fluorescent protein (ROSA-YFP) reporter gene was screened for with 5′-AAGACCGCGAAGAGTTTGTC-3′, 5′-AAAGTCGCTCTGAGTTGTTAT-3′, and 5′-GGAGCGGGAGAAATGGATATG-3′, giving a 320 bp EYFP and 600 bp wild-type band. For screening of global KOR knock-outs, the wild-type KOR allele was amplified using 5′-AATTGTCTTTTGGCCACTGC-3′ and 5′-GTTGGTTGCGGTCTTCATCT-3′ giving a 510 bp band, whereas the null KOR knock-out allele yielded a 450 bp band when screened in a separate reaction with 5′-AGGAAGCAAAAAGCCTCTCC-3′ and 5′-CTCGTCCTGCAGTTCATTCA-3′. For the floxed KOR mice, 5′-GCACCAAAGTCAGGGAAGGT-3′ and 5′CAGGGCATATACTCTTCTCGCT-3′ were used to screen for the wild-type and floxed KOR alleles, giving bands of 317 and 427 bp, respectively.
Stereotaxic injection.
Mice were anesthetized with isoflurane and mounted on a model 1900 stereotaxic alignment system (David Kopf Instruments). For VTA injections, a 32 gauge needle with a 45 degree beveled tip (Hamilton) was lowered bilaterally (anteroposterior −3.30 mm, mediolateral ±0.30 mm from bregma) and 0.5 μl was injected at dorsoventral −4.70 to 4.75 mm and at −4.40 to 4.45 mm from bregma. For DRN injections, a 25 gauge needle with beveled tip (Hamilton) was lowered unilaterally (anteroposterior −4.65 mm, mediolateral 0.00 mm from bregma) and 1 μl was injected at dorsoventral −3.90 to 4.00 mm from bregma. Injection locations were confirmed via postmortem immunohistochemistry.
Vector design and production.
Lenti-KOR and Lenti-KOR(S369A) (Lenti-KSA) were developed and produced as previously described (Land et al., 2008). AAV-DIO KOR-GFP and AAV-DIO KSA-GFP were developed using the AM/CBA-DIO-poly vector (Cardin et al., 2009). Rat KOR-GFP or KSA-GFP was subcloned into the AAV-DIO vector using AgeI and NotI in the reverse coding orientation so that expression would only be driven in the presence of Cre recombinase under the chicken β-actin promoter.
After confirming sequence fidelity, HEK293T cells were transfected with 10 μg AAV and 20 μg pDG1 vector per 10 cm plate. Three days after transfection, cells were harvested and subjected to three freeze–thaw cycles. The supernatant was transferred to a Beckman tube containing a 40% sucrose cushion and spun at 27,000 rpm overnight at 4°C. Pellets were resuspended in CsCl at a density of 1.37 g/ml and spun at 5000 rpm overnight at 4°C. The following day, 1 ml CsCl fractions were run on an agarose gel, and genome-containing fractions were selected and spun again at 5000 rpm overnight at 4°C. The 1 ml fractions were collected again, and genome containing fractions were dialyzed overnight. The filtered solution was transferred to a Beckman tube and spun at 27,000 rpm overnight at 4°C. The pellet (containing purified AAV) was resuspended in 100 μl 1× HBSS.
Conditioned place preference (CPP).
Mice were trained in a balanced three-chamber apparatus consisting of chambers of equal size with horizontal or vertical stripes, and a third smaller chamber in the center. The paradigm consisted of tests on day 1 and four and training on days 2 and 3. On test days, the mice were placed in the center chamber and then allowed to roam freely through all three chambers for 30 min. Time spent in each chamber was recorded with a video camera (Canon DC310) and analyzed using Ethovision (Noldus). On training days, mice received saline vehicle (10 ml/kg, i.p.) while restricted to one chamber in the morning and the prototypical κ opioid agonist U50,488 (2.5–5.0 mg/kg at 10 ml/kg, i.p.) or cocaine (15 mg/kg at 10 ml/kg, s.c.) in the other chamber in the afternoon. The drug-paired chamber was assigned based on the day 1 pretest to ensure that the average time spent in each chamber was the same for all groups. The final preference score was determined based on the difference in the amount of time spent in the drug-paired chamber on the day 4 post-test and the day 1 pretest. All mice were handled once or twice a day for 4 d the week before the day 1 pretest. For cocaine CPP experiments, locomotor activity was recorded and analyzed.
U50,488 locomotor experiments.
Following the 4 d U50,488 CPA experiment, on days 5 and 6, a subset of mice were injected with either saline or U50,488 (10 mg/kg) and allowed to roam the CPP chamber freely. To allow for a within-subjects comparison, injections were administered in an alternating fashion such that half of the mice in a given cohort received each injection on a given day.
Tail-flick analgesia.
Mice were held while the tip of their tail was submerged in 52 ± 1°C water, and the tail withdrawal latencies were timed. Each mouse was exposed twice: once as a baseline and a second time 30 min after administration of 15 mg/kg U50,488.
Immunohistochemistry.
Mice were anesthetized with sodium pentobarbital (120 mg/kg, i.p.) and intracardially perfused with ice-cold PBS and 4% PFA. Brains were stored overnight in 4% PFA, then cryoprotected in 30% sucrose (w/v) in phosphate buffer (PB) solution. At least 48 h later, brains were sliced at 30 μm using a microtome (Leica). Slices were stored at 4°C in PB with 0.1% sodium azide. VTA and DRN slices were then taken and washed for 3 × 10 min in PBS before blocking in PBS containing 5% normal goat serum and 0.3% Triton X-100. Slices were then incubated for 36–48 h in blocking solution containing pairs of the following primary antibodies: chicken anti-tyrosine hydroxylase (1:1000, AB9702, Millipore), rabbit anti-KOR (1:1000, from an affinity purified 1.32 μg/ml stock) (Drake et al., 1996), chicken anti-GFP (1:3000, AB13970, Abcam), mouse anti-tyrosine hydroxylase (1:500, T1299, Sigma), mouse anti-tryptophan hydroxylase (1:500, T0678, Sigma), and mouse anti-GAD67 (1:1000, MAB5406, Millipore). In addition, we used a rabbit polyclonal antibody generated against the phosphorylated-Tyr 12 residue of KIR 3.1 (7.5 μg/ml, GIRKp) (Ippolito et al., 2005; Lemos et al., 2012). The peptide sequence used to generate GIRKp is unique based on a blast search of the mouse genome, and GIRKp-ir was not evident in Kir3.1−/− mice (Ippolito et al., 2005). Standard immunohistochemical procedures were performed to obtain pY12-KIR 3.1 (GIRKp-ir) and yellow-fluorescent protein (YFP-ir) (Abcam, chicken polyclonal ab13970) staining. Slices were then washed for 6 × 15 min in PBS and incubated in blocking solution containing Alexa-488 and Alexa-555 (1:500, Invitrogen) tagged to IgG of the appropriate species. The slices were then washed for 3 × 10 min in PBS and 2 × 10 min in PB. Slices were then mounted onto Superfrost Plus slides (Fisher) with Vectashield mounting media and sealed with nail polish for microscopy. All slices were imaged at the W.M. Keck Center for Advanced Studies in Neural Signaling (Seattle) on either a Leica SL confocal or a Nikon Upright with QImaging Camera.
GIRKp-ir quantification.
For comparisons of GIRKp-ir from different behavioral treatment groups, two or three animals for each group were perfused, sectioned, and processed in parallel. Imaging of sections from animals exposed to different treatments was done at the same time with the same microscope and camera settings. As different rounds of staining often yield different immunoreactive (ir) intensities, images from each treatment group displayed in this study came from the same round of staining. The patterns of GIRKp-ir seen in the VTA across treatment groups were similar to that previously reported in the dorsal raphe (Lemos et al., 2012). The selectivity for dopamine neurons of the DAT-Cre driver present in our control and p38α CKODAT mice has been shown previously (Zhuang et al., 2005; Bruchas et al., 2011); and as such, we used YFP staining as a proxy for dopaminergic phenotype to directly compare Cre-expressing neurons in control and conditional knock-out mice. We quantified GIRKp-ir in putative dopamine neurons of the VTA using Photoshop software to calculate the percentage of YFP-ir-positive cells also displaying GIRKp-ir (i.e., colocalization) in fields imaged at 20× magnification. Cells were considered positive for the protein if the intensity of the immunoreactivity was 1 SD above the average background pixel intensity for that slice. Images used for quantification were not adjusted for brightness or contrast. Cell counts and quantification were performed by an observer blind to treatment and genotype.
Rotarod.
Mice were placed on a 4-cm-wide rotating rod (Rotarod, San Diego Instruments), which accelerated from 4 to 40 rpm over 4 min. The latency to fall off of or clasp onto the rotating rod was recorded.
In vitro voltammetry.
Mice were rapidly decapitated, the brain was dissected, and slices were prepared as previously described (Lemos et al., 2012). Microelectrodes were fabricated by pulling carbon fiber through a glass pipette using a Sutter P-97 puller to form a glass seal. The fiber was then cut to ∼100–120 μm past the seal. The potential at the working electrode was held at −0.4 V versus an Ag/AgCl reference electrode, then ramped to 1.3 V and then back to −0.4 V at 400 V/s at a rate of 10 Hz. A single biphasic electric pulse was applied using a platinum/iridium parallel bipolar stimulating electrode (FHC) at 300–500 μA to evoke dopamine release. Each animal contributed 1–3 slices to each group. Each slice was counted as an individual replicate.
In vivo voltammetry.
Mice were anesthetized with 1.5 g/kg urethane. Sixty minutes later, depth of anesthesia was confirmed and mice were mounted on a model 900 stereotaxic alignment system (David Kopf Instruments). A carbon fiber microelectrode (manufactured as described above) was implanted in the NAc (anteroposterior 1.52 mm, mediolateral ±1.15 mm, dorsoventral −3.9 to 4.3 mm from bregma) and a silver chloride reference electrode was implanted contralaterally. Bipolar parallel stimulation electrodes (Plastics One) were implanted either in the medial forebrain bundle (MFB) (anteroposterior −2.40 mm, mediolateral ±1.10 mm, dorsoventral −4.5 to 5.20 mm from bregma) or the pedunculopontine tegmental area (PPTg) (anteroposterior −0.68 mm fromλ, mediolateral ±0.70 mm, dorsoventral −2.50 to 3.20 mm from bregma). For MFB recordings, a stimulus of 6 pulses was applied at 30 Hz and 300 μA to evoke dopamine release. For PPTg recordings, a stimulus of 30 pulses was applied at 60 Hz and 150 μA. Stimuli were applied every 300 s. After 60 min, mice were injected with U50,488 (5 mg/kg, i.p.) and recording continued for 60 min. The six stimuli before drug administration were averaged to define the baseline. At the end of each experiment, the remaining urethane was injected into the mouse before inducing an electrolytic lesion at the site of the working electrode. The mouse was then intracardially perfused with ice-cold PBS 4% PFA. After cryoprotection with 30% sucrose, brains were sliced at 40 μm and then stained with cresyl violet to confirm the recording site (and, for PPTg stimulation experiments, the stimulation site) as previously described (Ehrich et al., 2014).
In vivo electrophysiology.
As previously described (Zweifel et al., 2011), microdrives were fabricated in house using 16-channel electrode interface boards (EIB-16; Neuralynx) and tetrodes made from 0.00099-inch-diameter tungsten wire (California Fine Wire). Mice were stereotaxically implanted into the VTA using coordinates x = −3.25 mm from bregma, y = 0.05 mm from midline, and z = −4.5 mm from dura. Two weeks following recovery, mice were screened for putative dopamine neurons. During recording sessions, data were acquired by connecting mice to a 16-channel Medusa Preamplifier. Signals were filtered at 300–5000 Hz using a RZ5 Signal Processor (Tucker-Davis Technologies). Tetrodes were lowered daily in ∼40 μm increments until unit activity was observed. For all recording sessions, mice were habituated in the recording chamber for 10 min before session start. Once recording began, a 10 min baseline period was acquired followed by administration of drugs. Putative dopamine neurons from N = 4 control and N = 5 p38α MAPK CKO mice were identified by firing rate (<12 Hz) and by sensitivity to quinpirole (a reduction in firing rate to ≤50% of baseline). Quinpirole-sensitive firing rates were calculated from a 10 min window starting 3 min after injection. Once dopaminergic cells were identified, cell activity was recorded over 4 consecutive days. One day following identification of quipirole-sensitive neurons, activity was recorded during a 10 min baseline followed by two successive injections of saline and 50 min of additional recording following each injection for a total recording session of 110 min. On the second, third, and fourth day, mice were administered saline (50 min recording) followed by U50,488 (5 mg/kg). For nor-binaltorphimine dihydrochloride (norBNI) pretreatment, five mice were injected with norBNI (10 mg/kg) 50 min before U50,488 injection. Single units were isolated by cluster analysis using Offline Sorter software (Plexon), and clustered waveforms were analyzed using MATLAB software (The MathWorks).
Drugs.
(±) U50,488H, (±) U69,593, nor-binaltorphimine dihydrochloride (norBNI), and cocaine hydrochloride were provided by the National Institute of Drug Abuse Drug Supply Program. SB203580 was obtained from Millipore. Quinpirole and urethane were obtained from Sigma-Aldrich. Sodium pentobarbital and Beuthanasia Special-D were obtained from University of Washington Medical Center Drug Services. All drugs were dissolved in saline for use in vivo or aCSF for use in vitro.
Analysis.
Data were compiled in Microsoft Excel, and data analyses were performed in Prism 5.01 for Windows (GraphPad). CPP data were analyzed by Student's t test or one-way ANOVA where appropriate. Locomotor, tail-flick analgesia, and voltammetry data were analyzed using two-way repeated-measures ANOVA. For in vivo voltammetry, the last six stimuli of each treatment (baseline vs U50,488) were averaged for each animal, and this average was used for statistical comparisons. For in vitro voltammetry, the last four stimuli of each treatment (baseline, SB203580/vehicle, U69,593, and norBNI) were averaged for each slice. When ANOVA revealed statistical significance, Bonferroni post hoc analyses were applied where appropriate; in certain cases, Bonferroni post hoc analyses were applied after no interaction was seen to confirm negative results. Data are presented as mean ± SEM. Significance was set at p < 0.05.
Results
KOR activation in both dopamine and serotonin neurons is required for place aversion
To compare the roles of KOR activation in dopamine neurons and serotonin neurons in aversion, we generated a conditional knock-out of KOR in dopaminergic (DAT-Cre-expressing) neurons and serotonergic (ePet-Cre-expressing) neurons using a Cre-induced excision of a floxed KOR allele (KOR CKODAT and KOR CKOPet, respectively). Male KORlox/lox littermate controls from both colonies showed CPA to U50,488, whereas neither KOR CKODAT nor KOR CKOPet showed U50,488 CPA (Fig. 1a; one-way ANOVA, F(2,40) = 7.338, p < 0.01; post hoc analyses: KORlox/lox vs KOR CKODAT, t(40) = 2.340, p < 0.05; KORlox/lox vs KOR CKOPet, t(40) = 3.623, p < 0.01). These results demonstrated that selective genetic ablation of KOR within either class of neurons was sufficient to block U50,488 CPA.
Figure 1.

Conditional knock-out of KOR from either dopaminergic or serotonergic neurons blocks U50,488-CPA, but only KOR CKO from dopaminergic neurons blocks inhibition of dopamine release. a, Conditioned place aversion following U50,488 (5 mg/kg) was evident in wild-type and KORlox/lox littermates but was blocked by conditional KOR gene deletion in either dopaminergic (CKO KORDAT) or serotonergic (CKO KORPet) neurons. Place preference scores showed that both CKO KORDAT and CKO KORPet lack U50,488 CPA relative to KORlox/lox littermates (n = 9–22). *p < 0.05 (Bonferroni post hoc after one-way ANOVA). **p < 0.01 (Bonferroni post hoc after one-way ANOVA). b, Representative image showing analysis of slice FSCV data. Bottom, Heat map represents current generated by oxidation and reduction at the site of the working electrode during application of the triangle waveform. Top, Current × time plot shows evoked dopamine release and uptake. Orange dashed line indicates magnitude of peak current, which was recorded for each stimulation event (marked by arrow: 1 pulse, 300–500 μA). Inset, Cyclic voltammagram from the time of peak current confirms that the current recorded was induced by oxidation and reduction of dopamine. Calibration: 2 nA, 0.1 V. c, Left, Diagram depicting placement of bipolar stimulating electrode (red) and recording electrode (blue) during slice FSCV experiments (modified from Paxinos and Franklin, 2008). Right, Representative traces showing raw current responses of KORlox/lox, KOR CKODAT, and KOR CKOPet to 1 μm U69,593 (green) and 1 μm norBNI (purple). Calibration: 4 nA, 2 s. Arrow indicates stimulation timing. d, Change from baseline locally stimulated peak [DA] during baseline and following administration of 1 μm U69,593 and 1 μm norBNI. Each drug was bath-applied during the interval signified by the bar. e, Summary data of percentage change in [DA] from baseline shows that KOR CKODAT significantly blocked U69,593-induced inhibition of evoked dopamine release relative to KORlox/lox littermates, whereas inhibition of DA release was unaltered in KOR CKOPet (n = 4 or 5 slices from 2 or 3 animals). ***p < 0.001 (Bonferroni post hoc after one-way ANOVA). All graphs represent data as mean ± SEM.
KOR CKO from dopaminergic neurons, but not serotonergic neurons, blocks inhibition of dopamine signaling
Prior studies concluded that the aversion response to KOR activation was caused by inhibition of dopamine release in the NAc (Shippenberg et al., 1993), which requires expression of KOR by dopamine neurons (Chefer et al., 2013; Tejeda et al., 2013). To determine whether KOR CKOPet blocked U50,488 CPA by altering the response of NAc dopamine signaling to KOR activation, we used FSCV to record evoked dopamine release from midbrain slices containing the NAc (Fig. 1b). The KOR agonist U69,593 (1 μm) produced robust inhibition of evoked dopamine in midbrain slices from KORlox/lox and KOR CKOPet mice (52.7% and 54.5% below baseline, respectively). This inhibition was reversed upon washout or addition of the KOR antagonist norBNI (1 μm) (Fig. 1c–e). In contrast, evoked dopamine response in slices from KOR CKODAT showed no significant change in the presence of either drug (Fig. 1c–e). A two-way repeated-measures ANOVA showed a significant interaction (F(2,11) = 18.10, p < 0.001). Further, post hoc analyses demonstrated that both KORlox/lox (t(11) = 4.309, p < 0.001) and KOR CKOPet (t(11) = 4.242, p < 0.001) mice showed significant inhibition of dopamine release relative to KOR CKODAT mice after U69,593. Thus, while KOR excision from either dopamine neurons or serotonin neurons blocked CPA, KOR excision from serotonin neurons blocked CPA despite continued inhibition of evoked NAc dopamine release.
KOR activation in the VTA is required for place aversion
Local inactivation of KOR by stereotaxic injection of the long-acting KOR antagonist norBNI (2.5 μg/μl) into the VTA blocked U50,488 CPA (Fig. 2a; unpaired t test, t(7) = 3.306, p < 0.05, compared with mice injected with an equal volume of saline in VTA). norBNI has previously been demonstrated to block KOR activation up to 21 d after administration (Bruchas et al., 2007b). This result demonstrated that activation of KORs within the VTA was necessary for U50,488 CPA. To assess sufficiency, we next injected KOR−/− (Oprk1−/−) mice with a lentiviral vector directing expression of the wild-type KOR (lenti-KOR) (viral design as previously described; Land et al., 2008). Lenti-KOR injection in the VTA selectively restored the aversive properties of KOR activation (one-way ANOVA, F(2,33) = 5.140, p < 0.05) (Fig. 2b). In contrast, uninjected KOR−/− or mice injected lentivirus encoding enhanced green fluorescent protein alone (lenti-eGFP) did not develop U50,488 CPA (Fig. 2b). A different group of KOR−/− mice were injected with a lentiviral vector directing expression of KOR with a serine to alanine substitution at serine 369 (lenti-KSA). Phosphorylation of KOR by GRK3 at serine 369 is necessary for arrestin-dependent activation of p38 MAPK while allowing for arrestin-independent signaling, such as activation of extracellular signal-regulated kinase (ERK1/2) (Bruchas et al., 2006). KOR−/− mice expressing lenti-KSA (t(33) = 2.891, p < 0.05) did not develop U50,488 aversion (Fig. 2b). KOR−/− mice injected with lenti-KOR in the DRN as a positive control also showed a restoration of U50,488 CPA (data not shown, one-sample t test, p < 0.01, t(14) = 3.538), replicating our previous findings (Land et al., 2008).
Figure 2.

KOR activation within the VTA is both necessary and sufficient for U50,488-induced CPA. a, Local inactivation of KOR by bilateral stereotaxic injection of norBNI into the VTA blocked U50,488 (5 mg/kg) induced CPA. Place preference scores for WT mice injected with either saline or 2.5 μg/μl norBNI in the VTA before U50,488 conditioning (n = 8). *p < 0.05, saline versus norBNI (t test). b, U50,488 produced significant place aversion in wild-type (KOR+/+) mice, but not in KOR−/− knock-out mice. Right, KOR−/− mice were injected with lenti-KOR, lenti-KSA, or lenti-eGFP in the VTA before U50,488 CPA. Lenti-KOR in the VTA significantly restored U50,488 CPA, whereas neither lenti-KSA nor lenti-eGFP did. Left, *p < 0.05 (t test). Right, *p < 0.05 (Bonferroni post hoc analysis after one-way ANOVA). n = 11–19. c, U50,488 (10 mg/kg) significantly reduced locomotor activity compared with saline injection in wild-type KOR+/+ mice, but not in KOR−/− or in mice receiving lenti-viral injections in VTA (n = 6–11). ***p < 0.001 (Bonferroni post hoc analysis after two-way ANOVA). x-axis labels are the same groups as in b. d, Similarly, U50,488 significantly reduced tail-withdrawal latencies compared with saline injection in wild-type KOR+/+ mice, but not in KOR−/− or in mice receiving lenti-viral injections in VTA. Withdrawal latencies were recorded before and 30 min after 15 mg/kg U50,488 (n = 11–19). ***p < 0.001 (Bonferroni post hoc after two-way ANOVA). x-axis labels are the same groups as in b. e, In contrast, latency to tail-flick was significantly increased by 15 mg/kg U50,488 in KOR−/− mice injected with lenti-KOR in DRN (n = 13). ***p < 0.001 (paired t test). f, Representative IHC images showing restoration of KOR immunoreactivity (red) in TH-ir (green) neurons of the VTA after injection of lenti-KOR or lenti-KSA into the VTA of KOR−/− mice, but not after injection of lenti-KOR into the DRN. Note the presence of double-labeled neurons (yellow) in uninjected KOR+/+ and in sections from KOR−/− mice injected with either lenti-KOR or lenti-KSA in VTA. Bottom-right inset (red rectangle), Location of the section of the parabrachial pigmented nucleus of the VTA from which images were taken (modified from Paxinos and Franklin, 2008).
Systemic injection of 10 mg/kg U50,488 induces locomotor hypoactivity, an effect that is mediated by KOR actions at substantia nigra dopamine neurons (Schnur and Walker, 1990). For 2 d following the CPA post-test, a subset of animals were alternately injected with either saline or U50,488 (10 mg/kg, i.p.) and allowed to freely explore the CPP chambers. Two-way ANOVA showed a significant interaction (F(4,37) = 8.480, p < 0.0001), and KOR+/+ mice showed a significant inhibition in locomotor activity after U50,488 relative to saline (t(37) = 7.259, p < 0.001). Neither KOR−/− mice nor any virally injected group showed an effect of U50,488 on locomotor activity (p > 0.05) (Fig. 2c), demonstrating that the restoration of KOR expression was restricted.
We previously reported that injection of lenti-KOR or lenti-KSA into the DRN restored U50,488-induced analgesia, leading to an increase in warm water tail-flick latency (Land et al., 2008). Two-way ANOVA showed a significant interaction (F(4,57) = 8.827, p < 0.0001), and subsequent post hoc analysis demonstrated that only KOR+/+ mice showed a significant increase in their tail-flick latency 30 min after injection with U50,488 (15 mg/kg, i.p.) (KOR+/+, p < 0.001). Neither KOR−/− mice without viral injection nor any group receiving lentiviral injections into the VTA showed an effect of U50,488 (p > 0.05, all groups) (Fig. 2d). As a positive control, we performed a tail-flick analgesia test on KOR−/− mice that had received lenti-KOR injected into the DRN. A paired t test showed that the lenti-KOR injection into the DRN restored U50,488-induced analgesia in these mice (t(12) = 4.409, p < 0.001) (Fig. 2e). This demonstrated that the restoration of U50,488 CPA in lenti-KOR VTA mice was not due to viral diffusion to the nearby DRN.
After the behavioral analyses were concluded, mice were killed, and immunohistochemistry (IHC) was performed to assess KOR expression in the VTA. KOR-immunoreactivity (ir) was evident in the VTA of wild-type mice, as well as KOR−/− mice injected with lenti-KOR or lenti-KSA in the VTA, but not in uninjected KOR−/− mice (Fig. 2f). KOR-ir was not evident in the VTA after lenti-KOR injection into the DRN (Fig. 2g), thus further demonstrating that the aversion seen after DRN injection was not due to viral diffusion to the VTA. KOR expression was not seen outside of the VTA in mice injected intra-VTA with either lenti-KSA or lenti-KOR (data not shown).
Selective restoration of KOR activation to VTA dopamine neurons of KOR−/− mice rescues aversion in a phosphorylation-dependent manner
Although the previous experiment demonstrated that KOR activation and phosphorylation within VTA neurons was sufficient to support U50,488 CPA, it did not clarify which cell types are required. To determine whether KOR expression within dopaminergic neurons of the VTA alone was sufficient to support KOR-CPA, we developed an adeno-associated double-floxed inverse open reading frame (AAV-DIO) viral construct (Cardin et al., 2009) directing expression of GFP-tagged KOR or KSA selectively in Cre-expressing neurons (Fig. 3a; DIO-KOR-GFP or DIO-KSA-GFP). The number of cells showing colocalization between GFP-ir and immunoreactivity for tyrosine hydroxylase (TH-ir), a selective marker for dopamine neurons, was significantly increased in DATCre/+ mice relative to DAT+/+ mice (Fig. 3b,c; two-way ANOVA, main effect of genotype, F(1,43) = 110.7, p < 0.0001), demonstrating selectivity of expression. Although WT control mice showed a normal CPA to U50,488, DATCre/+ KOR−/− mice showed U50,488 CPA after intra-VTA injection with DIO-KOR-GFP but not DIO-KSA-GFP (Fig. 3d; unpaired t test, t(29) = 2.767, p < 0.01). This demonstrates that KOR activation in VTA dopamine neurons alone is sufficient to support CPA, but this requires an intact GRK3 phosphorylation site.
Figure 3.

KOR activation selectively in VTA dopamine neurons is sufficient for KOR CPA. a, Schematic represents the design of AAV-DIO viral construct, along with inversion of KOR-GFP/KSA-GFP gene and excision of loxp/lox2722 sites in the presence of Cre. b, Representative IHC images showing DIO-KSA-GFP selectively expressed in VTA TH-ir neurons in DATCre/+ but not DAT+/+ animals after VTA injection: red represents TH-r; green represents GFP-ir; yellow represents colabeling. c, Cell counts after VTA injection of DIO-KOR-GFP or DIO-KSA-GFP show a significant increase in cells double-labeled with TH-ir and GFP-ir in DATCre/+ animals relative to DAT+/+ littermates (n = 7–16). ***p < 0.001 (Bonferroni post hoc after two-way ANOVA). d, U50,488-CPA was selectively restored after injection of DIO-KOR-GFP into the VTA of DATCre/+, KOR−/− mice, but not after injection of DIO-KSA-GFP (n = 14–17). **p < 0.01 (t test).
Expression of p38α MAPK in dopamine neurons is selectively required for KOR CPA
To test the hypothesis that p38 MAPK activation in dopaminergic neurons is required for the aversive properties of KOR activation, we generated a conditional knock-out of p38α MAPK in DAT-expressing neurons, using a Cre-induced recombination of a floxed p38α MAPK allele (Bruchas et al., 2011). A subset of animals carried a floxed-STOP ROSA-YFP reporter allele to ensure that Cre-induced recombination only occurred in dopamine neurons. Subsequent IHC demonstrated that YFP highly coexpressed with tyrosine hydroxylase (TH-ir) within the VTA only in Cre-expressing animals (Fig. 4a) but did not colabel with the 67 kDa isoform of glutamic acid decarboxylase, a marker for GABAergic somata (Fig. 4b). The DRN also contains dopaminergic neurons (Stratford and Wirtshafter, 1990; Hasue and Shammah-Lagnado, 2002; Fu et al., 2010), and we confirmed that Cre-driven YFP expression colabeled with TH-ir neurons in the A10c and A10dc populations within the DRN. However, no overlap was observed between YFP-ir cells and immunoreactivity for tryptophan hydroxylase (TPH), a marker for serotonergic neurons (Fig. 4c). Experiments were performed on three genotypes: WT (DAT+/+; p38α+/lox), Cre Δ (DATCre/+; p38α−/+), and p38α CKODAT (DATCre/+; p38α−/lox). The Cre Δ mice were used as controls to address any concerns regarding potential reductions in p38α or DAT expression (Giros et al., 1996).
Figure 4.
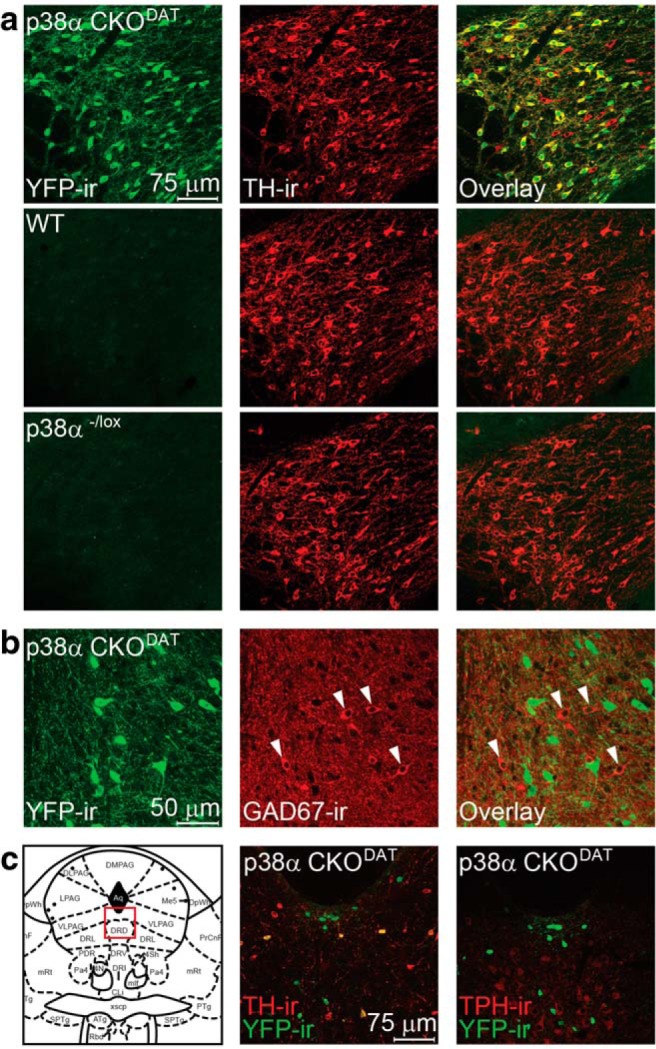
ROSA-YFP reporter shows that DAT-Cre selectively expresses in dopaminergic neurons. a, Representative IHC images showing that YFP-ir in an animal with DAT-Cre-directed ROSA-YFP expression extensively colabels with TH-ir throughout the VTA, yet YFP expression was absent in sections from mice lacking Cre: green represents YFP-ir; red represents TH-ir; yellow represents double-label. b, Representative IHC images showing that YFP-ir in sections from an animal with DAT-Cre-directed YFP expression in the VTA does not colabel with GAD67-ir. Arrows point to GABAergic neurons. c, Representative IHC images showing that YFP-ir in an animal with DAT-Cre-directed YFP expression in the DRN colabels with TH-ir but not TPH-ir. Left panel, Imaged section of the DRN (modified from Paxinos and Franklin, 2008).
As expected, WT and Cre Δ mice showed U50,488-induced CPA (Fig. 5a). However, p38α CKODAT mice failed to show CPA to the drug-paired context (one-way ANOVA, F(2,31) = 3.800, p < 0.05; post hoc vs WT, t(31) = 2.366, p < 0.05; post hoc vs Cre Δ, t(31) = 2.496, p < 0.05) (Fig. 5a). These results suggest that the aversive properties of KOR activation are dependent on the expression of p38α MAPK in dopaminergic neurons.
Figure 5.

Conditional knock-out of p38 MAPK from dopaminergic neurons selectively blocks the aversive properties of KOR activation. a, Preference scores show that p38α CKODAT mice do not show U50,488 CPA, unlike WT and Cre Δ littermates (n = 12–20). *p < 0.05 (Bonferroni post hoc after one-way ANOVA). b, Fall latency for each trial from rotarod performance assay shows no effect of p38α CKODAT (n = 10–16). c, WT, Cre Δ, and p38α CKODAT mice all show normal cocaine CPP (n = 8–11). d, WT, Cre Δ, and p38α CKODAT mice all show normal acute locomotor sensitization to cocaine (n = 8–11). **p < 0.01 (Bonferroni post hoc after two-way ANOVA). ***p < 0.001 (Bonferroni post hoc after two-way ANOVA).
We next tested mice with repeated exposure to the rotarod performance assay, a dopamine-dependent test of motor coordination in which mice learn to avoid a negative outcome over repeated trials (Zhou and Palmiter, 1995). p38α CKODAT mice showed no deficit in this task, as demonstrated by a statistically significant main effect of trial (Fig. 5b; two-way repeated-measures ANOVA, F(8,36) = 55.55, p < 0.0001) and no effect of genotype (F(2,36) = 1.059, p = 0.3573). Although a statistically significant interaction was seen (F(16,36) = 2.098, p < 0.01), post hoc analyses showed no significant difference between the performance of p38α CKODAT mice and WT (p > 0.05) or Cre Δ (p > 0.05) mice during any trial. Indeed, p38α CKODAT mice showed a slight trend toward improved performance relative to WT, suggesting that p38 inactivation in dopamine neurons does not disrupt basic motor learning and therefore is not grossly altering dopamine neuron function.
To determine whether conditional knock-out of p38α affected Pavlovian contextual learning, we measured cocaine CPP. p38α CKODAT mice developed robust cocaine CPP, and there was no significant difference between the three genotypes (one-way ANOVA, F(2,25) = 1.385, p = 0.2688) (Fig. 5c). Locomotor activity was recorded and analyzed during the cocaine conditioning sessions, and significant locomotor sensitization was evident in all three genotypes (Fig. 5d), as demonstrated by a main effect of training session (F(1,28) = 52.12, p < 0.0001), no interaction (F(2,28) = 0.1836, p = 0.8333) and no effect of genotype (F(2,28) = 1.314, p = 0.2849). Further, post hoc analyses for all groups showed a statistically significant increase in locomotor activity on the second day of cocaine administration relative to the first (WT: t(28) = 3.867, p < 0.01; Cre Δ: t(28) = 3.863, p < 0.01; p38α CKODAT: t(28) = 4.808, p < 0.001). Because acute sensitization to the locomotor effects of cocaine has been hypothesized to be mediated by induction of glutamatergic LTP in dopaminergic neurons (Borgland et al., 2006), these results further confirm that effects of p38 inactivation are specific to aversion relating to KOR activation and do not appear to reflect general deficits in dopamine neuron function.
Conditional knock-out of p38α from dopamine neurons fails to alter KOR-induced inhibition of dopamine release in vivo
To determine whether p38α MAPK was also required for the inhibition of dopamine release, we used FSCV to measure subsecond changes in the release of dopamine in anesthetized mice (Fig. 6a). U50,488 (5 mg/kg, i.p.) inhibited dopamine release evoked by MFB stimulation. U50,488 reduced evoked dopamine release equivalently in both NAc core (54.5 ± 4.2% inhibition from baseline) and shell (50.7 ± 2.3% inhibition from baseline) of wild-type mice, but did not do so in KOR−/− mice (Fig. 6b; two-way ANOVA, main effect of genotype: F(1,16) = 205.3, p < 0.0001).
Figure 6.

Conditional knock-out of p38α MAPK from dopaminergic neurons fails to alter inhibition of dopamine release by U50,488 recorded in vivo. a, Representative images demonstrating analysis of FSCV data before and after administration of U50,488. Bottom, Heat maps represent current generated by oxidation and reduction at the site of the working electrode during application of the triangle waveform. Red line indicates time of MFB stimulation (6 pulses at 30 Hz, 300 μA), Middle, Current × time plot shows evoked dopamine release and uptake. Orange dashed lines indicate magnitude of peak current, which was recorded following stimulation. Top, Cyclic voltammagram from the time of peak current confirms that the current recorded was induced by oxidation and reduction of dopamine. b, Change from baseline MFB-stimulated peak [DA] after administration of U50,488 in KOR+/+ and KOR−/− mice, as recorded from the NAc core and shell. n = 4–6. ***p < 0.001 (Bonferroni post hoc after two-way ANOVA). c, Diagram illustrating stimulation sites for MFB- and PPTg-evoked dopamine release. d, e, Representative traces showing raw current responses of MFB-stimulated (d) and PPTg-stimulated (e) dopamine recorded in WT, Cre Δ, and p38α CKODAT to 5 mg/kg U50,488 (green). Calibration: 2 nA, 1 s. Red line indicates stimulation. f, Change from baseline MFB-stimulated peak [DA] after administration of U50,488 (marked by arrow) in WT, Cre Δ, and p38α CKODAT mice. g, Change from baseline PPTg-stimulated peak [DA] after administration of U50,488 (marked by arrow) in WT, Cre Δ, and p38α CKODAT mice. Inset, PPTg-stimulated dopamine release before and after administration of U50,488. h, Summary data show that p38α CKODAT fails to alter U50,488-induced inhibition of dopamine relative to WT and Cre Δ littermates (n = 5–16). i, Summary data show that U50,488 does not significantly alter t1/2 in WT, Cre Δ, or p38α CKODAT mice. n = 5–16. n.s. (p > 0.05, Bonferroni post hoc analysis after one-way ANOVA confirmed the lack of statistical significance).
Because there was no trend toward a main effect of NAc subregion on the inhibition of dopamine release (F(1,16) = 0.9631, p = 0.3410) and no interaction (F(1,16) = 0.006200, p = 0.9382), time-resolved data collected from the NAc core and shell were collapsed together for comparison between MFB and PPTg stimulation. MFB stimulation allows for direct stimulation of the fibers projecting to the NAc and minimizes the potential impact of somatodendritic KOR activation, whereas PPTg stimulation synaptically activates VTA neurons (Zweifel et al., 2009) and may reveal somatodendritic effects of KOR activation on dopamine release in the NAc (Ford et al., 2006, 2007; Margolis et al., 2006; Ehrich et al., 2014). Positioning of the stimulating electrodes as shown (Fig. 6c) resulted in robust dopamine signal following either MFB or PPTg stimulation (Fig. 6d,e, respectively, inset). Release evoked by either MFB or PPTg stimulation was significantly inhibited by U50,488 (5 mg/kg, i.p.); however, conditional knock-out of p38α from dopamine neurons did not affect U50,488-induced inhibition of dopamine release following either MFB (Fig. 6d; unpaired t test, t(14) = 0.3018, p = 0.7673) or PPTg (Fig. 6e; unpaired t test, t(18) = 1.649, p = 0.1166) stimulation. Given the role of p38 MAPK in aversion and the prediction that the inhibition of dopamine release should be intrinsically aversive (Wise and Bozarth, 1982; Shippenberg et al., 1993; Di Chiara and Imperato, 2008; Chefer et al., 2013; Tejeda et al., 2013), the lack of effect of p38α deletion on evoked dopamine release was a surprise. This suggested that the aversive effects of KOR activation were not caused by either presynaptic or somatic inhibition of dopamine release within the NAc and is consistent with our observations that KOR-mediated aversion through serotonergic neurons is independent from inhibition of dopamine release.
Pharmacological inhibition of p38 MAPK by SB203580 fails to alter KOR-induced inhibition of dopamine release in vitro
To validate the results of the FSCV experiments performed in vivo in transgenic mice, we applied the selective p38 inhibitor SB203580 in an in vitro slice FSCV assay. Similar to results obtained in vivo, electrically evoked dopamine release detected in NAc slices was significantly reduced by the KOR agonist U69,593 (1 μm), and the inhibition was blocked by pretreatment with norBNI (10 μm) (Fig. 7a,b). Bath application of 10 μm SB203580 failed to alter basal dopamine release (two-way repeated-measures ANOVA showed no main effect of SB203580: F(1,14) = 1.297; p = 0.7794). SB203580 also failed to alter either inhibition of dopamine release by U69,593 or the reversal of inhibition by norBNI (main effect of KOR drug: F(2,14) = 51.95; p < 0.0001; no interaction: F(2,14) = 1.067; p = 0.3576). Despite the lack of a significant interaction, a post hoc was performed to confirm that SB203580 had no effect on U69,593-induced inhibition of dopamine release (t(14) = 0.7348, p > 0.05). To determine whether SB203580 had any effect on the velocity of the inhibition of dopamine release, we analyzed the slope of the linear phase during U69,593 onset and found no difference between the two (data not shown: t test, t(14) = 0.1010, p = 0.9210; vehicle = −6.0 ± 2.1, SB203580 = −6.2 ± 1.5).
Figure 7.
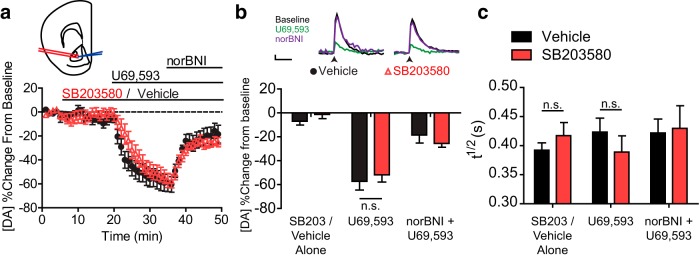
Pharmacological inhibition of p38 MAPK by SB203580 fails to alter KOR-mediated inhibition of dopamine release recorded in the nucleus accumbens of a mid-brain slice. a, Change from baseline locally stimulated peak [DA] during baseline and following administration of 10 μm SB203580 or aCSF vehicle, 1 μm U69,593 and 1 μm norBNI. Each drug was bath-applied during the interval signified by the bar. Inset, Diagram represents placement of bipolar stimulating electrode (red) and recording electrode (blue) during slice FSCV experiments (modified from Paxinos and Franklin, 2008). b, Summary data show that the p38 inhibitor SB203580 (10 μm) did not significantly affect U69,593-induced inhibition of dopamine release (n = 8 slices from 7 animals) (n.s., p > 0.05, Bonferroni post hoc after two-way repeated-measures ANOVA), despite lack of interaction. Inset, Locally stimulated dopamine release during administration of SB203580 or vehicle, during baseline (aCSF), U69,593, or U69,594 with norBNI. Calibration: 1 s, 2 nA. Arrow indicates stimulation. c, Clearance rates (T1/2) of evoked dopamine from peak during administration of 10 μm SB203580 or aCSF vehicle, 1 μm U69,593, and 1 μm norBNI (n = 8 slices from 7 animals) (n.s., p > 0.05, Bonferroni post hoc analysis after two-way repeated-measures ANOVA confirmed lack of statistical difference).
Because both KOR activation and direct p38 MAPK activation by anisomycin have been shown to alter the rate of dopamine uptake (Thompson et al., 2000; Zhu et al., 2005), we analyzed the half-life (t1/2) of released dopamine to determine whether there was any effect of either p38 MAPK inhibition or KOR activation in this assay (Fig. 7c). No effect of U69,593 was found on t1/2 of released dopamine (no main effect of KOR drug: F(2,14) = 1.018, p = 0.3745), which was also unaltered by SB203580 (main effect: F(2,14) = 0.0003, p = 0.9871). Despite the lack of a significant interaction, two post hoc analyses were performed to confirm that SB203580 had no effect on dopamine uptake either with or without the presence of U69,593 (SB203580 or vehicle alone: t(14) = 0.6755, p > 0.05; U69,593: t(14) = 0.9288, p > 0.05). These results indicate that the KOR-induced inhibition of dopamine release in the NAc did not require p38 activation either in vivo or in vitro.
In vivo recording of VTA neuron firing rates in freely moving mice
The lack of effect on evoked dopamine release suggested that KOR activation of p38α MAPK produced aversion by a different effect on VTA neuron excitability. Prior in vitro electrophysiological studies have demonstrated that acute KOR activation inhibits both presynaptic excitatory and inhibitory synaptic inputs to VTA dopamine neurons, as well postsynaptically reducing firing rates by activating outward potassium currents (Margolis et al., 2003, 2006; Ford et al., 2006, 2007). How VTA neuronal excitability is affected by repeated KOR activation at the doses and schedules used for conditioned place aversion is not clear from the in vitro analysis. To determine how the dose of U50,488 that promotes conditioned aversion in wild-type mice alters the firing of dopamine neurons in a p38-dependent manner, we recorded neural activity in the VTA of wild-type and p38α CKODAT in unanesthetized, freely behaving mice. Drivable tetrodes were implanted daily above the VTA (Fig. 8A) and lowered until putative dopamine cells were identified that displayed an inhibitory response to the dopamine D2 receptor agonist quinpirole, as previously described (Zweifel et al., 2009, 2011). Action potential waveforms of putative dopamine neurons and basic firing properties of these cells did not differ between control and p38α CKODAT mice (Fig. 8B–E).
Figure 8.

In vivo recording of VTA dopamine neurons in wild-type littermate and p38α CKODAT mice shows no significant differences in baseline firing properties. A, Brain atlas diagram illustrating placement of tetrodes for control (black) and p38α CKODAT (CKO) (red) mice. B, Average waveform of neurons responsive to quinpirole in control (black, N = 14) and CKO (red, N = 8) mice. C, Baseline firing rate of quinpirole-sensitive neurons. D, Baseline burst rate of quinpirole-sensitive neurons. E, Percentage spikes per burst of quinpirole-sensitive neurons.
On day 1, wild-type C57BL/6 male mice (control) and p38α CKODAT mice (CKO) were injected with saline following a 10 min baseline recording period. VTA neurons did not significantly change their firing rates (Fig. 9A,B). On the next 3 consecutive days, mice were injected with U50,488 (5 mg/kg, i.p., U50-1, U50-2, U50-3) following a 10 min baseline recording period to mimic place aversion conditioning. We were surprised to see that U50,488 injection did not significantly affect the average firing rate of putative dopamine neurons on the first day of injection in either control or CKO mice (Fig. 9A–C). On day 1, some cells increased and some decreased their firing rates (Fig. 9D), which resulted in no effect overall. However, on day 2 of U50,488, we observed a significant decrease in firing of neurons in both control and p38α CKODAT mice (Fig. 9A–C; repeated-measures ANOVA effect of day F(3,253) = 4.31; p < 0.01). Enhanced inhibitory responses to U50,488 were a temporary adaptation in control mice (day 2 t(35) = 2.53, p < 0.05), returning to day 1 levels of inhibition on the third day of U50,488 (Fig. 9A,C). In contrast to control mice, p38α CKODAT mice demonstrated inhibition on day 2 (t(35) = 2.04, p < 0.05) and day 3 (t(41) = 2.03, p < 0.05). p38α CKODAT mice maintained an increased inhibitory response on day 3, suggesting that p38α MAPK activation was responsible for the adaptation (Fig. 9B,C). To confirm the increased inhibitory response of neurons to a subsequent dose of U50,488 was dependent on KOR activation, we repeated our recordings in a separate cohort of control mice but pretreated with the KOR antagonist norBNI (10 mg/kg, i.p.) 50 min before U50,488 administration on the first day. Pretreatment with norBNI prevented the enhanced inhibitory response to U50,488 on the second day of administration, confirming KOR dependence (Fig. 9C).
Figure 9.
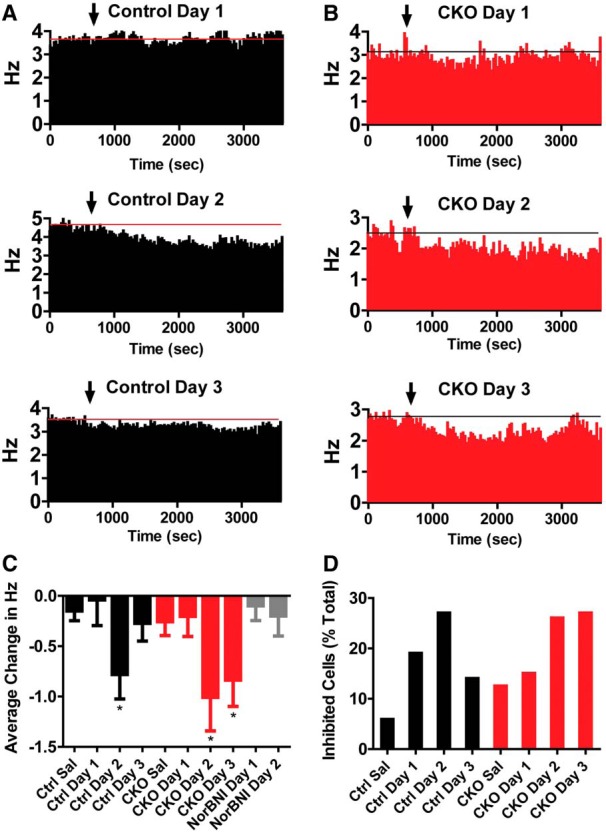
A, B, Change in firing rate following injection of saline or U50 on consecutive days for control (A) and CKO (B) mice (control saline, N = 31; U50 d 1, N = 21; U50 d 2, N = 26; U50 d 3, N = 35; p38αCKODAT saline, N = 33; U50 d 1, N = 40; U50 d 2, N = 35; U50 d 3, N = 41). Data are presented as the average change in firing rate (Hz) from a 10 min baseline period. Data are binned into 10 min blocks. C, Average change in firing rate during the last 40 min following injection of saline or U50,488 across groups. *p < 0.05, relative to corresponding saline (Bonferroni post-test). D, Change in firing rate following injection of U50 on successive days. Mice were pretreated with an injection of norBNI 50 min before U50 on day 1 (U50 d 1, N = 22; U50 d 2, N = 24).
p38 MAPK activation stimulates GIRK phosphorylation in VTA dopamine neurons
The change in excitability noted in the quinpirole-sensitive VTA neurons is consistent with an effect of p38α MAPK activation that we previously observed in DRN neurons (Lemos et al., 2012). KOR activation increases G-protein-gated inwardly rectifying potassium channel conductance in both DRN and VTA neurons (Margolis et al., 2003; Ford et al., 2006; Lemos et al., 2012), and this likely results in the reduced firing rates of the VTA neurons. KOR activation of p38 MAPK results in phosphorylation of these channels, which reduces their conductance (through an enhanced rate of deactivation) (Ippolito et al., 2002). Preventing this channel phosphorylation by p38α CKO would result in the pattern of sustained inhibition evident in the CKO mice on day 3. To test the hypothesis that KOR activation increases GIRK-phosphorylation in VTA neurons, we used a phospho-selective antibody previously shown to detect phospho-tyrosine 12 in Kir 3.1 (Ippolito et al., 2005). Treatment with the KOR agonist U50,488 was shown to increase pY12-Kir 3.1/GIRKp-ir in the dorsal raphe nucleus in a p38α-dependent manner (Lemos et al., 2012).
In the current study, we examined GIRKp-ir expression in ROSA26YFP-reporter labeled dopaminergic neurons of the VTA. In DAT-cre mice, ROSA26YFP expression was restricted to dopaminergic neurons of the VTA (Fig. 10A). Basal GIRKp-ir was low, but substantially increased following repeated U50,488 treatment (20 mg/kg i.p., once daily 3 d; Fig. 10A). Basal GIRKp-ir was also low in VTA sections from p38α CKODAT mice pretreated with saline; however, repeated U50,488 treatment increased GIRKp-ir only in nondopaminergic neurons (Fig. 10A). The number of dopaminergic neurons coexpressing GIRKp-ir was quantified in a blinded manner (Fig. 10B). The percentage of GIRKp-positive YFP-positive cells increased in control mice expressing wild-type levels of p38α (U50(3d) treated: 63 ± 1.5%, saline vehicle (3d): 35 ± 7%). The U50,488-induced increase in colocalization was not evident in p38α CKODAT conditional knock-out mice lacking p38α in dopaminergic neurons (U50(3d): 39 ± 4%, Sal(3d): 32 ± 3%, two-way ANOVA, F(1,40) = 4.91, main effect of treatment p = 0.0008; genotype p = 0.0074; interaction p = 0.0325, Bonferroni post hoc significant (t(40) = 4.521, p < 0.001) for control saline vs control U50, n = 3 or 4 slices per animal, 2 or 3 animals per group; Figure 10A,B). The increase in GIRK channel phosphorylation by KOR activated p38 is consistent with the suppression of U50,488 inhibitory effects on dopaminergic neurons firing observed on day 3, but how that results in CPA is not yet clear.
Figure 10.

U50,488 increases VTA dopamine neuron GIRKp-ir in a p38α MAPK-dependent manner. A, Representative merged fluorescent images (40×) illustrating the increased amount of GIRKp-ir (green fluorescence) in YFP-reporter-labeled dopamine neurons (red fluorescence) of the VTA following 3 d of U50,488 treatment, giving rise to yellow signal when colocalized. Scale bar, 50 μm. B, Quantification of the percentage of YFP-ir putative dopamine neurons of the VTA also positive for GIRKp-ir. In control mice that expressed wild-type levels of p38α, U50,488 treatment significantly increased the percentage (±SEM) of colabeled cells relative to saline vehicle, but this effect of U50 was absent in p38αCKODAT mice lacking p38α in the YFP-ir cells. ***p < 0.05, interaction (two-way ANOVA). N = 3 or 4 slices per animal, 2 or 3 animals per group. n.s., Not significant.
Discussion
The principal findings of this study are that KOR activation of p38 MAPK within the VTA is required for the aversive properties of KOR activation. Second, the aversive properties of KOR activation are rescued in KOR−/− mice by viral restoration of KOR signaling to VTA dopamine neurons. This restoration is dependent on the phosphorylation of KOR at serine 369, which is required for p38 activation but not for activation of ERK (Land et al., 2008). Third, the aversive properties of KOR activation are dependent on the expression of p38α in dopaminergic neurons, but the ability of KOR agonists to inhibit dopamine release is not dependent on p38 MAPK activation within dopaminergic neurons. Fourth, KOR activation has complex, time-dependent effects on VTA neuron firing rates, which likely involves p38 MAPK-induced GIRK phosphorylation and regulation of excitability. Together, these findings suggest that activation of p38 MAPK by KOR in dopaminergic neurons is required to elicit the aversive properties of KOR agonists, and this process occurs independently of KOR's ability to inhibit dopamine release.
KOR agonists induce dysphoria in humans (Pfeiffer et al., 1986), aversion in animal models (Shippenberg and Herz, 1986), and inhibition of dopamine signaling in the NAc in animal models (Di Chiara and Imperato, 1988; Spanagel et al., 1990). As such, it has been proposed that these effects are causally related: the aversive properties of KOR agonists are mediated by inhibition of NAc dopamine signaling (Di Chiara and Imperato, 1988; Spanagel et al., 1990; Tejeda et al., 2012; Carroll and Carlezon, 2013). However, this hypothesis and the data that support it are based on correlation, and dopamine receptor antagonists are not consistently aversive. The hypothesis was driven by the then-prevalent theory that NAc dopamine signaling mediated hedonic responses (Wise and Bozarth, 1982), whereas current theories focus more on the role of dopamine signaling in effort (Salamone et al., 2003), reward prediction error (Schultz, 1998), reinforcement, or incentive salience (Montague et al., 2004). The precise role played by reduction of dopamine in these models is varied and not entirely clear, as the experiments performed have focused on pharmacological or behavioral responses, which increase NAc dopamine or directly affect dopamine signaling at or downstream of the receptor. We confirm that KOR activation does inhibit dopamine release in the nucleus accumbens; however, KOR activation has other actions in the VTA-reward circuit and aversion seems unlikely to be a consequence of a simple reduction in dopamine transmission.
Reinterpretation of the KOR/dynorphin aversive circuit
Previous experiments have focused on the role of a single monoaminergic circuit as mediating the aversive properties of KOR activation, predominately via effects on dopamine signaling (Shippenberg et al., 1993) or via effects on serotonergic signaling (Land et al., 2008; Bruchas et al., 2011). Other experiments have focused on the ability of KOR agonists to alter glutamatergic or GABAergic input onto dopamine neurons (Margolis et al., 2005; Polter et al., 2014). In wild-type KOR+/+ animals, p38 MAPK activation is necessary in both classes of neurons; further, KOR activation is necessary in the NAc and DRN (Land et al., 2008; Bruchas et al., 2011), the VTA (present results), and the mPFC (Tejeda et al., 2013). This implies the actions of KOR at multiple sites in the neuronal circuit, requiring either simultaneous or feedforward activation of KORs and subsequently of p38 MAPK in an extended circuit to cause aversion. The current results suggest that KOR-mediated aversion is encoded in a complex chain of neuronal processing events involving multiple brain regions and that it can be disrupted by breaking any one of the links. Alternately, KOR activation in different circuits could mediate different modes of aversion, operationally mediated by disparate mood states, such as anhedonia, depression, dysphoria, or anxiety. The CPA model used in this and other studies may be incapable of parsing between these different states. Future experiments could focus on trying to distinguish between these different states to determine whether each is disparately affected by manipulation of KOR and p38 MAPK activation in these circuits.
Potential downstream effects of p38 MAPK in dopaminergic neurons
Although the results presented in this study strongly implicate p38α MAPK-mediated signaling in the VTA dopaminergic neurons, this kinase has a broad range of potential substrates (Cuadrado and Nebreda, 2010). In serotonergic neurons, KOR activation of p38α MAPK caused translocation of the serotonin transporter (SERT, SLC6A4) from an intracellular, endosomal compartment to the cell surface to increase the rate of serotonin reuptake (Bruchas et al., 2011; Schindler et al., 2012). KOR activation of p38α MAPK could potentially have affected the DAT (SLC6A3) because in vivo administration of the κ agonist U69,593 increased dopamine uptake (Thompson et al., 2000); however, p38 MAPK did not affect dopamine uptake in DAT-expressing COS cells (Lin et al., 2003), and we did not observe an effect of p38 activation on dopamine uptake in the present study.
In contrast, repeated KOR activation produced p38-dependent effects on VTA dopamine neuron firing rates in freely moving, unanesthetized mice in the present study. Prior analyses using whole-cell voltage-clamp methods demonstrated that KOR regulates both presynaptic and postsynaptic currents (Margolis et al., 2003, 2005, 2006; Ford et al., 2006, 2007). Consistent with the in vitro findings, we observed a complex response following acute U50,488 administration on day 1, with some cells increasing and some cells decreasing their firing rates. The net effect on excitability would be expected to depend on the balance between changes in excitatory, inhibitory, and postsynaptic effects of KOR activation. This balance shifted on day 2, and the p38α MAPK-sensitive effect was only evident after repeated KOR stimulation, which is consistent with prior demonstration that p38 activation requires sustained KOR activation and arrestin recruitment (Bruchas et al., 2006, 2007a, 2011). Although this is somewhat controversial, KOR activation may acutely increase the G-protein-gated inwardly rectifying potassium channel in VTA dopamine cells (Margolis et al., 2003; Ford et al., 2006), as it does in other neurons. Activation of GIRK would hyperpolarize the membrane and might contribute to the inhibition of neuronal firing observed in these freely moving mice. GIRK is a hetrotetrameric potassium channel containing a pair of highly conserved tyrosine residues in the amino termini of its Kir3.1 and Kir3.4 components (Rogalski et al., 2000). Tyrosine phosphorylation of these residues inactivates the channel by increasing the rate of Gβγ dissociation, as measured in transfected cells (Ippolito et al., 2002, 2005). Kappa receptor activation of p38 MAPK stimulates GIRK phosphorylation by activating an src-like kinase in spinal cord (Rogalski et al., 2000; Clayton et al., 2009), and GIRK currents are similarly inhibited by stress-induced KOR activation of p38 MAPK in DRN neurons (Lemos et al., 2012). The present study suggests that stress-induced aversion results from sustained dynorphin activation of KOR, causing arrestin-dependent p38α MAPK phosphorylation of Y12-Kir3.1. The change in postsynaptic effects would alter the balance of excitatory and inhibitory responses, but exactly how this results in the behavioral response is not yet clear. What is clear is that p38α MAPK signaling downstream of KOR activation impacts the phosphorylation status of at least one protein, which is likely just a sample of many post-translational modifications that are altered by activation of this kinase. Given the broad substrate range of p38α MAPK, it is very likely that the ability of KOR activation to mediate aversive effects is through changes in the phosphorylation status and function of these proteins that operate independently of detectable changes in dopamine release.
These experiments suggest that p38 MAPK in dopamine neurons plays a key role in the aversive properties of KOR. The actions of dynorphin and KOR-induced activation of p38 MAPK have been proposed to mediate the dysphoric component of stress (Land et al., 2008) and play a key role in depressive behaviors (Bruchas et al., 2011). Understanding the cellular mechanisms of these behaviors provides novel insights into the stress response. In addition, results suggesting that the dysphoric effects of KOR agonists require arrestin-dependent, p38 MAPK signaling supports the hypothesis that a G-protein-biased KOR agonist might be an analgesic with low abuse potential (Chavkin, 2011).
Footnotes
This work was supported by National Institute of Health Grants R01DA030074 to C.C., T32DA07278 to J.M.E., J.R.K., and S.S.S., K99DA025182 to M.R.B., K05DA020570 to C.C., and F31DA038932 to J.M.E. We thank Chad Zietz, Allisa Song, Brooke Jarvie, David Blaszka, and Scott Ng-Evans for technical support.
The authors declare no competing financial interests.
References
- Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. doi: 10.1016/j.neuron.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Brady KT, Sinha R. Co-occurring mental and substance use disorders: the neurobiological effects of chronic stress. Am J Psychiatry. 2005;162:1483–1493. doi: 10.1176/appi.ajp.162.8.1483. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci. 2007a;27:11614–11623. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, Li S, Chavkin C. Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J Biol Chem. 2007b;282:29803–29811. doi: 10.1074/jbc.M705540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, Lemos JC, Hagan CE, Neumaier JF, Quintana A, Palmiter RD, Chavkin C. Selective p38α MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron. 2011;71:498–511. doi: 10.1016/j.neuron.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll FI, Carlezon WA., Jr Development of kappa opioid receptor antagonists. J Med Chem. 2013;56:2178–2195. doi: 10.1021/jm301783x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C. The therapeutic potential of kappa-opioids for treatment of pain and addiction. Neuropsychopharmacology. 2011;36:369–370. doi: 10.1038/npp.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, Schattauer SS, Levin JR. Arrestin-mediated activation of p38 MAPK: molecular mechanisms and behavioral consequences. Handb Exp Pharmacol. 2014;219:281–292. doi: 10.1007/978-3-642-41199-1_14. [DOI] [PubMed] [Google Scholar]
- Chefer VI, Bäckman CM, Gigante ED, Shippenberg TS. Kappa opioid receptors on dopaminergic neurons are necessary for kappa-mediated place aversion. Neuropsychopharmacology. 2013;38:2623–2631. doi: 10.1038/npp.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton CC, Xu M, Chavkin C. Tyrosine phosphorylation of Kir3 following kappa-opioid receptor activation of p38 MAPK causes heterologous desensitization. J Biol Chem. 2009;284:31872–31881. doi: 10.1074/jbc.M109.053793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signaling. Biochem J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CT, Patterson TA, Simmons ML, Chavkin C, Milner TA. Kappa opioid receptor-like immunoreactivity in guinea pig brain: ultrastructural localization in presynaptic terminals in hippocampal formation. J Comp Neurol. 1996;370:377–395. doi: 10.1002/(SICI)1096-9861(19960701)370:3<377::AID-CNE8>3.3.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Ehrich E, Turncliff R, Du Y, Leigh-Pemberton R, Fernandez E, Jones R, Fava M. Evaluation of opioid modulation in major depressive disorder. Neuropsychopharmacology. 2015;40:1448–1455. doi: 10.1038/npp.2014.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrich JM, Phillips PE, Chavkin C. Kappa opioid receptor activation potentiates the cocaine-induced increase in evoked dopamine release recorded in vivo in the mouse nucleus accumbens. Neuropsychopharmacology. 2014;39:3036–3048. doi: 10.1038/npp.2014.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Beckstead MJ, Williams JT. Kappa opioid inhibition of somatodendritic dopamine inhibitory postsynaptic currents. J Neurophysiol. 2007;97:883–891. doi: 10.1152/jn.00963.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, Le Maître E, Fabre V, Bernard JF, David Xu ZQ, Hökfelt T. Chemical neuroanatomy of the dorsal raphe nucleus and adjacent structures of the mouse brain. J Comp Neurol. 2010;518:3464–3494. doi: 10.1002/cne.22407. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Hasue RH, Shammah-Lagnado SJ. Origin of the dopaminergic innervation of the central extended amygdala and accumbens shell: a combined retrograde tracing and immunohistochemical study in the rat. J Comp Neurol. 2002;454:15–33. doi: 10.1002/cne.10420. [DOI] [PubMed] [Google Scholar]
- Hough LB, Nalwalk JW, Chen Y, Schuller A, Zhu Y, Zhang J, Menge WM, Leurs R, Timmerman H, Pintar JE. Improgan, a cimetidine analog, induces morphine-like antinociception in opioid receptor-knockout mice. Brain Res. 2000;880:102–108. doi: 10.1016/S0006-8993(00)02776-1. [DOI] [PubMed] [Google Scholar]
- Ippolito DL, Temkin PA, Rogalski SL, Chavkin C. N-terminal tyrosine residues within the potassium channel Kir3 modulate GTPase activity of Galphai. J Biol Chem. 2002;277:32692–32696. doi: 10.1074/jbc.M204407200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito DL, Xu M, Bruchas MR, Wickman K, Chavkin C. Tyrosine phosphorylation of K(ir)31 in spinal cord is induced by acute inflammation, chronic neuropathic pain, and behavioral stress. J Biol Chem. 2005;280:41683–41693. doi: 10.1074/jbc.M507069200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry. 1999;156:837–841. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- Kormos V, Gaszner B. Role of neuropeptides in anxiety, stress, and depression: from animals to humans. Neuropeptides. 2013;47:401–419. doi: 10.1016/j.npep.2013.10.014. [DOI] [PubMed] [Google Scholar]
- Lalanne L, Ayranci G, Kieffer BL, Lutz PE. The kappa opioid receptor: from addiction to depression, and back. Front Psychiatry. 2014;5:170. doi: 10.3389/fpsyt.2014.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JC, Roth CA, Messinger DI, Gill HK, Phillips PE, Chavkin C. Repeated stress dysregulates kappa opioid receptor signaling in the dorsal raphe through a p38α MAPK-dependent mechanism. J Neurosci. 2012;32:12325–12336. doi: 10.1523/JNEUROSCI.2053-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Zhang PW, Zhu X, Melgari JM, Huff R, Spieldoch RL, Uhl GR. Phosphatidylinositol 3-kinase, protein kinase C, and MEK1/2 kinase regulation of dopamine transporters (DAT) require N-terminal DAT phosphoacceptor sites. J Biol Chem. 2003;278:20162–20170. doi: 10.1074/jbc.M209584200. [DOI] [PubMed] [Google Scholar]
- Mantsch JR, Baker DA, Funk D, Lê AD, Shaham Y. Stress-induced reinstatement of drug seeking: 20 years of progress. Neuropsychopharmacology. 2015 doi: 10.1038/npp.2015.142. doi: 10.1038/npp.2015.142. Advance online publication. Retrieved May 15, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Kappa-opioid agonists directly inhibit midbrain dopaminergic neurons. J Neurosci. 2003;23:9981–9986. doi: 10.1523/JNEUROSCI.23-31-09981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Both kappa and mu opioid agonists inhibit glutamatergic input to ventral tegmental area neurons. J Neurophysiol. 2005;93:3086–3093. doi: 10.1152/jn.00855.2004. [DOI] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, Fields HL. Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc Natl Acad Sci U S A. 2006;103:2938–2942. doi: 10.1073/pnas.0511159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ. Kappa-opioid receptors and analgesia. Trends Pharmacol Sci. 1990;11:70–76. doi: 10.1016/0165-6147(90)90321-X. [DOI] [PubMed] [Google Scholar]
- Montague PR, Hyman SE, Cohen JD. Computational roles for dopamine in behavioural control. Nature. 2004;431:760–767. doi: 10.1038/nature03015. [DOI] [PubMed] [Google Scholar]
- Neumann ID, Wegener G, Homberg JR, Cohen H, Slattery DA, Zohar J, Olivier JD, Mathé AA. Animal models of depression and anxiety: what do they tell us about human condition? Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:1357–1375. doi: 10.1016/j.pnpbp.2010.11.028. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB. The mouse brain in stereotaxic coordinates. Ed 3. San Diego: Academic; 2008. [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233:774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- Polter AM, Bishop RA, Briand LA, Graziane NM, Pierce RC, Kauer JA. Poststress block of kappa opioid receptors rescues long-term potentiation of inhibitory synapses and prevents reinstatement of cocaine seeking. Biol Psychiatry. 2014;76:785–793. doi: 10.1016/j.biopsych.2014.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski SL, Appleyard SM, Pattillo A, Terman GW, Chavkin C. TrkB activation by brain-derived neurotrophic factor inhibits the G protein-gated inward rectifier Kir3 by tyrosine phosphorylation of the channel. J Biol Chem. 2000;275:25082–25088. doi: 10.1074/jbc.M000183200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone JD, Correa M, Mingote S, Weber SM. Nucleus accumbens dopamine and the regulation of effort in food-seeking behavior: implications for studies of natural motivation, psychiatry, and drug abuse. J Pharmacol Exp Ther. 2003;305:1–8. doi: 10.1124/jpet.102.035063. [DOI] [PubMed] [Google Scholar]
- Saxon A, Ling W, Mooney L, Hillhouse M, Thomas C, Hasson A, Ang A, Nielsen D. Cocaine use reduction with buprenorphine (CURB) preliminary study findings. Kappa Therapeutics. 2015;2015:39. conference proceedings. [Google Scholar]
- Schindler AG, et al. Stress produces aversion and potentiates cocaine reward by releasing endogenous dynorphins in the ventral striatum to locally stimulate serotonin reuptake. J Neurosci. 2012;32:17582–17596. doi: 10.1523/JNEUROSCI.3220-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnur P, Walker JM. Effects of U50,488H on locomotor activity in the hamster. Pharmacol Biochem Behav. 1990;36:813–816. doi: 10.1016/0091-3057(90)90082-S. [DOI] [PubMed] [Google Scholar]
- Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- Scott MM, Wylie CJ, Lerch JK, Murphy R, Lobur K, Herlitze S, Jiang W, Conlon RA, Strowbridge BW, Deneris ES. A genetic approach to access serotonin neurons for in vivo and in vitro studies. Proc Natl Acad Sci U S A. 2005;102:16472–16477. doi: 10.1073/pnas.0504510102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shippenberg TS, Herz A. Differential effects of mu and kappa opioid systems on motivational processes. NIDA Res Monogr. 1986;75:563–566. [PubMed] [Google Scholar]
- Shippenberg TS, Bals-Kubik R, Herz A. Examination of the neurochemical substrates mediating the motivational effects of opioids: role of the mesolimbic dopamine system and D-1 vs D-2 dopamine receptors. J Pharmacol Exp Ther. 1993;265:53–59. [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS. The effects of opioid peptides on dopamine release in the nucleus accumbens: an in vivo microdialysis study. J Neurochem. 1990;55:1734–1740. doi: 10.1111/j.1471-4159.1990.tb04963.x. [DOI] [PubMed] [Google Scholar]
- Stratford TR, Wirtshafter D. Ascending dopaminergic projections from the dorsal raphe nucleus in the rat. Brain Res. 1990;511:173–176. doi: 10.1016/0006-8993(90)90239-8. [DOI] [PubMed] [Google Scholar]
- Tejeda HA, Shippenberg TS, Henriksson R. The dynorphin/κ-opioid receptor system and its role in psychiatric disorders. Cell Mol Life Sci. 2012;69:857–896. doi: 10.1007/s00018-011-0844-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda HA, Hanks AN, Scott L, Mejias-Aponte C, Hughes ZA, O'Donnell P. Prefrontal cortical kappa-opioid receptor modulation of local neurotransmission and conditioned place aversion. Neuropsychopharmacology. 2013;38:1770–1779. doi: 10.1038/npp.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AC, Zapata A, Justice JB, Jr, Vaughan RA, Sharpe LG, Shippenberg TS. Kappa opioid receptor activation modifies dopamine uptake in the nucleus accumbens and opposes the effects of cocaine. J Neurosci. 2000;20:9333–9340. doi: 10.1523/JNEUROSCI.20-24-09333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van't Veer A, Carlezon WA., Jr Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology (Berl) 2013;229:435–452. doi: 10.1007/s00213-013-3195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vortherms TA, Roth BL. Salvinorin A: from natural product to human therapeutics. Mol Interv. 2006;6:257–265. doi: 10.1124/mi.6.5.7. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. Action of drugs of abuse on brain reward systems: an update with specific attention to opiates. Pharmacol Biochem Behav. 1982;17:239–243. doi: 10.1016/0091-3057(82)90076-4. [DOI] [PubMed] [Google Scholar]
- Zhou QY, Palmiter RD. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell. 1995;83:1197–1209. doi: 10.1016/0092-8674(95)90145-0. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Masson J, Gingrich JA, Rayport S Hen R. Targeted gene expression in dopamine and serotonin neurons of the mouse brain. J Neurosci Methods. 2005;143:27–32. doi: 10.1016/j.jneumeth.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Zhu CB, Carneiro AM, Dostmann WR, Hewlett WA, Blakely RD. p38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2A-dependent process. J Biol Chem. 2005;280:15649–15658. doi: 10.1074/jbc.M410858200. [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, Darvas M, Kim MJ, Mizumori SJ, Paladini CA, Phillips PE, Palmiter RD. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci U S A. 2009;106:7281–7288. doi: 10.1073/pnas.0813415106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel LS, Fadok JP, Argilli E, Garelick MG, Jones GL, Dickerson TM, Allen JM, Mizumori SJ, Bonci A, Palmiter RD. Activation of dopamine neurons is critical for aversive conditioning and prevention of generalized anxiety. Nat Neurosci. 2011;14:620–626. doi: 10.1038/nn.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]