

Abstract
Bacteriocins are toxins produced by bacteria to kill competitors of the same species. Theory and laboratory experiments suggest that bacteriocin production and immunity play a key role in the competitive dynamics of bacterial strains. The extent to which this is the case in natural populations, especially human pathogens, remains to be tested. We examined the role of bacteriocins in competition using Pseudomonas aeruginosa strains infecting lungs of humans with cystic fibrosis (CF). We assessed the ability of different strains to kill each other using phenotypic assays, and sequenced their genomes to determine what bacteriocins (pyocins) they carry. We found that (i) isolates from later infection stages inhibited earlier infecting strains less, but were more inhibited by pyocins produced by earlier infecting strains and carried fewer pyocin types; (ii) this difference between early and late infections appears to be caused by a difference in pyocin diversity between competing genotypes and not by loss of pyocin genes within a lineage over time; (iii) pyocin inhibition does not explain why certain strains outcompete others within lung infections; (iv) strains frequently carry the pyocin-killing gene, but not the immunity gene, suggesting resistance occurs via other unknown mechanisms. Our results show that, in contrast to patterns observed in experimental studies, pyocin production does not appear to have a major influence on strain competition during CF lung infections.
Keywords: bacteriocins, pyocins, competition, cystic fibrosis, Pseudomonas aeruginosa
1. Introduction
Bacteria produce antimicrobial toxins, termed bacteriocins, to kill competitors [1]. A number of experimental studies have shown that bacteriocins can play a key role in competitive dynamics among closely related bacterial strains [2,3]. Bacteriocin ‘producers’ can invade and outcompete ‘sensitive’ strains that are killed by that toxin. These producers can, in turn, be outcompeted by ‘resistant’ strains that are immune to the bacteriocin, but do not produce it, and hence do not pay the metabolic cost of producing it. While the ‘resistant’ strains can be outcompeted by ‘sensitive’ strains because sensitive strains do not invest in the metabolic production of expressing the immunity protein that neutralizes the toxin. In spatially structured populations this can lead to population dynamic cycling with all three strains coexisting, analogous to the rock–paper–scissors (RPS) game, because each strain can be invaded by another strain—producers outcompete sensitive, resistant outcompete producers and sensitive outcompete resistant [1,4,5].
The extent to which bacteriocins determine the dynamics of competing Pseudomonas aeruginosa strains in nature, or lead to RPS dynamics, is not known. In addition, previous work has focused on Escherichia coli, which carry bacteriocins with relatively simple toxin–antitoxin systems, where one protein is responsible for the killing action (toxin) and another immunity protein for neutralizing the killing protein [4,6]. However, in natural populations of bacteria like P. aeruginosa one strain usually carries and releases multiple types of bacteriocins known as pyocins, which have a diversity of killing and resistance mechanisms in addition to the toxin–antitoxin systems [2,7]. For example, three pyocin types exist (S, R and F), with each of these having multiple subtypes with varying toxicity levels [2,7–10]. The soluble S-type is similar to colicins of E. coli and is composed of killing and immunity genes. While the R and F type are phage-derived and have different killing and resistance mechanisms, where both consist of only a killing component with no specified immunity that neutralizes the toxin but instead resist via alteration or absence of cell-surface receptors [2,9–15].
We examine bacteriocin-mediated population dynamics in natural populations using cystic fibrosis (CF) lung isolates of P. aeruginosa, an opportunistic human pathogen. Early colonization of the lung is characterized by a diversity of strains that intermittently co-infect from environmental sources or other patients, while later stages are characterized by chronic infections usually dominated by one strain [13,16–19]. Our first aim was to test whether the competitive success of different strains, within hosts, can be explained by variation in pyocin production and resistance. In particular, whether the production of bacteriocins has allowed certain strains to eliminate competitors and become dominant. We examined the extent to which different strains where able to produce pyocins that inhibited the growth of other strains, comparing: (i) isolates sampled from non-lung environments and 13 CF Danish patients at different stages of infection and (ii) isolates sampled from a longitudinal study following the infection dynamics of strains within eight CF Danish patients. We sequenced the genomes of all these strains to identify the pyocin genes, examine how pyocin diversity changes during lung infections and examine the genetic basis of the inhibition patterns that we observed phenotypically. We tested the generality of our results by comparing our sequence data with that from previously sequenced epidemic strains, and from 55 isolates of a transmissible Danish isolate that has been sampled for over 38 years [20].
Our second aim was to test whether RPS dynamics occur in natural P. aeruginosa populations. We used all the isolates to assess the frequency of occurrence of the different types of producer, resistant and sensitive strains. Theory predicts that structured populations allow all these types to be maintained at appreciable frequencies, whereas sensitives will dominate in unstructured (panmictic) populations [4,21–23]. We do not know the extent to which human hosts represent relatively structured or unstructured populations. We focus on S pyocins because they are composed of a killing and immunity gene, and in particular, the five S pyocin types for which both killing and immunity genes are known. Our third aim was to assess how often the S pyocin immunity gene was responsible for cell resistance to the killing gene. While it has been observed that some strains appear to not be killed by S pyocins, despite lacking the immunity gene for that pyocin, the relative occurrence and importance of this in nature remains unknown [24].
2. Material and methods
We obtained three collections of lung isolates from the Department of Clinical Microbiology, Rigshospitalet, Copenhagen, where they had been isolated from CF patients treated at the Copenhagen CF clinic (collections 1, 2 and 3 below).
(a). Pseudomonas aeruginosa isolates
Collection 1 consisted of 41 isolates, which we grouped into categories according to their source of origin: non-lung, or from infected lungs and the stage of lung infection. Specifically, we divided these isolates into non-lung strains from the natural environment or from patient burn wounds (laboratory PAO1), acute isolates that cause a single intermittent colonization in a child, chronic isolates that cause a single one-off chronic infection for less than six months in an adult, and multiply chronic isolates that are highly transmissible and cause multiple chronic infections for longer than six months in adults (electronic supplementary material, table A1). We sequenced all isolates (see the electronic supplementary material) and based on the bacterial genome multilocus sequence typing (MLST) profiles the 41 isolates belong to 29 distinct independent genotypes. We used collection 1 to determine whether the ability to produce, inhibit and resist pyocins, measured both phenotypically and genotypically, correlates with how long a strain has been in the lung.
Collection 2 consisted of the genome sequences of 55 P. aeruginosa clinical CF lung isolates [20]. The 55 isolates were of the same genotype and belonged to the DK2 clone type, a transmissible Danish lineage of P. aeruginosa and were sampled from 21 Danish patients with multiply chronic CF infections over 38 years between 1972 and 2008 (electronic supplementary material, table A2; [20]). We used these genome sequences to screen for the bacteriocin genes and analyse whether they are lost or mutated over time during infections.
Collection 3 consisted of longitudinal isolates that had been sampled from the onset of infection and over 6–10 years from eight different patients, three of which suffer from chronic infections and five from acute infections as defined by Marvig et al. [18] (electronic supplementary material, table A3). The isolates had already been sequenced at the Department of Clinical Microbiology, Rigshospitalet, Copenhagen [18]. Altogether there were 122 isolates that, based on genome MLST profiles, belonged to 17 different genotypes. We used this collection to track changes in bacteriocin production over time in isolates of the same genotype and to track competitive dynamics of co-infecting genotypes within the same lung environment.
Collection 4 consisted of P. aeruginosa isolates, which had been previously sequenced and we found in GenBank. These isolates include CF lung isolates (the Liverpool strain LESB58, an isolate from a chronically infected patient in Boston PA2192, the Manchester epidemic strain C3719 and PACS2; accession numbers: NC_011770, NZ_CH482384, NZ_CH482383, NZ_AAQW01000001, respectively) and non-lung isolates (PA14: from a burn wound patient and PA7: a human clinical non-respiratory isolate from Argentina; accession numbers: NC_008463 and NC_009656, respectively). We used these genome sequences to compare the pyocin gene profiles of other epidemic strains to the Danish strains and non-lung strains of our collection.
(b). Materials
For all the experiments we used King's broth (KB) media and agar to culture bacteria (20 g peptone, 1.5 g K2HPO4·3H2O and 1.5 g MgSO4·7H2O for the media and an additional 12 g agar powder for KB agar, per litre of distilled water (dH2O)). We used M9 minimal salts solution to dilute cell cultures (6.8 g Na2HPO4, 3 g KH2PO4, 0.5 g NaCl and 10 g NH4Cl per litre of dH2O). We used the Magellan plate reader v. 7.0 and the fluorimeter SpectraMax M2, Molecular Devices, to take endpoint OD reads for constructing optical density growth curves. The camera box (Syngene Ltd, UK) G:box EF2 was used for all pictures of bacterial agar plates.
(c). Experimental protocols
(i). Bacterial growth curves
We grew each isolate for 24 h at 37°C at 200 r.p.m. in 6 ml of KB media inoculated from a frozen stock in 20 ml glass vials. We measured optical density (OD) at 600 nm (A600), and standardized all cultures to an OD of 1.0 using M9 solution to dilute. We used 1 µl of standardized culture to inoculate 200 µl of KB media in a 96-well plate. To construct growth curves we set the plate to incubate at 37°C for 44 h with automatic periodic shaking every half hour for 15 s prior to OD measurements totalling 89 OD reads. We then plotted the growth curves to determine when each isolate reached stationary phase.
(d). Phenotypic characterization of pyocin production/resistance
(i). Preparation of supernatant
We cultured all isolates for 24 h at 37°C at 200 r.p.m. in 6 ml of KB media inoculated from a frozen stock in 20 ml glass vials. We measured cell density at A600 and standardized all cultures to an OD of 1.0. We then inoculated 30 µl of each standardized culture into 6 ml of fresh KB media in 20 ml glass vials and allowed them to grow at 37°C at 200 r.p.m. until they reach stationary phase. We then centrifuged the cultures for 10 min at 5000g to obtain a clear supernatant, which we filter sterilized (0.2 µm pore size) into sterile tubes and placed in the freezer at −20°C. We spotted a control KB agar plate with 20 µl of each culture's supernatant and incubated them overnight to ensure they were free of bacterial growth. Although inhibition of strains by supernatants could be because of small peptides secreted into the supernatant, the broad killing zones that we observe will primarily be because of pyocins. We also excluded inhibition potentially caused by phage lysis (see the electronic supplementary material).
(ii). Preparation of bacterial lawns
We cultured isolates for 20 or 30 h (for slower growing strains) at 37°C at 200 r.p.m. in 6 ml of KB media inoculated from a frozen stock in 20 ml glass vials. We standardized cultures to OD of 1.0, and diluted them 100-fold. We then spread 50 µl of each diluted culture on the supernatant-spotted KB plates.
(iii). Experimental design—using the agar spot assay
We measured the ability of different strains to inhibit each other by testing whether a strain's pyocin-carrying supernatant inhibits a bacterial culture of another strain. We spotted KB agar plates with the supernatants, and then spread a lawn of each culture (electronic supplementary material, figure A1). We placed a 20 µl spot of each supernatant onto KB agar plates. We allowed the spots to dry into the agar before spreading a lawn of 50 µl of each diluted culture. We then set the plates to incubate at 37°C for 12 h until a uniform lawn had appeared. We replicated the same lawn with each set of spots three times. Each plate consisted of control sterile KB media spots and a spot of self-supernatant for each isolate's lawn.
We used semiquantitative assays to examine the lawns for zones of inhibition, or reduced growth on all supernatant spots for collections 1 and 3. We took pictures of the plates using a camera with contrasting colour images in order to better visualize and detect reduced growth when the inhibition areas were not clear zones (electronic supplementary material, figure A1). As a control, we confirmed that sterile KB media and self-supernatant spots did not cause any inhibition of the lawns. We then recorded which strains inhibited each other and compiled a 41 × 41 matrix organized into four isolate categories of non-lung and three different stages of a CF infection (acute, chronic one-off and multiply chronic) across the patients. We also compiled a separate matrix for within-patient isolate interactions for the eight different patients of collection 3.
(e). Data analysis
(i). Genome sequence analysis—screening for pyocin genes
All isolate genome sequences were assembled de novo using the assembly software Velvet, the contigs from which were uploaded to a database running the BIGSdb platform [25]. We assessed the diversity of isolates using multilocus sequence typing (MLST) where the genomes were screened for alleles of the seven key housekeeping loci, acs, aro, mut, nuo, pps, trp and gua, of P. aeruginosa available at the www.pubmlst.org/paeruginosa website. The MLST concept is extended to the whole genome scale screening for allele variants of a diversity of other loci and we analysed this data using a genome comparator tool to identify distinct genotypes among the isolates [25]. For further data analysis, we grouped isolates with identical genome MLST profiles together as one independent sample to minimize pseudoreplication.
The BIGSdb software includes ‘autogagger’ and ‘autodefiner’ tools that scan deposited whole genome sequences against loci identifying exact matches or allele variants with 98% sequence identity for which new alleles are generated. Isolates' records are updated with specified allele numbers marking the regions on assembled contiguous sequences (contigs) for defined loci. We blasted the genomes for pyocin genes and tagged the resulting allelic variants before proceeding with further analysis.
We screened all isolate genomes for the soluble S pyocin (S1, S2, S3, S4, S5 and AP41) loci and their allele variants for the killing and corresponding immunity gene using a known sequence as a reference available from previously sequenced genomes in NCBI GenBank (electronic supplementary material, table A4). We also screened the genomes for regulatory (prtN and prtR) and lysis genes (hol and lys) as well as the R and F pyocin structural genes (electronic supplementary material, table A4, figure A2). We used pyocin loci R2 and F2 as reference genes to screen for other potential alleles that belong to other R and F pyocin subtypes. The R pyocin is characterized by the presence of the whole suite of genes between the hol-lys and the F pyocin by the presence of the toxin loci between lys and trpG (electronic supplementary material, figure A2). We tagged the genomes of all isolates and annotated each of the loci and its identified alleles (electronic supplementary material, figure A3).
For some of the F2 toxin loci, alleles could not be identified because of high sequence dissimilarity. In such cases, we checked for the presence of open reading frames (ORF) in the expected location of the F-specific region and, if present, we marked them as present but with an unidentified allele. In all isolates the genes for R and F pyocins are found between trpE and trpG in one region lying on one contig, facilitating the search for ORFs in the missing R or F regions.
For our analysis a gene was considered to be present in the isolate when it is present in its fully functional coding sequence. When a sequence is truncated and partly found in the genome because it lies at the edge of a contig it is impossible to determine whether it exists in the genome as a fully functional coding sequence or a mutated non-functional gene. We considered genes lying at the edge of a contig as present and functional for further analysis. Sequences with premature stop codon mutations are considered non-functional. If one of the genes in the cassette composing the R- or F-pyocins is missing in an isolate we consider the pyocin to be non-functional and as absent. However, this was never the case, they were either all present together or none at all.
To ensure that the R- and F-pyocin loci are in fact absent from the genomes and the results are not due to a sequencing error, we checked whether any other DNA sequence lies in the R- and F-specific region of the genome. The R-pyocin genes are in a lysis cassette, between the holin and lys genes, and the F-specific region is between lys and trpG genes. We found that for all isolates not caryring R and F pyocins, there are no ORFs between the holin-lys and lys-trpG loci respectively, confirming the absence of R- and F-pyocin genes from the isolates.
(ii). Statistical analysis of phenotypic inhibition profiles
For the 41 isolates taken from different stages of infection (collection 1), we merged and averaged the data of all isolates with identical MLST genotypic profiles in each isolate category resulting in a total of 29 independent samples. To avoid pseudoreplication, we estimated the number of times isolates of identical genotypes are inhibited or inhibiting and used the averaged value for the analysis (electronic supplementary material, figure A4). We tested for significant differences in inhibition with time spent in the lung (as estimated by type of CF lung infection) using a generalized linear model (GLM) with level of inhibition as a response variable and two explanatory variables: time spent in the lung and type of P. aeruginosa isolate category. We tested for changes in the proportion of inhibition caused by the isolate's supernatant as a function of time isolates spent in the lung and the proportion of lawns inhibited by supernatants as a function of time bacterial lawn isolates spent in the lung. We also specifically tested for: (i) differences in the level of inhibition caused by isolates' supernatants of each category on the isolate lawns within each category and (ii) differences in the level of inhibition in the isolate lawns of each category caused by the isolates' supernatants of each category.
For the longitudinal samples from the eight CF patients (collection 3), we did not merge isolates of the same genotype as one independent sample because the aim of these experiments is to test for significant differences in inhibition with time of infection across isolates infecting the same lung. We used GLM analysis to test for significant differences in inhibition levels with time (in years) each isolate has spent infecting the lung. In the analysis we modelled the proportion of killing caused by isolates' supernatant as a response variable with time of infection as a continuous variable and genotype as a discrete variable. This analysis was done on isolates per patient and isolates across all patients for more general patterns. The data from both isolate collections are proportion data and so we normalized for residuals prior to analysis using an arcsine square-root transformation.
(iii). Analysis of S pyocins
To assess the frequency in which producer, resistant and sensitive S pyocin isolates occur we used the genetic data to assess how often an S pyocin occurred with both its killing and immunity gene, and how often either gene was found alone across all independent genotypes. When the isolates that share the same genome MLST type showed variation in their S pyocin profile, we considered the isolates as independent samples. This resulted in a total of 53 independent samples, with a total of 80 occurrences of S pyocin genes found across all.
To assess whether the S immunity gene is responsible for cell protection from S pyocins we used the pyocin gene profiles in all isolates to predict how often one cell carrying the S pyocin will kill a cell that does not carry that S pyocin (electronic supplementary material, figure A5). Then we determine how often killing is observed from the phenotypic experimental data. We then record how often resistance of the target cell occurs in the presence of the immunity gene. When the same genotype showed variation in its S pyocin profile, we also considered the isolates as independent samples here. We recorded resistance or susceptibility from the experimental competitive interactions when more than 50% of the isolates of each independent genotype are susceptible or resistant. If exactly 50% of the isolates of a genotype show resistance and the other 50% susceptibility, we record that as data that cannot be interpreted. This occurred in 25 of 635 competitive interactions that were therefore excluded from further analysis. In the majority of cases, there was no discrepancy among isolates within a genotype, and so the criteria we use above do not significantly change the results when not applied.
For the above analyses we analysed data of S1, S2, S3, S5 and AP41 pyocins, but we excluded the S4 pyocin because it is still unknown whether there is an immunity gene for it, and if there is, it has not been identified or annotated yet. We used proportion χ2-squared to test for significant differences in the frequency of genes occurring together or alone and to test for significant differences in the frequency of strains that resist via alternative resistance mechanisms and strains that resist using the immunity gene. We implemented all analyses in R statistical software (http://www.R-project.org).
3. Results
(a). Pyocin production and resistance
(i). Comparison across patients sampled at different stages of infection
Considering the 41 isolates taken from different stages of infections (collection 1), we found that strains that were isolated from later stage infections were both significantly less likely to inhibit and more likely to be inhibited by other strains (figure 1; electronic supplementary material, figure A4; inhibiting: F7,108 = 10.63, p = 4.03 × 10−10; inhibited: F7,108 = 11.14, p = 1.53 × 10−10; N = 29 distinct genotypes). The interaction terms between the type of lawn inhibited, and the type of supernatant causing inhibition were not significant. Therefore, excluding the effect of interaction, there is a significant decrease in the supernatants inhibiting (F4,111 = 16.91, p = 7.43 × 10−11) and a significant increase in the lawns being inhibited (F4,111 = 18.9, p = 7.10 × 10−12) with increasing infection time in the lung.
Figure 1.
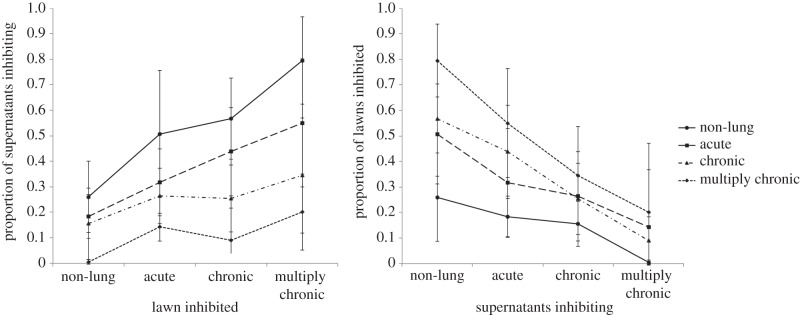
Proportion of supernatants inhibiting and lawns inhibited. Strains that were isolated from later-stage infections were both (a) less likely to inhibit and (b) more likely to be inhibited by other strains. The error bars indicate ±95% CI bars around mean values.
We found that strains had a significantly lower diversity of pyocins as infections progress to chronic stages (figure 2; GLM, F1,27 = 5.003, p = 0.034). Lower pyocin diversity in multiply chronic transmissible strains can be either because of gene loss over infection time within a lineage, or that strain lineage never having had them in the genomes. To determine whether the pyocin genes are lost from the genomes when a strain infects for long periods we examined the 55 isolates taken over 38 years from one of the Danish multiply chronic transmissible genotypes (DK2; collection 2). These isolates do not show loss of or mutation accumulation in the pyocin genes over time. These data indicate that pyocin genes of transmissible isolates are not lost with increasing infection time but were initially never present in the genomes; such that chronic transmissible strains are characterized by a lower pyocin diversity relative to non-chronic isolates.
Figure 2.
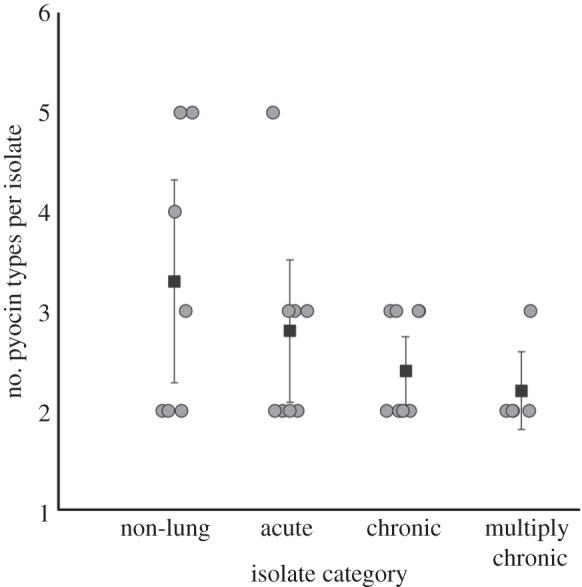
Pyocin diversity across different stages of infection. Isolates from later-stage infections carry fewer pyocin types than isolates from early infections and non-lung environments. Circles represent isolates and squares represent the means of number of pyocin types for each isolate category. The error bars indicate ±95% CI bars around mean values.
To compare our results to previously published strains, particularly that Danish multiply chronic strains carry a reduced diversity of pyocins, we analysed the genomic pyocin data on six P. aeruginosa isolates from GenBank, four chronic or epidemic CF strains from the UK, Boston and an unknown source, and two clinical non-lung isolates (collection 3). We found the same pattern in these isolates that we had found in our Danish isolates. Similar to the multiply chronic isolates of our collection, the CF isolates (except for the Liverpool strain) also show absence of all the R pyocin genes but retain the F genes, while the clinical non-lung isolates carry both the R and F pyocin genes, similar to the non-lung isolate category of our collection (electronic supplementary material, figure A3).
(ii). Comparison over time within patients
We then analysed longitudinal isolates, taken over a 6–10 year period, from eight patients. We found that, within patients, isolates' inhibition levels did not change over time infected (T7 = −0.70, p = 0.505, N = 8)—inhibition increased in two patients, decreased in four, and there was no inhibition in two patients. Examining the genomic data of isolates in patients with multiple infecting genotypes, pyocin diversity does not vary significantly with the likelihood of persisting in the lung (T7.73 = −0.43, p = 0.676, N = 5).
Considering the eight patients, five had two or more genotypes infecting the lung. In two of these patients there was no inhibition between genotypes. In the remaining three patients, at least one of the genotypes was able to inhibit the growth of another genotype in that patient. In two out of these three patients, the inhibiting genotype persisted longer in the infection, consistent with a role of bacteriocin-mediated competition (figure 3). In contrast, in the third patient, the inhibited genotype persisted longer in the infection.
Figure 3.
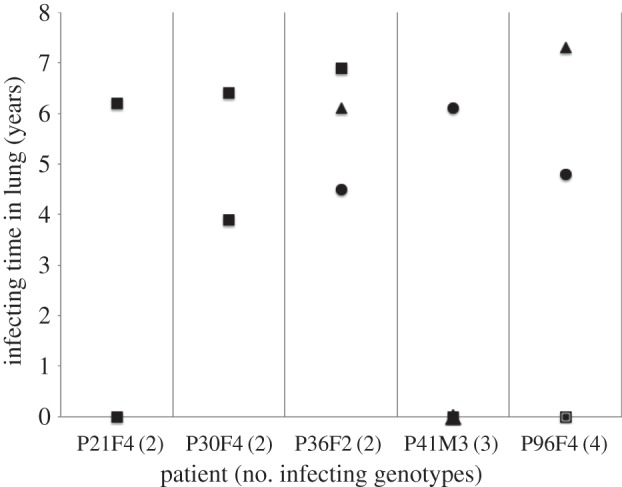
Within patient competitive dynamics. Considering the eight patients where we were able to follow strain dynamics over time, five were infected by multiple strains. These patients contained 14 distinct genotypes, and none of the patients were infected with the same genotype. In only three of these five patients at least one of the strains was able to inhibit another strain in the infection. Squares represent genotypes that are not inhibited nor cause any inhibition; triangles are genotypes that inhibit the susceptible circle genotypes. In two out of these three patients (P36F2 and P96F4), the inhibiting genotype (triangle) persisted longer in the infection, consistent with a role of bacteriocin-mediated competition. In the other patient (P41M3), the inhibited genotype (circle) persisted longer in the infection, contrary to the prediction from bacteriocin dynamics.
(b). Linkage between S pyocin killing and immunity
We then examined the extent to which killing and immunity co-occurred for the five S-type pyocins. We focused on S-type pyocins because they are composed of two genes, one responsible for the killing activity and an immunity gene that protects against the killing. Natural populations have been observed to contain strains that carry both, the toxin and immunity genes (toxin producers), just the immunity gene (resistant) or neither (sensitive).
Overall, 98% (53/54) of all independent isolates in all collections contained at least one S-pyocin gene. The average number of pyocin types per isolate was 1.5, giving 80 (53 × 1.5) occurrences of pyocin genes. Amongst those, 51% (41/80) contained both the toxin and the immunity genes, 35% (28/80) only the killing gene and 14% (11/80) only the immunity gene, (figure 4; 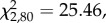 p = 2.96 × 10−6). The relatively high occurrence of only the killing gene occurs only in the nuclease S pyocins (S1, S2, S3 and AP41). This is surprising, because this would lead to the isolate killing itself, however, this is not phenotypically observed for any of the strains tested in the inhibition assays. Whereas the S5 pyocin, the only known pore-forming S toxin, which is therefore not lethal when expressed, occurs a total of 13 times across genotypes but is always present with the killing and immunity gene together.
p = 2.96 × 10−6). The relatively high occurrence of only the killing gene occurs only in the nuclease S pyocins (S1, S2, S3 and AP41). This is surprising, because this would lead to the isolate killing itself, however, this is not phenotypically observed for any of the strains tested in the inhibition assays. Whereas the S5 pyocin, the only known pore-forming S toxin, which is therefore not lethal when expressed, occurs a total of 13 times across genotypes but is always present with the killing and immunity gene together.
Figure 4.
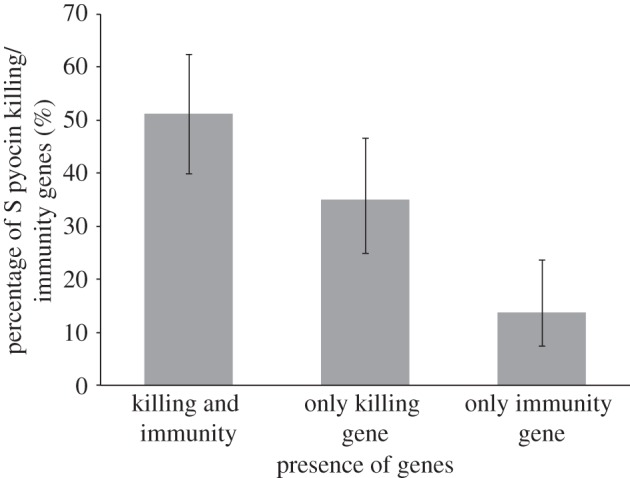
Soluble pyocin killing and immunity genes. Killing and immunity genes are found together (51%), or either the killing gene (35%) or the immunity gene (14%) alone. The error bars indicate ±95% CIs.
We examined the extent to which strains might have resistance mechanisms other than the specific immunity genes (electronic supplementary material, figure A5). We considered 877 strain interactions (competitions), including the isolates from the different stages of infection and the longitudinal isolates from the eight patients (collections 1 and 3). Examining our genetic data, we found that in 635 of these competitions, one strain carried at least one S-type pyocin that the other strain did not. In 33% (207/635) of these cases, we found that the toxin carrying strain was able to inhibit the growth of the other strain. In the 403 interactions where growth was not inhibited, we found that in 19% (77/403) of cases the strain not inhibited contained the immunity gene to the pyocin(s) being produced by the killing strain. In the remaining 326 cases (81%, 326/403), the strain did not have the immunity gene(s) and so must be using some alternative mechanism to resist the pyocin. Alternative resistance mechanisms protect strains significantly more frequently than the immunity gene (figure 5;  p < 2.2 × 10−16).
p < 2.2 × 10−16).
Figure 5.
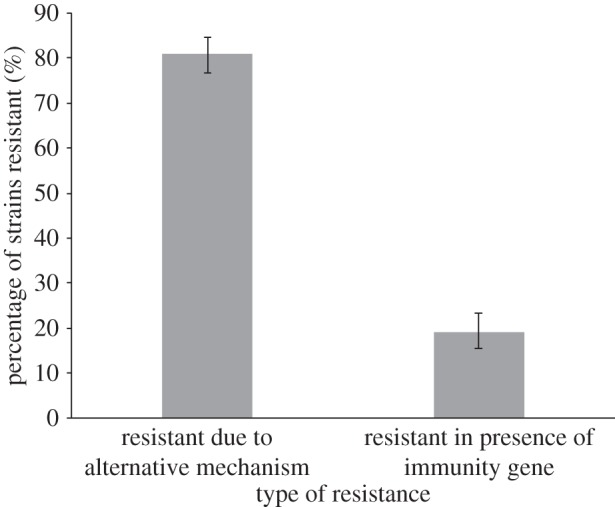
Protection from S pyocins by unknown resistance mechanisms. Considering cases where one strain does not carry a pyocin type carried by another (403 strain pairs), resistance via some other mechanism is much more common (81%) than resistance likely due to the immunity gene (19%). The error bars indicate ±95% CIs with correction for continuity around proportion values.
4. Discussion
We found that, in P. aeruginosa infections of the CF lung: (i) isolates from later infection stages caused less inhibition of other strains, were significantly more inhibited by the pyocins produced by other strains (figure 1) and carried a significantly lower diversity of pyocin types (figure 2); (ii) pyocin genes were not lost from a chronic transmissible strain over time; (iii) within infections, over time, there is no change in inhibition/inhibiting levels or pyocin diversity; (iv) pyocin inhibition profiles do not significantly explain which strains persist longer within lung infections (figure 3); (v) 35% of S pyocins occurred only as the killing gene, without the immunity gene to that pyocin (figure 4); (vi) in cases where one strain does not carry an S pyocin type carried by another, resistance via other mechanisms is much more common (81%) than resistance via the immunity gene (19%; figure 5).
(a). Bacteriocins and strain dominance
We found no support for the hypothesis that certain strains are able to achieve dominance in the CF lung through bacteriocins (figures 1 and 3). In fact, we found the opposite pattern, that more dominant/persistent strains, taken from later infection stages are less able to kill and more easily killed by other strains (figure 1). Our genetic analysis suggests that this pattern arises because isolates from later infections carry fewer pyocin genes (figure 2). This pattern could occur through strains losing pyocin genes overtime, or through strain competition, with more competitive strains producing less pyocins [26–30]. Our results suggest the latter with more competitive strains producing fewer pyocins (figure 2) rather than pyocin genes being lost from strains during infection.
Why do more dominant strains which cause chronic infections carry a lower diversity of pyocins (figure 2; [29])? A number of possible selective forces, which are not mutually exclusive, may be involved. Higher strain diversity, both within and across species, may favour a higher pyocin diversity required for competition during early stages of infection [19,31–37]. Another possibility is that higher pyocin diversity may be more costly in chronic infections if it leads to a higher antibiotic susceptibility—for example, expression of the R/F region increases strain susceptibility to fluoroquinolone antibiotics due to induction of the lysis system [38]. However, this does not explain maintenance of the F pyocin in the multiply chronic isolates of our collection, unless carrying either R or F rather than both, also makes the cell less inducible and therefore more resistant to antibiotics.
Within patient infections, strains show no competitive dominance as a result of bacteriocins and this is because any given cell is interacting more often with clonemates that share the same genotype. In which case the bacteriocins are useless. However, even when interacting with strains of different genotypes that are co-infecting, bacteriocins do not always explain strain persistence and dominance (figure 3). More often it is the strains that do no inhibit nor are inhibited by others that persist longest, implying that toxins are not useful in the CF lung environment. Despite this, strains retain a diversity of toxins, most probably as a result of adaptation to a range of environments they previously colonized.
It is important to note that it is possible that pyocin production can vary with the conditions in the CF lung that would show inhibition profiles different from our experiments. For example, stress-inducing conditions, such as anaerobic or oxidative stress, usually associated with pathogenesis in the lung, as well as mutagenic agents inducing production of all pyocin types but repressing expression of the S immunity [37–40]. It has also been speculated that the CF lung is an iron-limited environment [41], which can make cells more susceptible to S pyocin killing due to upregulated expression of ferri-pyoverdine and pyochelin receptors, which are required for iron uptake but also serve as receptors for S pyocins [2,42,43]. However, we note that our genetic analysis supports the general trends observed in the phenotype inhibition assays.
(b). Killing and immunity genes
We found that 35% of S pyocins are found as the killing gene without the corresponding immunity gene (figure 4). This is surprising because these are nuclease toxins, which suggests that these strains would kill themselves when expressed. However, we also found that strain resistance often occurs without the presence of the immunity gene, suggesting that whether or not you produce the specified immunity gene in P. aeruginosa is irrelevant. Considering cases where one strain does not carry a pyocin type carried by another, resistance via some other mechanism is much more common (81%) than resistance via the immunity gene (19%; figure 5). Possible alternative resistance mechanisms could be production of another ‘orphan’ immunity protein—as found in other pseudomonad species [44], that a secondary receptor is needed in addition to the primary receptor to allow entry of the pyocin into the cell or cells have mutations in or are lacking the primary S pyocin receptors [24,42,43].
We did not find evidence for bacteriocin-mediated competition explaining strain dynamics and/or producing RPS dynamics. Such dynamics are still expected even when there are multiple bacteriocin types in a population [21]. One possible explanation is that P. aeruginosa bacteriocins do not appear to involve single production/immunity genes, as we have shown immunity can occur via alternative mechanisms. This contrasts with E. coli where bacteriocins are neutralized by the immunity proteins [45]. Our results suggest that the outcome of strain competition is being driven primarily by other factors, which influence the ability to survive and grow in the CF lung. These could include susceptibility to antibiotics or attack from the immune system. Another explanation is that bacteriocins may be useless in the CF environment because they may not penetrate through the mucus layers and reach target cells, therefore, selecting against high-producing bacteriocin strains.
To conclude, we have found no evidence that killing by bacteriocins allows strains to outcompete other strains in the CF lung. Instead, we found the opposite result that multiply chronic Danish strains, which are better at surviving and persisting, as well as other epidemic CF strains carry fewer bactericions. Screening of bacteriocin profiles may therefore offer a method for predicting which infections are more likely to persist as chronic. A major task is to determine why lower bacteriocin diversity is correlated with the ability to persist in infections of the CF lung.
Supplementary Material
Acknowledgments
We thank Keith A. Jolley and James E. Bray for their help with uploading the genome sequences into the rMLST platform and training M.G. to use the BIGSdb software. We would also like to thank Craig Maclean, Steve Paterson, Jay Biernaski, Rene Niehus and Karl Heilbron for their useful comments.
Ethics
The use of samples was approved by the local ethics committee at the Capital Region of Denmark Region Hovedstaden: registration number H-1-2013-032. All patients have given informed consent. In patients under 18 years of age, informed consent was obtained from their parents. All patient samples were anonymized prior to any experimental use or analysis.
Competing interests
We declare we have no competing interests.
Funding
Funding was provided by the European Research Council (ERC), Royal Society and L'Oreal/UNESCO Fellowship for Women in Science to A.S.G.
References
- 1.Riley MA, Gordon DM. 1999. The ecological role of bacteriocins in bacterial competition. Trends Microbiol. 7, 129–133. ( 10.1016/S0966-842x(99)01459-6) [DOI] [PubMed] [Google Scholar]
- 2.Michel-Briand Y, Baysse C. 2002. The pyocins of Pseudomonas aeruginosa. Biochimie 84, 499–510. ( 10.1016/S0300-9084(02)01422-0) [DOI] [PubMed] [Google Scholar]
- 3.Riley MA, Wertz JE. 2002. Bacteriocins: evolution, ecology, and application. Annu. Rev. Microbiol. 56, 117–137. ( 10.1146/Annurev.Micro.56.012302.161024) [DOI] [PubMed] [Google Scholar]
- 4.Kerr B, Riley MA, Feldman MW, Bohannan BJM. 2002. Local dispersal promotes biodiversity in a real-life game of rock–paper–scissors. Nature 418, 171–174. ( 10.1038/Nature00823) [DOI] [PubMed] [Google Scholar]
- 5.Durrett R, Levin S. 1997. Allelopathy in spatially distributed populations. J. Theor. Biol. 185, 165–171. ( 10.1006/Jtbi.1996.0292) [DOI] [PubMed] [Google Scholar]
- 6.Kirkup BC, Riley MA. 2004. Antibiotic-mediated antagonism leads to a bacterial game of rock–paper–scissors in vivo. Nature 428, 412–414. ( 10.1038/nature02429) [DOI] [PubMed] [Google Scholar]
- 7.Parret AHA, De Mot R. 2002. Bacteria killing their own kind: novel bacteriocins of pseudomonas and other gamma-proteobacteria. Trends Microbiol. 10, 107–112. ( 10.1016/S0966-842X(02)02307-7) [DOI] [PubMed] [Google Scholar]
- 8.Heo YJ, Chung IY, Choi KB, Cho YH. 2007. R-type pyocin is required for competitive growth advantage between Pseudomonas aeruginosa strains. J. Microbiol. Biotechnol. 17, 180–185. [PubMed] [Google Scholar]
- 9.Kohler T, Donner V, van Delden C. 2010. Lipopolysaccharide as shield and receptor for R-pyocin-mediated killing in Pseudomonas aeruginosa. J. Bacteriol. 192, 1921–1928. ( 10.1128/Jb.01459-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uratani Y, Hoshino T. 1984. Pyocin-R1 inhibits active-transport in Pseudomonas aeruginosa and depolarizes membrane potential. J. Bacteriol. 157, 632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sano Y, Matsui H, Kobayashi M, Kageyama M. 1993. Molecular structures and functions of pyocin-S1 and pyocin-S2 in Pseudomonas aeruginosa. J. Bacteriol. 175, 2907–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sano Y, Kageyama M. 1993. A novel transposon-like structure carries the genes for pyocin-Ap41, a Pseudomonas aeruginosa bacteriocin with a DNase domain homology to E2 group colicins. Mol. Gen. Genet. 237, 161–170. [DOI] [PubMed] [Google Scholar]
- 13.Duport C, Baysse C, Michelbriand Y. 1995. Molecular characterization of pyocin S3, a novel S-type pyocin from Pseudomonas aeruginosa. J. Biol. Chem. 270, 8920–8927. ( 10.1074/jbc.270.15.8920) [DOI] [PubMed] [Google Scholar]
- 14.Fyfe JAM, Harris G, Govan JRW. 1984. Revised pyocin typing method for Pseudomonas aeruginosa. J. Clin. Microbiol. 20, 47–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scholl D, Cooley M, Williams SR, Gebhart D, Martin D, Bates A, Mandrell R. 2009. An engineered R-type pyocin is a highly specific and sensitive bactericidal agent for the food-borne pathogen Escherichia coli O157:H7. Antimicrob. Agents Ch. 53, 3074–3080. ( 10.1128/Aac.01660-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fegan M, Francis P, Hayward AC, Fuerst JA. 1991. Heterogeneity, persistence, and distribution of Pseudomonas aeruginosa genotypes in cystic fibrosis patients. J. Clin. Microbiol. 29, 2151–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Godard C, Plesiat P, Michel-Briand Y. 1993. Persistence of Pseudomonas aeruginosa strains in seven cystic fibrosis patients followed over 20 months. Eur. J. Med. 2, 117–120. [PubMed] [Google Scholar]
- 18.Marvig RL, Sommer LM, Molin S, Johansen HK. 2015. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 47, 57–64. ( 10.1038/ng.3148) [DOI] [PubMed] [Google Scholar]
- 19.Jelsbak L, et al. 2007. Molecular epidemiology and dynamics of Pseudomonas aeruginosa populations in lungs of cystic fibrosis patients. Infect. Immun. 75, 2214–2224. ( 10.1128/Iai.01282-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marvig RL, Johansen HK, Molin S, Jelsbak L. 2013. Genome analysis of a transmissible lineage of Pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet. 9, pArtn E1003741 ( 10.1371/journal.pgen.1003741) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biernaskie JM, Gardner A, West SA. 2013. Multicoloured greenbeards, bacteriocin diversity and the rock–paper–scissors game. J. Evol. Biol. 26, 2081–2094. ( 10.1111/Jeb.12222) [DOI] [PubMed] [Google Scholar]
- 22.Chao L, Levin BR. 1981. Structured habitats and the evolution of anticompetitor toxins in bacteria. Proc. Natl Acad. Sci. USA 78, 6324–6328. ( 10.1073/Pnas.78.10.6324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Czaran TL, Hoekstra RF, Pagie L. 2002. Chemical warfare between microbes promotes biodiversity. Proc. Natl Acad. Sci. USA 99, 786–790. ( 10.1073/Pnas.012399899) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denayer S, Matthijs S, Cornelis P. 2007. Pyocin S2 (Sa) kills Pseudomonas aeruginosa strains via the FpvA type I ferripyoverdine receptor. J. Bacteriol. 189, 7663–7668. ( 10.1128/Jb.00992-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jolley KA, Maiden MCJ. 2010. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 11 pArtn 595 ( 10.1186/1471-2105-11-595) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ernst RK, et al. 2003. Genome mosaicism is conserved but not unique in Pseudomonas aeruginosa isolates from the airways of young children with cystic fibrosis. Environ. Microbiol. 5, 1341–1349. ( 10.1046/J.1462-2920.2003.00518.X) [DOI] [PubMed] [Google Scholar]
- 27.Eberl L, Tummler B. 2004. Pseudomonas aeruginosa and Burkholderia cepacia in cystic fibrosis: genome evolution, interactions and adaptation. Int. J. Med. Microbiol. 294, 123–131. ( 10.1016/j.ijmm.2004.06.022) [DOI] [PubMed] [Google Scholar]
- 28.Romling U, Fiedler B, Bosshammer J, Grothues D, Greipel J, von der Hardt H, Tummler B. 1994. Epidemiology of chronic Pseudomonas aeruginosa infections in cystic fibrosis. J. Infectious Dis. 170, 1616–1621. ( 10.1093/infdis/170.6.1616) [DOI] [PubMed] [Google Scholar]
- 29.Tredgett MW, Doherty C, Govan JRW. 1990. Incidence of common pyocin types of Pseudomonas aeruginosa from patients with cystic fibrosis and chronic airways diseases. J. Med. Microbiol. 32, 169–172. ( 10.1099/00222615-32-3-169) [DOI] [PubMed] [Google Scholar]
- 30.Tummler B, Kiewitz C. 1999. Cystic fibrosis: an inherited susceptibility to bacterial respiratory infections. Mol. Med. Today 5, 351–358. ( 10.1016/S1357-4310(99)01506-3) [DOI] [PubMed] [Google Scholar]
- 31.Yang L, Jelsbak L, Molin S. 2011. Microbial ecology and adaptation in cystic fibrosis airways. Environ. Microbiol. 13, 1682–1689. ( 10.1111/J.1462-2920.2011.02459.X) [DOI] [PubMed] [Google Scholar]
- 32.Bakkal S, Robinson SM, Ordonez CL, Waltz DA, Riley MA. 2010. Role of bacteriocins in mediating interactions of bacterial isolates taken from cystic fibrosis patients. Microbiology 156, 2058–2067. ( 10.1099/Mic.0.036848-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blackwell CC, Winstanley FP, Brunton WAT. 1982. Sensitivity of thermophilic campylobacters to R-type pyocins of Pseudomonas aeruginosa. J. Med. Microbiol. 15, 247–251. ( 10.1099/00222615-15-2-247) [DOI] [PubMed] [Google Scholar]
- 34.Campagnari AA, Karalus R, Apicella M, Melaugh W, Lesse AJ, Gibson BW. 1994. Use of pyocin to select a Haemophilus ducreyi variant defective in lipooligosaccharide biosynthesis. Infect. Immun. 62, 2379–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morse SA, Jones BV, Lysko PG. 1980. Pyocin inhibition of Neisseria gonorrhoeae—mechanism of action. Antimicrob. Agents Ch. 18, 416–423. ( 10.1128/AAC.18.3.416) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams SR, Gebhart D, Martin DW, Scholl D. 2008. Retargeting R-type pyocins to generate novel bactericidal protein complexes. Appl. Environ. Microb. 74, 3868–3876. ( 10.1128/Aem.00141-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waite RD, Curtis MA. 2009. Pseudomonas aeruginosa PAO1 pyocin production affects population dynamics within mixed-culture biofilms. J. Bacteriol. 191, 1349–1354. ( 10.1128/Jb.01458-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brazas MD, Hancock REW. 2005. Ciprofloxacin induction of a susceptibility determinant in Pseudomonas aeruginosa. Antimicrob. Agents Ch. 49, 3222–3227. ( 10.1128/Aac.49.8.3222-3227.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang W, Small DA, Toghrol F, Bentley WE. 2005. Microarray analysis of Pseudomonas aeruginosa reveals induction of pyocin genes in response to hydrogen peroxide. BMC Genom. 6, pArtn 115 ( 10.1186/1471-2164-6-115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacob F. 1954. Biosynthèse induite et mode d'action d'une pyocine, antibiotique de Pseudomonas pyocyanea. Ann. I Pasteur Paris 86, 149–160. [PubMed] [Google Scholar]
- 41.Brown MRW, Anwar H, Lambert PA. 1984. Evidence that mucoid Pseudomonas aeruginosa in the cystic fibrosis lung grows under iron-restricted conditions. FEMS Microbiol. Lett. 21, 113–117. ( 10.1111/J.1574-6968.1984.Tb00195.X) [DOI] [Google Scholar]
- 42.Baysse C, Meyer JM, Plesiat P, Geoffroy V, Michel-Briand Y, Cornelis P. 1999. Uptake of pyocin S3 occurs through the outer membrane ferripyoverdine type II receptor of Pseudomonas aeruginosa. J. Bacteriol. 181, 3849–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elfarash A, Dingemans J, Ye L, Hassan AA, Craggs M, Reimmann C, Thomas MS, Cornelis P. 2014. Pore-forming pyocin S5 utilizes the FptA ferripyochelin receptor to kill Pseudomonas aeruginosa. Microbiology 160, 261–269. ( 10.1099/mic.0.070672-0) [DOI] [PubMed] [Google Scholar]
- 44.Ghequire MG, De Mot R. 2014. Ribosomally encoded antibacterial proteins and peptides from Pseudomonas. FEMS Microbiol. Rev. 38, 523–568. ( 10.1111/1574-6976.12079) [DOI] [PubMed] [Google Scholar]
- 45.Cascales E, Buchanan SK, Duche D, Kleanthous C, Lloubes R, Postle K, Riley M, Slatin S, Cavard D. 2007. Colicin biology. Microbiol. Mol. Biol. Rev. 71, 158–229. ( 10.1128/Mmbr.00036-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.