

Abstract
Tasmanian devil facial tumour disease (DFTD) is a clonally transmissible cancer threatening the Tasmanian devil (Sarcophilus harrisii) with extinction. Live cancer cells are the infectious agent, transmitted to new hosts when individuals bite each other. Over the 18 years since DFTD was first observed, distinct genetic and karyotypic sublineages have evolved. In this longitudinal study, we investigate the associations between tumour karyotype, epidemic patterns and host demographic response to the disease. Reduced host population effects and low DFTD infection rates were associated with high prevalence of tetraploid tumours. Subsequent replacement by a diploid variant of DFTD coincided with a rapid increase in disease prevalence, population decline and reduced mean age of the population. Our results suggest a role for tumour genetics in DFTD transmission dynamics and epidemic outcome. Future research, for this and other highly pathogenic emerging infectious diseases, should focus on understanding the evolution of host and pathogen genotypes, their effects on susceptibility and tolerance to infection, and their implications for designing novel genetic management strategies. This study provides evidence for a rapid localized lineage replacement occurring within a transmissible cancer epidemic and highlights the possibility that distinct DFTD genetic lineages may harbour traits that influence pathogen fitness.
Keywords: Tasmanian devil facial tumour disease, aneuploidy, cancer evolution, disease ecology, emerging infectious disease
1. Introduction
Pathogens are ubiquitous and important drivers of host population dynamics and evolution in natural populations [1–3]. While the ecology and population dynamics of host–pathogen systems are well characterized [1,2,4,5], understanding of evolutionary dynamics is limited by lack of empirical knowledge of the interplay between epidemic processes and host and pathogen genetics at individual and population levels [6]. The rate at which naive individuals adapt to new pathogens and whether host–pathogen interactions evolve towards commensalism remain unresolved questions in epidemiology. Virulence (pathogen-induced reduction in host fitness) is a fundamental trait that is frequently under strong selective pressure in a trade-off between host fitness and pathogen transmissibility [7,8]. Predicting pathogen virulence, evolutionary trajectories and how these influence transmission and epidemic outcomes in wild populations is a major scientific challenge for managing emerging wildlife diseases and for predicting conservation outcomes [6,9–11].
Cancers in wildlife are increasingly recognized as having substantial effects on host populations [12,13]. Directly transmissible cancers, in which the pathogen is a clonal infectious cell line spread through injurious contact [14], are particularly rare in nature. They are known only in the recently emerged Tasmanian devil facial tumour disease (DFTD) [15], which threatens its unique host, the Tasmanian devil (Sarcophilus harrisii), with extinction [16], a leukaemia-type disease affecting soft-shell clams in North America [17], and the much older (approx. 11 000 years) and evolutionarily stable canine transmissible venereal tumour (CTVT) [18]. The devil transmissible tumour is spread by biting [19], resulting in tumours inside and around the mouth, and is almost always fatal 6–12 months from the appearance of clinical signs [20]. Both DFTD and CTVT have evolved mechanisms of immune escape in allogeneic hosts, in part via epigenetic downregulation of major histocompatibility complex (MHC) gene expression in tumour cells [14,21].
We have reported reduced impacts of the disease at one study site, West Pencil Pine (hereafter WPP) in the northwest of Tasmania, compared with the impact of the disease observed at other sites [20]. The epidemiological pattern of DFTD at sites across eastern Tasmania, as the disease spread from its northeastern origin where it was first detected in 1996, is of a rapid increase in force of infection reaching a prevalence of more than 50% within 2–4 years after local epidemic outbreak [16]. This is accompanied by severe declines in individual survival rates and population growth rates, leading to regional-scale decline in population size of more than 90% [22], as well as collapse of age structure [23,24]. At WPP, disease prevalence remained low (less than 20% in adults; less than 10% in juveniles), there was no population decline, and age structure remained unaltered for up to 4 years following the outbreak [20]. Inexplicably, DFTD-infected individuals at WPP had higher survival rates than those in eastern populations [20].
We propose three possible explanations for this differing population response: (i) lower host susceptibility to DFTD at WPP compared with other sites, possibly conferred by host genetic factors or environmental variables; (ii) lower rate of injurious biting at WPP compared with other sites and/or differences in devil contact networks at WPP compared with other sites; and (iii) lower DFTD virulence at WPP compared with other sites due to differences in tumour genetics.
To explore the first hypothesis, we compared variation in MHC copy number among individuals with different disease status within the WPP population. Despite higher MHC copy number in devils in northwestern Tasmania than in eastern Tasmania [25], we found no differences in MHC copy number variation between individuals that acquired DFTD and those that never acquired DFTD in their lifetime [26]. Host resilience to infection at this site cannot be entirely rejected, however, given that MHC sequence diversity in DFTD-affected and -unaffected animals in the area has not yet been fully characterized. Furthermore, other non-MHC host genetic factors could influence susceptibility to infection. It is also possible that environmental variables, stress levels, parasite burden and co-infection with other pathogens may influence disease resilience in WPP devils.
To investigate the second hypothesis, we quantified intraspecific bite rates, a behaviour that could influence transmission, in WPP and in another DFTD-affected devil population in eastern Tasmania [19]. We did not observe any differences in bite rates across sites [19]. Variance among sites in social network dynamics and the implications of this for transmission are factors to be explored.
Our third hypothesis is that the reduced impacts of DFTD that we observed at WPP compared with other sites are explained by differences in the epidemiology and effects of the DFTD lineage found at WPP compared with DFTD lineages found at other sites. During the period in which we observed reduced disease impacts at WPP, the most prevalent DFTD lineage found at the site was tetraploid. Tetraploidy is reported to reduce cell proliferation and growth rates in tumours [27–29]. Reduced tumour growth rates could, in principle, offer a selective advantage to host and pathogen by increasing survival rates of infected individuals and thus lifetime transmission of DFTD. Over the course of its epidemic, DFTD has been evolving both genetically [30] and karyotypically, and local changes in ploidy have been observed [31]. Competition between tumour lineages is therefore expected to occur. The dominant tumour karyotype observed in most affected populations has been diploid, and tetraploid tumours have rarely been found to be prevalent at a host or tumour population level [31]. Diploid karyotypes have typified severely affected populations in eastern Tasmania early in the epidemic outbreak [16,31].
In this paper, we test for associations between the ploidy of facial tumours, epidemic dynamics and population effects at WPP. We describe our observation at WPP of the replacement of a tetraploid variant of DFTD with a diploid form, coinciding with a change of disease impact at the site. DFTD provides an opportunity to disentangle the interaction between competing pathogen phenotypes and their effects on infection and host population dynamics in a wild animal in the early epidemic stages of an emerging disease, and more specifically of a transmissible cancer. We discuss the implications of our results for managing emerging wildlife diseases in an adaptive framework.
2. Material and methods
(a). Field methods
Our study site, WPP, is a 25 km2 area of private forest production land in northwest Tasmania (41°31′ S, 145°46′ E). We have monitored this site since DFTD was first detected there in May 2006, at which time WPP was the known western epidemic extent. We have systematically sampled this population every three months, to coincide with devil life-history events: in February (juvenile weaning and dispersal; just prior to the mating season), in May (after the mating season), in August (females with young in the pouch) and in November (females in late lactation with young in dens). Trapping sessions are carried out with 40 traps set over 10 consecutive nights in a capture–mark–recapture framework. We use custom-built carnivore traps (constructed of 300 mm polypipe) baited with meat. Traps are checked daily starting at dawn.
All devils are permanently and individually identified with microchip transponders (Allflex NZ Ltd, Palmerstone North, New Zealand). Devils are aged using a combination of head width (a linear measure of body size), molar eruption, molar tooth wear and canine over-eruption (M.E.J. 2010, unpublished data), a method considered to be precise up to 3 years of age (wild devil lifespan is 6 years). In our longitudinal study, most individuals are captured as juveniles and are of definitively known age. DFTD status is categorized from histopathological confirmation of tumour biopsies, or when this is not possible, by the highest rank of confidence in visual examination and scoring of the tumour [32]. Fine needle aspirates from tumours are taken using 5 ml syringes and 23G needles. Live tumour cells are immediately transferred into liquid transport medium (RPMI 1640 and 10% fetal calf serum—Sigma-Aldrich 0.2 ml of penicillin–streptomycin solution and 2 µg ml−1 amphotericin B) and transported within 48 h to the Mt Pleasant Diagnostic Laboratories in Launceston, Tasmania for cell culture and karyotype analyses.
(b). Cell culture and cytogenetic analyses
Tumour cell culture and cytogenetic analyses were undertaken as described by Pearse et al. [31]. Fine needle aspirates were centrifuged to separate the tumour cells. Tissue biopsies were disaggregated under sterile conditions by first washing three times with 0.1 ml penicillin–streptomycin solution and 1 µg ml−1 amphotericin B in Dulbecco's phosphate-buffered saline (Invitrogen), then disaggregating in 3 ml of AmnioMax C-100 media with a scalpel blade, and finally by pushing remaining lumps through a 3 ml syringe with an 18G needle until a milky single-cell suspension was formed. The tumour cells were aliquoted into culture flasks containing 8 ml of AmnioMax C-100 cell culture medium (or RPMI 1640—Sigma-Aldrich with 10% fetal calf serum), 0.1 ml of penicillin–streptomycin solution and 1 µg ml−1 amphotericin B, and incubated at 35°C.
Representative aliquots of each sample were harvested after 24–48 h in culture. Before harvesting, 0.1 ml of 10 µg ml−1 demecolcine (Sigma-Aldrich) was added to each culture, and the flask was returned to the incubator (3 or 4 h for fast- or slow-growing cultures, respectively). The cells were then centrifuged for 10 min at 1000 r.p.m., and the cell pellet slowly resuspended in 7 ml of hypotonic 0.075 M KCl and placed in a water bath at 37°C for 18 min. Chilled Carnoy's fixative (2 ml; 3 : 1 ratio of methanol and acetic acid) was added, the tubes centrifuged for 10 min at 1000 r.p.m. and the pellet gently resuspended in fixative and stored at 4°C overnight. The following day, the cells were resuspended in four changes of fresh fixative. Finally, the cells were resuspended in fixative to a milky suspension. This suspension was dropped onto clean frozen microscope slides from 10 cm to ensure chromosome spread. Slides were allowed to dry and then placed in an oven at 57°C for 3 days before banding. G-banding was conducted by treating slides with a 0.15% solution of trypsin for up to 30 s before staining with Leishmann's stain for 2.5 min, then mounting with Leica mounting medium for analysis.
G-banding analysis was performed using a Leica DM 2000 microscope and photographed with a Leica DFC 420 C camera. Karyotypes were made, originally by hand and later (from 2008) using Karyo v. 3.1 software (VideoTesT Company). Tumours with at least 20 DFTD metaphases carrying 13–14 chromosomes were defined as diploid and those with at least 20 DFTD metaphases with 24–28 chromosomes were defined as tetraploid. At least 20 DFTD metaphases were analysed for each tumour. Tumours for which we had no data (not sampled due to logistic constraints) or with evidence of both diploid and tetraploid DFTD cells were classed as ‘unknown karyotype’.
(c). Statistical analyses
We used multinomial logit models [33] with a binomial error distribution to test whether prevalence of different DFTD karyotypes was a function of age class, trend (time since disease arrival), sex, and the interaction of age class and trend. There were insufficient data to include any interactions with sex; however, no study has previously reported an effect of sex in the epidemiology of DFTD or in the likelihood of infection [16,20,22]. Tumours of unknown karyotypes and those with evidence of both diploid and tetraploid karyotypes were pooled as there were insufficient data to model them separately, and animals were only included in the analysis on the first occasion that they were captured with a tumour—given that all tumours sampled more than once (11 tetraploid and 18 diploid) kept their original karyotype, subsequent recaptures were omitted. Multinomial models were fitted using the ‘multinom’ function implemented in ‘nnet’ package [34], with fitted values visualized using ‘ggplot2’ [35] within R v. 3.01.1 [36]. Effects were tested using likelihood ratios.
Changes in disease prevalence as a function of age class and trapping session were analysed using generalized linear mixed models with a logit link function, implemented in the function ‘lmer’ in R package ‘lme4’ [37].
Changes in age structure were analysed by modelling the proportion of independent and young devils (less than 3 years old) relative to older devils (age 3 years or older) in the population. We used generalized linear models with quasi-binomial error distribution, seasonality as a factor and trend (time since DFTD arrival) as a continuous predictor variable.
Adult population size for each field trip was estimated in a capture–mark–recapture framework applying open population models (POPAN) with constant recapture rates, time variation in survival rates and probability on entry (PENT) parameters, using the program MARK [38] implemented in the ‘RMark’ package in R. We excluded data from February trapping sessions, when large numbers of recently weaned and dispersing juveniles decreased the probability of capturing adults. Differences in the rate of population change with time and progression of the DFTD epidemic were analysed using linear models in which season was a factor, time since DFTD arrival was a continuous predictor variable, and the response (estimated population size) was weighted by the inverse of its squared standard error.
3. Results
The probability of becoming diseased remained low in WPP for the first 5 years following DFTD outbreak; at this time, the tetraploid DFTD karyotype was prevalent in the population. Probability of becoming diseased increased significantly after the diploid karyotype appeared in the sixth and seventh year after disease outbreak (figures 1 and 2a–c). Multinomial modelling showed that sex was not needed in the model (χ22 = 3.25, p
= 0.20), but time since disease arrival interacted with age class (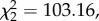 p
= 0.03), which showed that the profile of probabilities of becoming diseased changed over time but with differing patterns for at least one age class (figure 1). The number of cases with unknown karyotypes decreased significantly with time. Among the cases of unknown karyotypes, 90.3% (n = 65) were tumours with no data available, whereas 9.7% (n = 7) were tumours with evidence of both diploid and tetraploid karyotypes. The tetraploid variant of DFTD was not observed in the population 6 years after disease arrival (figure 1).
p
= 0.03), which showed that the profile of probabilities of becoming diseased changed over time but with differing patterns for at least one age class (figure 1). The number of cases with unknown karyotypes decreased significantly with time. Among the cases of unknown karyotypes, 90.3% (n = 65) were tumours with no data available, whereas 9.7% (n = 7) were tumours with evidence of both diploid and tetraploid karyotypes. The tetraploid variant of DFTD was not observed in the population 6 years after disease arrival (figure 1).
Figure 1.

Multinomial logit model showing the probability of having each type of DFTD karyotype in the Tasmanian devil population as a function of age class and time since disease arrival at the WPP site, Tasmania. Dots above the line are raw data vertically jittered around p = 1, but not jittered around the values of times since disease arrival.
Figure 2.

DFTD prevalence and age structure of the Tasmanian devil population at WPP through time, before the replacement of predominant tumour karyotypes from tetraploid (2006–2011) to diploid (2012–2013) karyotypes (as indicated by the two bars below the x-axis). (a–c) Prevalence of DFTD in three age classes over time. Error bars are exact binomial confidence intervals and the dashed line represents the best fit of a logistic regression model. (d) Proportion of Tasmanian devils in four different age categories: 1 year (white), 2 years (grey), 3 years (brown) and 4+ years old (black). (Online version in colour.)
Disease prevalence remained low (less than 25% in 2–3 and 3+ year old devils; less than 10% in juveniles) during the first 5 years of the epidemic when the observed tumour lineage was predominantly tetraploid (figure 2a–c). Prevalence increased up to 80% in the adult age classes and to 20% in juveniles when the diploid form of DFTD replaced the tetraploid form in years six to seven (figure 2a–c). The final model included the additive effects of trend (time since disease arrival) and age class (see table 1 for parameter estimates). Overall, disease prevalence increased significantly with time since disease arrival (p ≤ 0.001) in all age classes.
Table 1.
Changes in disease prevalence. Parameter estimate values of the generalized linear mixed model for changes in DFTD prevalence at WPP. Age class effects are relative to 1–2-year-old devils and ‘trend’ is defined as time since DFTD arrival.
| parameter | estimate | s.e. | p-value |
|---|---|---|---|
| intercept | −6.64 | 0.44 | <0.001 |
| age. class 2–3 | 2.31 | 0.31 | <0.001 |
| age. class 3+ | 2.32 | 0.29 | <0.001 |
| trend | 0.67 | 0.06 | <0.001 |
Population age structure remained unaltered, with all age classes present in relatively similar proportions up to 5 years after disease outbreak (figure 2d). However, in the sixth year the population was composed mostly of 1–2-year-old devils (more than 80%); there were very few individuals more than 3 years old (10% of the population) and only 5% of the population was aged 4 years or older (figure 2d). By 2013, more than 80% of the population was 1–2 years old and there were no devils of 4 years or older. There was a significant effect of time since disease arrival (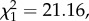 p
= 0.01; parameter estimate for the final model = 0.143, s.e. ± 0.05) in the proportion of young devils versus age 3 years or older in the population.
p
= 0.01; parameter estimate for the final model = 0.143, s.e. ± 0.05) in the proportion of young devils versus age 3 years or older in the population.
Adult population size remained stable during the first 5 years, but started to decline during the sixth and seventh years after DFTD outbreak, coinciding with the change of dominant DFTD karyotype, from tetraploid to diploid (figure 3). There was a significant effect of time since disease arrival (p ≤ 0.001; parameter estimate for the final model = −10.04, s.e. ± 1.48). There was no effect of season in the rate of change in population size (F2,denomd.f. = 0.107, p = 0.898).
Figure 3.
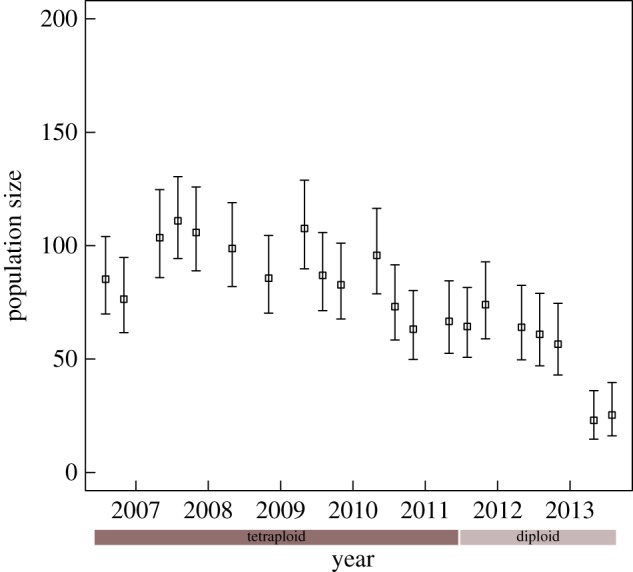
Population size estimates (POPAN) for adult Tasmanian devils at the WPP study site in Tasmania since the beginning of the epidemic outbreak. The timing of replacement of the dominant karyotypes is indicated by the two bars below the x-axis. Error bars are 95% confidence intervals. (Online version in colour.)
4. Discussion
We report the replacement of a tetraploid lineage of a transmissible cancer, DFTD, with a diploid form, coincident with a significant change in epidemic dynamics and host population effects. At our WPP study site in northwest Tasmania, the tetraploid tumour was associated with a low force of infection and no changes in population size or age structure in the first 5 years after disease outbreak. Replacement of the tetraploid with a diploid tumour form occurred rapidly, over a 12-month period, and coincided with a significant increase in disease prevalence, as well as major population decline and reduction of mean age in the subsequent year. The greatest impacts were on mature adults, which are more likely to engage in biting activity [19], consistent with DFTD impacts at other sites [16,20,22].
Lineage replacement through time involving ploidy has been reported at one site in Tasmania [30], with other examples not well documented spatially or temporally [30,31]. Lineage replacement can be attributed to two hypotheses: local transformation in ploidy within the host (in situ evolutionary transformation), or replacement of the tetraploid lineage with the arrival of a diploid lineage from adjacent populations (geographical spread of karyotypes). Although we cannot at present distinguish between these possibilities, we note that the diploid form of DFTD is the most common karyotype in surrounding areas of Tasmania, and thus migration from adjacent populations is a plausible origin for the diploid DFTD lineage in WPP. Moreover, if diploid tumours did indeed evolve in situ at WPP, it is likely, given the complexity of chromosomal changes that must be acquired for a cancer cell to transform from tetraploidy to diploidy, that the diploid form of DFTD in WPP has a clonal origin or arrived from adjacent populations. In addition, we have not seen any tumours change in ploidy at an individual host level in our dataset (all tumours sampled more than once have kept their original karyotype; 11 tetraploid, 18 diploid tumours). The hypothesis of geographical spread of a diploid karyotype from adjacent populations is therefore the most parsimonious explanation. The association of a rapid switch in ploidy with changes in disease epidemiological parameters suggests that the diploid lineage may have a selective advantage, although we cannot rule out the possibility that this strain became more prevalent through neutral drift or that the patterns that we are seeing relate to other attributes of the tumour that are correlated with ploidy. Plausible causal mechanisms based on lineage competition include links between tumour ploidy, virulence and total transmission.
Mechanisms that could alter the fitness of tumour lineages are grounded in the trade-off model of optimal pathogen virulence, which provides a useful theoretical framework to understand how competing pathogen strains can interact and shape epidemiological processes [8,39]. This model proposes that pathogens with reduced virulence (such as a slower-growing tetraploid DFTD tumour), which do not kill their hosts too fast and so increase the infective period, would be favoured because they can achieve greater total transmission over the host's lifespan. That our results contradict the expectations of this model is interesting because we are studying a disease early in its evolutionary history and the disease is caused by a transmissible cancer, rather than a virus or protozoan. Our results suggest that both ploidy forms of DFTD are competing, but that other selective pressures, rather than slower epidemic impacts on the host influencing total pathogen transmission, may be more important. Given that at this early stage of the tumour–host interaction (7–8 years since epidemic outbreak) there are sufficient susceptible hosts in the population, and that devils do not have immunity to either initial or subsequent reinfections to DFTD, the less virulent form of DFTD would be expected to be replaced by the more virulent in the time frame of our study (8 years). This pattern of ploidy in relation to time since disease outbreak is reflected throughout Tasmania, as diploid forms of DFTD are usually found at sites 2–4 years after disease arrival where severe population declines have resulted, whereas tetraploid tumours are uncommon and spatially sporadic. Therefore, virulent genetic lineages of DFTD could continue to be selected for even after severe population declines and at low population densities. On the other hand, host selective adaptations towards higher tolerance to infection are expected to occur in long-term DFTD-affected populations. Transmissible cancers are rare (only three cases in nature: DFTD, emerged almost 20 years ago; CTVT, emerged at least 10 000 years ago [18]; and the recently emerged leukaemia-like cancer in soft-shell clams [17]), and there is much to learn about early and long-term selective pressures on virulence (reduction in host fitness) and malignancy (metastasis, genomic instability) that may be better explored within a genomic sequencing framework.
Although we have found evidence for a DFTD lineage replacement at WPP that correlates with changing epidemic patterns, we cannot discount the possibility that host genetic or environmental factors may have influenced the tumour dynamics observed at our site. Although Lane et al. [26] found no association between MHC copy number and individual disease history at WPP, it is possible that other genetic or environmental factors may influence susceptibility to infection and/or tolerance to the disease. Thus, it may be that at the WPP site a higher force of infection is required to establish the infection patterns and DFTD impact on hosts that we have observed at other sites. We also cannot discount differences in social contacts as a causal explanation for the different epidemic patterns observed at WPP compared with other sites, although the fact that we did not observe any difference in rates of biting injuries, a key component of DFTD transmission, between WPP (tetraploid tumours, low population effects) and another site in northern Tasmania that was severely affected by DFTD from disease outbreak (Wisedale, diploid tumour, rapid population decline) [19] makes this explanation less likely. To further elucidate the role of social contacts and biting behaviour in transmission dynamics and population effects of DFTD requires longitudinal study of social contacts and biting rates in diseased and non-diseased populations.
Predicting the evolutionary trajectory of pathogen virulence, host tolerance to disease and epidemic outcomes in wild populations is central to predicting conservation outcomes and for managing emerging wildlife diseases [6,10,39,40]. Local adaptation is expected, driven by coevolutionary processes involved in transmissibility and virulence [41]. When the pathogen is a transmissible cancer, we can expect variation, competition and selection of tumour phenotypes in time and space, given the stochasticity in chromosomal segregation errors and the nature by which aneuploid karyotypes are formed [29]. Our results from WPP suggest that tumour variants may shape epidemic patterns in the wild, and therefore potentially the conservation outcome of DFTD for wild devil populations. Previous studies, including epidemiological models of the Tasmanian devil–DFTD system, suggest that host–pathogen interactions are unlikely to evolve towards endemicity and long-term coexistence [42]. However, these devil–DFTD models assumed constant values for key epidemiological parameters such as transmissibility and disease-induced mortality with each generation of infection. Future research should include tumour lineage dynamics within an evolutionary epidemiological analytical framework, as well as assessing variation in host tolerance to different DFTD genetic variants under different ecological or immunological conditions.
Transmissible clonal cancers in wildlife have only recently been recognized as a threat to biodiversity conservation [12,43], including both direct effects on species decline and the trophic cascades that result from the loss of keystone species and ecological functions, such as is occurring in Tasmania with the severe decline of its top mammalian predator [44]. The Tasmanian devil's facial tumour disease is a new transmissible cancer that we have studied in the wild almost since its emergence. This host–pathogen system promises to reveal new insights into the evolutionary ecology of cancers and, more generally, of emerging infectious wildlife diseases.
Acknowledgements
We are grateful to Mark and Claire Walsh from Discovery Holiday Parks—Cradle Mountain for providing accommodation and logistic support during fieldwork. Gunns Ltd. facilitated land access during our study. We thank Sarah Peck for veterinary support in the field, Bobby Hua for technical and logistical support during fieldwork, and a large number of volunteers who helped to collect data. The Save the Tasmanian Devil Program of the Tasmanian Department of Primary Industries, Parks, Water and the Environment provided in-kind logistic support.
Ethics
This research was carried out with approval from the University of Tasmania's Animal Ethics Committee (A0013326).
Data accessibility
All data are available on request from R.K.H. at rkhamede@utas.edu.au.
Authors' contributions
R.K.H. carried out fieldwork and statistical analyses, designed the study and drafted the manuscript. A.-M.P. and K.S. performed cell culture in the laboratory and cytogenetic analyses. L.A.B. helped with statistical analyses and interpretation of data. E.P.M. assisted with interpretation of results and helped draft the manuscript. M.E.J. participated in the study design, data analyses and interpretation of results, and helped draft the manuscript. All authors gave final approval for publication.
Competing interests
We declare we have no competing interests
Funding
This research project was funded by an ARC Discovery grant (DP110102656) to M.E.J., an Eric Guiler Tasmanian Devil Research Grant through the Save the Tasmanian Devil Appeal of the University of Tasmania Foundation to M.E.J. and R.K.H., and a National Science Foundation grant to M.E.J. (NSF DEB-1316549). M.E.J. was supported by an ARC Future Fellowship (FT100100250).
References
- 1.Anderson RM, May RM. 1979. Population biology of infectious diseases: part I. Nature 280, 361–367. ( 10.1038/280361a0) [DOI] [PubMed] [Google Scholar]
- 2.May RM, Anderson RM. 1979. Population biology of infectious diseases: part II. Nature 280, 455–461. ( 10.1038/280455a0) [DOI] [PubMed] [Google Scholar]
- 3.Andre JB, Ferdy JB, Godelle B. 2003. Within-host parasite dynamics, emerging trade-off, and evolution of virulence with immune system. Evolution 57, 1489–1497. ( 10.1554/02-667) [DOI] [PubMed] [Google Scholar]
- 4.Grenfell BT, Dobson AP. 1995. Ecology of infectious diseases in natural populations. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 5.Hudson PJ. 2002. The ecology of wildlife diseases. New York, NY: Oxford University Press. [Google Scholar]
- 6.Grenfell BT, Pybus OG, Gog JR, Wood JL, Daly JM, Mumford JA, Holmes EC. 2004. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303, 327–332. ( 10.1126/science.1090727) [DOI] [PubMed] [Google Scholar]
- 7.Andre JB, Hochberg ME. 2005. Virulence evolution in emerging infectious diseases. Evolution 59, 1406–1412. ( 10.1111/j.0014-3820.2005.tb01791.x) [DOI] [PubMed] [Google Scholar]
- 8.Ebert D, Bull JJ. 2003. Challenging the trade-off model for the evolution of virulence: is virulence management feasible? Trends Microbiol. 11, 15–20. ( 10.1016/s0966-842x(02)00003-3) [DOI] [PubMed] [Google Scholar]
- 9.Rigaud T, Perrot-Minnot MJ, Brown MJF. 2010. Parasite and host assemblages: embracing the reality will improve our knowledge of parasite transmission and virulence. Proc. R. Soc. B 277, 3693–3702. ( 10.1098/rspb.2010.1163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alizon S, de Roode JC, Michalakis Y. 2013. Multiple infections and the evolution of virulence. Ecol. Lett. 16, 556–567. ( 10.1111/ele.12076) [DOI] [PubMed] [Google Scholar]
- 11.Galvani AP. 2003. Epidemiology meets evolutionary ecology. Trends Ecol. Evol. 18, 132–139. ( 10.1016/s0169-5347(02)00050-2) [DOI] [Google Scholar]
- 12.McAloose D, Newton AL. 2009. Wildlife cancer: a conservation perspective. Nat. Rev. Cancer 9, 517–526. ( 10.1038/nrc2665) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vittecoq M et al. . 2013. Cancer: a missing link in ecosystem functioning? Trends Ecol. Evol. 28, 628–635. ( 10.1016/j.tree.2013.07.005) [DOI] [PubMed] [Google Scholar]
- 14.Murchison EP. 2009. Clonally transmissible cancers in dogs and Tasmanian devils. Oncogene 27, S19–S30. ( 10.1038/onc.2009.350) [DOI] [PubMed] [Google Scholar]
- 15.Pearse AM, Swift K. 2006. Allograft theory: transmission of devil facial-tumour disease. Nature 439, 549 ( 10.1038/439549a) [DOI] [PubMed] [Google Scholar]
- 16.McCallum H, Jones M, Hawkins C, Hamede R, Lachish S, Sinn DL, Beeton N, Lazenby B. 2009. Transmission dynamics of Tasmanian devil facial tumor disease may lead to disease-induced extinction. Ecology 90, 3379–3392. ( 10.1890/08-1763.1) [DOI] [PubMed] [Google Scholar]
- 17.Metzger MJ, Reinisch C, Sherry J, Goff SP. 2015. Horizontal transmission of clonal cancer cells causes leukemia in soft-shell clams. Cell 161, 255–263. ( 10.1016/j.cell.2015.02.042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murchison EP, et al. 2014. Transmissable dog cancer genome reveals the origin and history of an ancient cell lineage. Science 343, 437–440. ( 10.1126/science.1247167) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamede RK, McCallum H, Jones M. 2013. Biting injuries and transmission of Tasmanian devil facial tumour disease. J. Anim. Ecol. 82, 182–190. ( 10.1111/j.1365-2656.2012.02025.x) [DOI] [PubMed] [Google Scholar]
- 20.Hamede R, Lachish S, Belov K, Woods G, Kreiss A, Pearse AM, Lazenby B, Jones M, McCallum H. 2012. Reduced effect of Tasmanian devil facial tumor disease at the disease front. Conserv. Biol. J. Soc. Conserv. Biol. 26, 124–134. ( 10.1111/j.1523-1739.2011.01747.x) [DOI] [PubMed] [Google Scholar]
- 21.Siddle HV, et al. 2013. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc. Natl Acad. Sci. USA 110, 5103–5108. ( 10.1073/pnas.1219920110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lachish S, Jones M, McCallum H. 2007. The impact of disease on the survival and population growth rate of the Tasmanian devil. J. Anim. Ecol. 76, 926–936. ( 10.1111/j.1365-2656.2007.01272.x) [DOI] [PubMed] [Google Scholar]
- 23.Jones ME, Cockburn A, Hamede R, Hawkins C, Hesterman H, Lachish S, Mann D, McCallum H, Pemberton D. 2008. Life-history change in disease-ravaged Tasmanian devil populations. Proc. Natl Acad. Sci. USA 105, 10 023–10 027. ( 10.1073/pnas.0711236105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lachish S, McCallum H, Jones M. 2009. Demography, disease and the devil: life-history changes in a disease-affected population of Tasmanian devils (Sarcophilus harrisii). J. Anim. Ecol. 78, 427–436. ( 10.1111/j.1365-2656.2008.01494.x) [DOI] [PubMed] [Google Scholar]
- 25.Siddle HV, Marzec J, Cheng Y, Jones M, Belov K. 2010. MHC gene copy number variation in Tasmanian devils: implications for the spread of a contagious cancer. Proc. R. Soc. B 277, 2001–2006. ( 10.1098/rspb.2009.2362) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lane A, Cheng Y, Wright B, Hamede R, Levan L, Jones M, Ujvari B, Belov K. 2012. New insights into the role of MHC diversity in devil facial tumour disease. PLoS ONE 7, e36955 ( 10.1371/journal.pone.0036955) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Storchova Z, Pellman D. 2004. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 5, 45–54. ( 10.1038/nrm1276) [DOI] [PubMed] [Google Scholar]
- 28.Storchova Z, Kuffer C. 2008. The consequences of tetraploidy and aneuploidy. J. Cell Sci. 121, 3859–3866. ( 10.1242/jcs.039537) [DOI] [PubMed] [Google Scholar]
- 29.Pavelka N, Rancati G. 2013. Never in neutral: a systems biology and evolutionary perspective on how aneuploidy contributes to human diseases. Cytogenet. Genome Res. 139, 193–205. ( 10.1159/000348303) [DOI] [PubMed] [Google Scholar]
- 30.Murchison EP, et al. 2012. Genome sequencing and analysis of the Tasmanian devil and its transmissible cancer. Cell 148, 780–791. ( 10.1016/j.cell.2011.11.065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pearse AM, Swift K, Hodson P, Hua B, McCallum H, Pyecroft S, Taylor R, Eldridge MD, Belov K. 2012. Evolution in a transmissible cancer: a study of the chromosomal changes in devil facial tumor (DFT) as it spreads through the wild Tasmanian devil population. Cancer Genet. 205, 101–112. ( 10.1016/j.cancergen.2011.12.001) [DOI] [PubMed] [Google Scholar]
- 32.Hawkins CE, et al. 2006. Emerging disease and population decline of an island endemic, the Tasmanian devil Sarcophilus harrisii. Biol. Conserv. 131, 307–324. ( 10.1016/j.biocon.2006.04.010) [DOI] [Google Scholar]
- 33.Faraway J. 2006. Extending the linear model with R: generalised linear, mixed effects and nonparametric regression models. London, UK: Chapman & Hall. [Google Scholar]
- 34.Venables W. 2002. Modern applied statistics with S, 4th edn New York, NY: Springer. [Google Scholar]
- 35.Wickham H. 2009. ggplot2: elegant graphics for data analysis. Berlin, Germany: Springer. [Google Scholar]
- 36.R Core Team. 2013. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 37.Bates D, Maechler M, Bolker B. 2013. lme4: linear mixed-effects models using S4 classes. R package v. 0.999999–2. See https://cran.r-project.org/web/packages/lme4/index.html.
- 38.Cooch E, White G. 2002. Program MARK: a gentle introduction. See http://www.phidot.org/software/mark/docs/book.
- 39.Carval D, Ferriere R. 2010. A unified model for the coevolution of resistance, tolerance, and virulence. Evolution 64, 2988–3009. ( 10.1111/j.1558-5646.2010.01035.x) [DOI] [PubMed] [Google Scholar]
- 40.Venesky MD, Mendelson Iii JR, Sears BF, Stiling P, Rohr JR. 2012. Selecting for tolerance against pathogens and herbivores to enhance success of reintroduction and translocation. Conserv. Biol. 26, 586–592. ( 10.1111/j.1523-1739.2012.01854.x) [DOI] [PubMed] [Google Scholar]
- 41.Dybdahl MF, Storfer A. 2003. Parasite local adaptation: Red Queen versus Suicide King. Trends Ecol. Evol. 18, 523–530. ( 10.1016/s0169-5347(03)00223-4) [DOI] [Google Scholar]
- 42.Hamede R, Bashford J, Jones M, McCallum H. 2012. Simulating devil facial tumour disease outbreaks across empirically derived contact networks. J. Appl. Ecol. 49, 447–456. ( 10.1111/j.1365-2664.2011.02103.x) [DOI] [Google Scholar]
- 43.McCallum H, Jones ME. 2012. Infectious cancer in wildlife. In New directions in Conservation Medicine: applied cases of ecological health (eds Aguirre A, Daszak P, Ostfeld RS), pp. 270–283. Oxford, UK: Oxford University Press. [Google Scholar]
- 44.Hollings T, Jones M, Mooney N, McCallum H. 2014. Trophic cascades following the disease-induced decline of an apex predator, the Tasmanian devil. Conserv. Biol. 28, 63–75. ( 10.1111/cobi.12152) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available on request from R.K.H. at rkhamede@utas.edu.au.